

Abstract
Pregnancy is a complex physiological state, in which the metabolism of endogenous as well as exogenous agents is ostensibly altered. One exogenous agent of concern is the hepatocarcinogen aflatoxin B1 (AFB1), a foodborne fungal toxin, that requires phase I metabolic oxidation for conversion to its toxic and carcinogenic form, the AFB1-8,9-exo-epoxide. The epoxide interacts with cellular targets causing toxicity and cell death; these targets include the covalent modification of DNA leading to mutations that can initiate malignant transformation. The main detoxification pathway of the AFB1-epoxide involves phase II metabolic enzymes including the glutathione-S-transferase (GST) family. Pregnancy can modulate both phase I and II metabolism and alter the biological potency of AFB1. The present work investigated the impact of pregnancy on AFB1 exposure in mice. A single IP dose of 6 mg/kg AFB1 was administered to pregnant C57BL/6 J mice at gestation day 14 and matched non-pregnant controls. Pregnant mice accumulated 2-fold higher AFB1-N7-guanine DNA adducts in the liver when compared with nonpregnant controls 6 h post-exposure. Enhanced DNA adduct formation in pregnant animals paralleled elevated hepatic protein expression of mouse CYP1A2 and mouse homologs of human CYP3A4, phase I enzymes capable of bioactivating AFB1. Although phase II enzymes GSTA1/2 showed decreased protein expression, GSTA3, the primary enzymatic protection against the AFB1-epoxide, was unaffected at the protein level. Taken together, our results reveal that pregnancy may constitute a critical window of susceptibility for maternal health, and provide insight into the biochemical factors that could explain the underlying risks.
Keywords: Aflatoxin B1, DNA adducts, early life exposure, maternal exposure, maternal fetal health axis
In excess of 750 000 people die of hepatocellular carcinoma (HCC) each year, making HCC the second leading cause of cancer deaths worldwide (Ferlay et al., 2013). Major risk factors for HCC include chronic infection with hepatitis B or C viruses and dietary exposures to aflatoxin B1 (AFB1) (WHO, 2013; Wild and Gong, 2010). Individual exposure to AFB1 or HBV results in a human liver cancer risk enhancement of 3.4- and 7-fold, respectively (Kensler et al., 2011; Qian et al., 1994); that risk rises to 60-fold when both factors are present concurrently. The International Agency for Research on Cancer classifies AFB1 as a Group 1 agent—known human carcinogen—based on evidence from studies of AFB1-treated animals, epidemiological studies in exposed populations and studies involving functional biomarkers that mechanistically predict disease (IARC, 1993).
The aflatoxins are a group of mycotoxins produced by the fungi Aspergillus flavus and Aspergillus parasiticus (Moss, 1998) that readily contaminate many staple foods including nuts, corn and cereals. Contamination occurs most frequently in developing countries with tropical and subtropical climates, where food storage and quality control practices are inadequate (Strosnider et al., 2006). Up to 4.5 billion people are believed to be at risk of aflatoxin exposure (Liu and Wu, 2010). Among the aflatoxins, AFB1 is the most common and most toxic variant (Edwards et al., 1971; Wogan et al., 1971). Its primary mechanism of toxicity and carcinogenicity is the bioactivation of AFB1 by phase I enzymes (ie, cytochromes P450) to a highly reactive epoxide (AFB1-8,9-exo-epoxide) that can covalently modify cellular components, including DNA. Epidemiological studies have identified AFB1-DNA adducts as biomarkers of AFB1 exposure, and predictors of HCC risk in humans (Egner et al., 2006; Groopman et al., 2008).
The B6C3F1 mouse remains a versatile and commonly deployed model for AFB1 exposure and carcinogenicity (Vesselinovitch et al., 1972). During the neonatal period, administration of a single dose of AFB1 results in 90–100% incidence of liver cancer in male B6C3F1 mice, whereas females have a 10% incidence (Chawanthayatham et al., 2015, 2017; Vesselinovitch et al., 1972; Wattanawaraporn et al., 2012). The biological basis for the differential AFB1 sensitivity between sexes is not completely understood. Previous work shows that both male and female neonates form similar levels of the strongly mutagenic AFB1-guanine adducts measured 6 h post-toxin administration, with both sexes demonstrating similar levels of characteristic AFB1 mutations at 10 weeks after dosing (Chawanthayatham et al., 2015; Wattanawaraporn et al., 2012; Woo et al., 2011). Similarly, the 10-week mutation frequencies of male and female mice treated in utero are identical (Chawanthayatham et al., 2015). These data suggest that postpubescent sex-specific biochemical factors may determine the differential sensitivity of males to AFB1-induced HCC.
In humans, AFB1 is metabolized by cytochromes P450 3A4 (CYP3A4) and 1A2 (CYP1A2), forming the AFB1-epoxide (Gallagher et al., 1994). Reaction of the epoxide with DNA produces a characteristic biomarker, the AFB1-N7-guanine adduct (Bennett et al., 1981; Croy and Wogan, 1981; Egner et al., 2006; Groopman et al., 1981). The AFB1-epoxide can be detoxified by the glutathione-S-transferases (GSTs), via conjugation to glutathione (GSH) (Figure 1) (Dohnal et al., 2014). The metabolic activation of AFB1 has not been fully elucidated in mice; specifically, studies that examine the particular P450 enzymes involved in bioactivation are lacking. Mice express an orthologue of human Cyp1A2, which ostensibly can oxidize AFB1 into the AFB1-epoxide, but they lack a direct orthologue of Cyp3A4. However, mice do have a plethora of potential functional homologs among their many metabolic cytochromes (Nelson et al., 2004). Indeed, a phylogenetic analysis reveals several members of the mouse CYP3A family that are closely related to human CYP3A4 (Supplementary Figure 1). The murine detoxification pathways for aflatoxin are better understood, with mouse GSTA3 being primarily responsible for the detoxification of the AFB1-epoxide (Ilic et al., 2010; Kensler et al., 2014). The initial period of sensitivity of neonatal mice to AFB1 is rapidly overcome via increasing the expression of GSTA3 in the postnatal period (Ilic et al., 2010; Shupe and Sell, 2004).
Figure 1.

Overview of AFB1 metabolism. AFB1 is metabolically activated by phase I enzymes, including cytochromes P450 CYP1A2 or CYP3A family members to the reactive AFB1 epoxide, which can damage DNA by forming the AFB1-N7-guanine covalent adduct. The main pathway to detoxify the epoxide involves the phase II enzymes GSTs, which chemically inactivate it by conjugation to GSH (Kensler et al., 2003).
Pregnancy alters the expression of metabolic enzymes, including cytochrome P450s and GSTs, that process endogenous and exogenous chemicals (Koh et al., 2011; Wen et al., 2013; Zhang et al., 2008). These metabolic changes may stem from pregnancy-induced alterations in the levels of female sex hormones such as estradiol, progesterone, and placental growth hormone (Selevan et al., 2000; Zhang et al., 2008). Therefore, in principle, pregnancy-associated metabolic changes could enhance bioactivation, decrease detoxification of xenobiotics, and thus compromise maternal health. To test that hypothesis, the present study evaluated the effects of pregnancy as a susceptibility factor to the toxicological impact of AFB1 exposure. We discovered that following single-dose AFB1 exposure, pregnant females demonstrated higher levels of AFB1-epoxide damage to DNA. The increased levels of DNA damage define pregnancy as a critical window of susceptibility to an important environmental carcinogen. These data also open the possibility that the more susceptible pregnant female may enable greater than expected levels of AFB1 exposure to the fetus, potentially increasing its life-long risk of developing cancer due to early life exposure.
MATERIALS AND METHODS
Chemicals
AFB1 (≥98% purity) and dimethylsulfoxide (DMSO; ≥99.9%) were purchased from Sigma-Aldrich Corp. (St Louis, Missouri). All other chemicals were at least of ACS reagent grade and obtained from commercial suppliers.
Animals
Female C57BL/6 gpt delta transgenic mice (a gift from Dr Nohmi et al., 1996) were mated with C3H/HeJ males purchased from the Jackson Laboratories (Bar Harbor, Maine). GPT mice were utilized in order to maintain congruency with previous studies within the lab, as well as enable future mutagenesis assays. Pregnant mice (n = 8) were administered AFB1 (6 mg/kg dissolved in 100 μl DMSO) on GD14 via IP injection. Age-matched nonpregnant C57BL/6 gpt delta mice were treated identically. Six hours after AFB1 administration, mice were sacrificed via CO2 inhalation; their livers were surgically extracted, snap frozen and stored at −80 °C. Liver tissue samples from AFB1-treated animals were subsequently used for DNA adduct analysis. Liver tissue samples from age-matched groups of pregnant and nonpregnant animals that were not treated with AFB1 were harvested similarly and used for transcript and protein analysis. All experiments were conducted in accordance with protocols approved by the MIT Committee on Animal Care.
DNA adduct analysis in liver tissues
DNA was isolated from liver tissue samples (n = 8) using previously described methods (Woo et al., 2011). AFB1-DNA adducts were released from DNA by hydrolysis in 1.0 N HCl at 95 °C for 15 min (Groopman et al., 1981). 15N5-Guanine-derived AFB1-adduct internal standards were added to the hydrolytically released AFB1-N7-guanine adducts to permit quantitative analysis by isotope dilution mass spectrometry. Prior to analysis by mass spectrometry, AFB1-DNA adduct mixtures were separated by ultra-high performance liquid chromatography (UPLC). The protonated parent ion of the AFB1-N7-guanine adduct (m/z 480.1) was selected and subjected to collision-induced fragmentation producing a m/z 152 product ion that was monitored to quantify adduct levels (Egner et al., 2006).
Western blot analysis
Liver tissue (n = 4) was homogenized in radio-immunoprecipitation assay buffer (RIPA, Santa Cruz Biotechnology, Dallas Texas), which was supplemented with protease inhibitors (phenylmethylsulfonyl fluoride (PMSF; 10 mM) and Leupeptin, (0.01 mg/ml). Laemmli buffer was added, followed by denaturation via boiling for 5 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to an Immobilon-P PVDF membrane (Millipore, Billerica, Massachusetts). Membranes were blocked with 5% BSA (Jackson Immuno-research, West Grove, Pennsylvania) in a Tris-buffered saline solution containing 0.1% Tween 20 followed by treatment with a primary antibody diluted in blocking solution. AntiGSTA3 was a kind gift from Dr John D. Hayes, University of Dundee, UK. AntiGSTA1/2 (No. ABS1651) was from Millipore, AntiCYP3A4 (No. 18227 1 AP0) was from Proteintech (Rosemont, Illinois). AntiCYP1A2 (No. AP11325c) was purchased from Abgent (San Diego, California). Antiβ-Actin (sc No. 69879) was from Santa Cruz Biotechnology, and antiGAPDH (No. NB300-221) was from Novus Biologicals (Littleton, Colorado). Immune complexes were allowed to react with the appropriate secondary antibodies conjugated to horseradish peroxidase (Cell Signaling No. 7074 S, Danvers, Massachusetts) and were visualized with enhanced chemiluminescence (Cell Signaling No. 6883P3).
Quantitative real-time PCR
Total RNA was extracted from snap frozen liver samples from untreated pregnant and non-pregnant mice using Tri reagent (Sigma Aldrich) (n = 6). RNA was purified using the RNeasy Mini Kit and treated with DNase to remove any potential genomic DNA contamination (Qiagen, Germantown, Maryland). RNA quantity and quality was assessed via spectrophotometric measurement at A260nm and A260nm/A280nm. cDNA was synthesized from 1 µg of RNA using the Quantitect reverse transcription kit (Qiagen). Primer sequences (see Supplementary Table 1) were obtained from PrimerBank or custom designed and ordered from Integrated DNA technologies (Coralville, Iowa) (Spandidos et al., 2010). qRT-PCR reactions were run in quadruplicate utilizing Quantitect SYBR green (Qiagen) on a Roche Light Cycler 480. The purity of PCR products was assessed by melting temperature and size determined by agarose gel. PCR efficiency was calculated by reference to a standard curve using serial sample dilutions. Relative transcript levels were determined using the Pfaffl (2001) method.
Statistical analyses
Student’s 2-tailed t tests were used to determine the significance of differences in DNA adduct levels, Western analyses of proteins and mRNA levels assayed by RT-PCR between experimental groups.
RESULTS
AFB1-DNA Adduct Formation Is Enhanced During Pregnancy
We used the AFB1-N7-guanine adduct as a biomarker to investigate the efficiency of both phase I and II metabolism of AFB1 in pregnant and nonpregnant mice (Figure 1). Six hours post AFB1 administration, mice were sacrificed and DNA was isolated from liver tissue. Subsequently, the AFB1-N7-guanine adduct levels were analyzed by quantitative isotope dilution mass spectrometry (Egner et al., 2006). We found that AFB1-N7-guanine adduct levels were approximately 2-fold greater in the livers of GD14 pregnant mice compared with adduct levels in nonpregnant mice (19.2 ± 4.22 pmol adduct/mg DNA vs 8.61 ± 0.70 pmol adduct/mg DNA; p = .027; Figure 2).
Figure 2.

Pregnancy confers an elevated risk of AFB1-induced DNA damage. AFB1-N7-Gua levels in liver DNA from nonpregnant (n = 8) and C57BL/6 J pregnant mice (n = 8). Mice treated with 6 mg kg−1 of AFB1 were sacrificed 6 h later for DNA adduct analysis. (*denotes p < .05). Plotted data are mean ± SEM.
Pregnancy Modulates the Expression of AFB1 Metabolizing Enzymes in a Manner That Favors Toxicity
Following the observation of a 2-fold increase in AFB1-N7-guanine adducts in the livers of pregnant over nonpregnant mice, we examined the gene expression of key metabolic enzymes in the aflatoxin activation and detoxification pathways. In untreated animals, phase I proteins that have been shown to be responsible for the bioactivation of AFB1 were found to have differentially increased expression in pregnant versus nonpregnant mice. Specifically, murine CYP(s) recognized by the antibody to human CYP3A4 showed a approximately 3-fold increase, and CYP1A2 showed a 5-fold increase (Figure 3). In our experiments below, we cannot be certain that the human antibody used detects specifically the corresponding mouse homolog; hence, we consider the murine response to a given human antibody (eg, human CYP3A4 or CYP1A2) to be taken with the caveat that the antibody may show unexpected crossreactivity. Given this operational definition, the observed upregulation of these CYPs, CYP3A4, and CYP1A2, may be responsible for the increased AFB1-N7-guanine adduct burden in pregnant mice. To further probe the regulation of CYPs during pregnancy we determined transcript levels via RT-PCR. No change was detected in the transcript levels of Cyp1a2 between the pregnant and nonpregnant mice (Figure 4). Because of uncertainty in the specificity of the human 3A4 antibody for mouse CYP3A family proteins, we performed a Protein Blast that identified 8 mouse CYPs having >70% homology to human CYP3A4. These CYP3A4 homologs were probed via RT-PCR, with Cyp3a57 showing a significant increase, and Cyp3a16 and Cyp3a44 showing trends of increased transcript expression in pregnant mice (Figure 4). GSTA3, the primary enzyme responsible for phase II metabolism of AFB1 in mice (Kensler et al., 2014; Van Ness et al., 1998), showed no significant change in expression between pregnant and nonpregnant animals at either protein (Figure 3) or transcript levels (Figure 3). However, we observed a 2-fold reduction in the protein levels of GSTA1/2, (Figure 3), with a concurrent reduction in transcript levels (Figure 4). This reduction in GSTA1/2 is consistent with a compromised ability to mitigate oxidative and other stresses, and could increase the half-life of the genotoxic AFB1 epoxide.
Figure 3.

Pregnancy yields an increase in expression of bioactivating enzymes of AFB1. Western blot analysis of representative phases I and II metabolic enzymes in untreated nonpregnant and GD14 pregnant mice (biological replicates shown for each condition). Fold change was measured via densitometry. Relative protein levels were normalized to the levels of β-actin and GAPDH. *denoted p < .05 (mean ± SEM; n = 4).
Figure 4.

Pregnancy alters expression of Cyp450 genes in the mouse. A host of phase I and II enzyme expression was analyzed via RT-PCR. A significant reduction in expression is present in GSTA1 (p < .05), with trends of increased expression for both Cyp3A13 and Cyp3A44. Relative transcript expression was normalized to GAPDH levels. *denoted p < .05 Data shown are mean ± SEM; n = 6.
DISCUSSION
Our study is consistent with the view that pregnancy can increase aflatoxin genotoxicity (increased AFB1 DNA adducts) by enhancing the metabolic activation of the toxin (Figure 5). In support of this notion, we found that pregnancy, specifically at GD14, was associated with significantly elevated AFB1 DNA adducts 6 h post-exposure. GD14 of pregnancy also demonstrated increased protein levels of the phase I metabolism enzymes CYP1A2 and mouse homologs of human CYP3A4, which are known to bioactivate AFB1. Additionally, pregnancy did not affect the levels of GSTA3, the native powerful countermeasure to the AFB1 epoxide. As a result, it is possible that the increase in metabolic activation afforded by the phase I enzymes shifts the equilibrium of AFB1 activation/detoxification in favor of activation, yielding an increase in the levels of DNA adducts. Moreover, the reduction in the protein levels of GSTA1/2 may further increase stress on the GSH peroxidase pathway, yielding a reduction in the intracellular reduced GSH pool. In turn, the decreased levels of reduced GSH may hinder the ability of GSTA3 to rapidly detoxify the AFB1 epoxide. Furthermore, diminished levels of GSTA1/2 could lead to greater susceptibility to other toxicological insults, including oxidative stress, altering cellular homeostasis to the detriment of efficient AFB1 detoxification. More research, however, is needed to fully understand the implications of decreased GSTA1/2 levels on the toxicological impact of AFB1.
Figure 5.
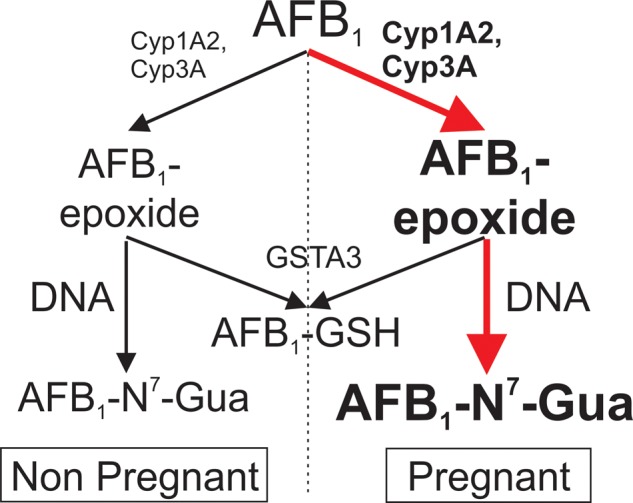
Pregnancy enhances the toxicological impact of AFB1. The observed increase in phase I metabolic enzymes (CYP1A2, CYP3A) in pregnant mice leads to increased levels of AFB1-epoxide, while amounts of the phase II detoxifying enzyme (GSTA3) remain unchanged. This effect results in enhanced genotoxic stress as measured by the levels of AFB1-N7-Gua adducts, a biomarker of AFB1 exposure.
AFB1 causes mutations in both male and female mice when administered either prenatally or in the early life stage. This exposure, however, is better tolerated in female mice, as demonstrated by lower rates of cancer incidence later in life when compared with males. Both sexes are resistant to the effects of AFB1 after reaching adulthood, withstanding doses of up to 60 mg/kg (Vesselinovitch et al., 1972; Wogan, 1969). This effect is due to the induction of expression of GSTA3 approximately one week after birth (Shupe and Sell, 2004). Similarly to aflatoxin, the DNA ethylating agent N-nitrosodiethylamine is a potent inducer of HCC in mice, with a sex bias that disproportionately favors female survival (Nakatani et al., 2001). Studies from the DEN literature show that estrogens inhibit production of inflammatory cytokines, such as interleukin-6 (IL-6) from Kupffer cells. Hence, in estrogen-producing females, a reduction in IL-6 decreases the inflammatory response in the liver following toxin exposure and thus could reduce liver cancer risk (Naugler et al., 2007). By contrast, male mice have lower estrogen levels, which translates to high levels of IL-6, which stimulates inflammation and leads to cytotoxicity as evidenced by elevated serum aminotransferases and apoptosis (Naugler et al., 2007). The proinflammatory environment acts as a tumor promoter, stimulating hepatocyte proliferation and leading to an increased incidence of HCC (Naugler et al., 2007). This phenotype may be mirrored in male mice exposed to aflatoxin early in life.
This work did not directly investigate the mechanistic connection between the pregnancy-related steroid hormones such as β-estradiol and progesterone and the genotoxic effects of aflatoxin but our results suggest that such studies represent a logical next step. The modulatory effects of pregnancy on metabolic enzymes have been examined previously. Wen and co-workers have demonstrated that pregnancy leads to alterations in phase II metabolism, specifically reductions in Uridine 5'-diphospho-glucuronosyltransferases at both the transcript and protein levels, as well as reductions in GSTA1 and GSTA4 transcripts at GD14 (Wen et al., 2013). Contrary to our results, they did not observe a reduction in GSTA1 protein levels.
Pregnancy-related hormones can act as transcriptional regulators of cytochrome P450 expression (Choi et al., 2013). Inprimary human hepatocyte cultures, estradiol enhances the expression of CYP3A4, CYP2A6, and CYP2B6, while progesterone induces the expression of CYP3A4, CYP2A6, CYP2B6, CYP2C8, and CYP3A5 (Choi et al., 2013). In mice, estradiol and glucocorticoid hormones can potentiate the effects of growth hormones on induction of homologs of CYP3A4 (CYP3A41 and CYP3A11) expression (Sakuma et al., 2004). In fact, increased protein expression and activity of CYP3A family members during pregnancy has been reported by several studies, with concurrent increased transcripts of Cyp3a16, Cyp3a41, and Cyp3a44 (Isoherranen and Thummel, 2013; Zhang et al., 2008). With regard to CYP1A2, the literature precedent is less definitive. One study, in which rats were exposed to estradiol, reported elevation of CYP1A2 expression, which is in agreement with the present study, and enhancement in the expression of CYP2C (CYP2C6, CYP2C7, and CYP2C12) (Choi et al., 2011). Other studies, which are at odds with ours, show that the hormones produced during pregnancy, including estrogens, progesterone and growth hormone, reduce the levels of CYP1A2 (Isoherranen and Thummel, 2013; Koh et al., 2011). Further work is needed to reconcile these divergent observations.
Pregnancy-dependent increased aflatoxin burden on the mother may have downstream consequences to fetal health and development. Studies have demonstrated aflatoxin as a transplacental xenobiotic both in mice (Chawanthayatham et al., 2015) and in humans (Abdulrazzaq et al., 2002; Denning et al., 1990; Hsieh and Hsieh, 1993). Although the exact method of bioactivation and fetal transmission is not fully elucidated, the developing fetus represents a critical window of susceptibility to AFB1. Our lab has demonstrated that in utero exposure to AFB1 leads to an increase in mutations (Chawanthayatham et al., 2015), and others have shown that humans exposed to AFB1in utero leads to reduced birthweight (Abdulrazzaq et al., 2002; Turner et al., 2007), alteration of methylation profiles of white blood cells (Hernandez-Vargas et al., 2015) and other effects.
In conclusion, our study establishes pregnancy as a risk factor for AFB1 genotoxicity; 6 h post-exposure to the toxin, the pregnant mice accumulate twice as many AFB1-N7-guanine adducts as the nonpregnant control group. This result may be partially explained by the increased levels of phase I metabolizing enzymes in pregnant animals, coupled with the lack of a compensatory change in the phase II enzymes (Figure 3). Further lines of research into the effects of chronic AFB1 exposure throughout pregnancy may reveal significantly greater impact on both maternal and fetal health.
Perhaps of greater importance is the revelation of pregnancy as a generalizable risk factor for toxicological impact. The observed reduction in the expression of a representative phase II enzyme, GSTA1/2, in pregnant animals versus nonpregnant controls suggests that pregnancy may confer increased risk to a host of endogenous and exogenous toxicants, in addition to AFB1. The genotoxic vulnerability conferred by pregnancy emphasizes the need not only to protect susceptible prenatal life, but also to lessen the exposure of the mother to environmental chemicals. Indeed, the knowledge of the need and subsequent interventions to protect a more vulnerable mother may reasonably offer the secondary benefit of protection to the fetus.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
This work was supported by the National Institutes of Health (T32-ES007020, P30-ES002109, R01-ES016313, R01-CA080024, P30-CA006973).
Supplementary Material
REFERENCES
- Abdulrazzaq Y. M., Osman N., Ibrahim A. (2002). Fetal exposure to aflatoxins in the United Arab Emirates. Ann. Trop. Paediatr. 22, 3–9. [DOI] [PubMed] [Google Scholar]
- Bennett R. A., Essigmann J. M., Wogan G. N. (1981). Excretion of an aflatoxin-guanine adduct in the urine of aflatoxin entreated rats. Cancer Res. 41, 650–654. [PubMed] [Google Scholar]
- Chawanthayatham S., Thiantanawat A., Egner P. A., Groopman J. D., Wogan G. N., Croy R. G., Essigmann J. M. (2015). Prenatal exposure of mice to the human liver carcinogen aflatoxin B1 reveals a critical window of susceptibility to genetic change. Int. J. Cancer 136, 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawanthayatham S., Valentine C. C. 3rd, Fedeles B. I., Fox E. J., Loeb L. A., Levine S. S., Slocum S. L., Wogan G. N., Croy R. G., Essigmann J. M. (2017). Mutational spectra of aflatoxin B1 invivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A. 114, E3101–E3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. Y., Fischer L., Yang K., Chung H., Jeong H. (2011). Isoform-specific regulation of cytochrome P450 expression and activity by estradiol in female rats. Biochem. Pharmacol. 81, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. Y., Koh K. H., Jeong H. (2013). Isoform-specific regulation of cytochromes P450 expression by estradiol and progesterone. Drug Metab. Dispos. 41, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croy R. G., Wogan G. N. (1981). Temporal patterns of covalent DNA adducts in rat liver after single and multiple doses of aflatoxin B1. Cancer Res. 41, 197–203. [PubMed] [Google Scholar]
- Denning D. W., Allen R., Wilkinson A. P., Morgan M. R. (1990). Transplacental transfer of aflatoxin in humans. Carcinogenesis 11, 1033–1035. [DOI] [PubMed] [Google Scholar]
- Dohnal V., Wu Q., Kuča K. (2014). Metabolism of aflatoxins: Key enzymes and interindividual as well as interspecies differences. Arch. Toxicol. 88, 1635–1644. [DOI] [PubMed] [Google Scholar]
- Edwards G. S., Wogan G. N., Sporn M. B., Pong R. S. (1971). Structure-activity relationships in DNA binding and nuclear effects of aflatoxin and analogs. Cancer Res. 31, 1943–1950. [PubMed] [Google Scholar]
- Egner P. A., Groopman J. D., Wang J. S. (2006). Quantification of aflatoxin-B1-N7-guanine in human urine by high-performance liquid chromatography and isotope dilution tandem mass spectrometry. Chem. Res. Toxicol. 19, 1191–1195. [DOI] [PubMed] [Google Scholar]
- Ferlay J., Soerjomataram I., Ervik M., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D. M., Forman D., Bray F. (2013). GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer; Available from: http://globocan.iarc.fr, last accessed Novemberon 13, 2015.
- Gallagher E., Wienkers L., Stapleton P., Kunze K., Eaton D. L. (1994). Role of human microsomal and human complementary DNA-expressed cytochromes P4501A2 and P4503A4 in the bioactivation of aflatoxin B1. Cancer Res. 54, 101–108. [PubMed] [Google Scholar]
- Groopman J. D., Croy R. G., Wogan G. N. (1981). In vitro reactions of aflatoxin B1-adducted DNA. Proc. Natl. Acad. Sci. U. S. A. 78, 5445–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groopman J. D., Kensler T. W., Wild C. P. (2008). Protective interventions to prevent aflatoxin-induced carcinogenesis in developing countries. Annu. Rev. Pub. Health 29, 187–203. [DOI] [PubMed] [Google Scholar]
- Hernandez-Vargas H., Castelino J., Silver M. J., Dominguez-Salas P., Cros M. P., Durand G., Le Calvez-Kelm F., Prentice A. M., Wild C. P., Moore S. E., et al. (2015). Exposure to aflatoxin B1 in utero is associated with DNA methylation in white blood cells of infants in the Gambia. Int. J. Epidemiol. 44, 1238–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh L. L., Hsieh T. T. (1993). Detection of aflatoxin B1-DNA adducts in human placenta and cord blood. Cancer Res. 53, 1278–1280. [PubMed] [Google Scholar]
- IARC (1993). Some naturally occuring substances: Food items and constituents heterocyclin aromatic amines and mycotoxins. IARC Monogr. Eval. Carcino. Risks Hum. 56, 245–540. [Google Scholar]
- Ilic Z., Crawford D., Vakharia D., Egner P. A., Sell S. (2010). Glutathione-S-transferase A3 knockout mice are sensitive to acute cytotoxic and genotoxic effects of aflatoxin B1. Toxicol. Appl. Pharmacol. 242, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoherranen N., Thummel K. E. (2013). Drug metabolism and transport during pregnancy: How does drug disposition change during pregnancy and what are the mechanisms that cause such changes?. Drug Metab. Dispos. 41, 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler K. H., Slocum S. L., Chartoumpekis D. V., Dolan P., Johnson N. M., Ilic Z., Crawford D., Sell S., Groopman J. D., Kensler T. W., et al. (2014). Genetic or pharmacologic activation of Nrf2 signaling fails to protect against aflatoxin genotoxicity in hypersensitive GSTA3 knockout mice. Toxicol. Sci. 139, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler T. W., Qian G. S., Chen J. G., Groopman J. D. (2003). Translational strategies for cancer prevention in liver. Nat. Rev. Cancer 3, 321–329. [DOI] [PubMed] [Google Scholar]
- Kensler T. W., Roebuck B. D., Wogan G. N., Groopman J. D. (2011). Aflatoxin: A 50-year odyssey of mechanistic and translational toxicology. Toxicol. Sci. 120, S28–S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K. H., Xie H., Yu A. M., Jeong H. (2011). Altered cytochrome P450 expression in mice during pregnancy. Drug Metab. Dispos. 39, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wu F. (2010). Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 118, 818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss M. O. (1998). Recent studies of mycotoxins. J. Appl. Microbiol. 84, 62S–76S. [DOI] [PubMed] [Google Scholar]
- Nakatani T., Roy G., Fujimoto N., Asahara T., Ito A. (2001). Sex hormone dependency of diethylnitrosamine-induced liver tumors in mice and chemoprevention by leuprorelin. Jpn. J. Cancer Res. 92, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler W. E., Sakura T., Kim S., Maeda S., Kim K., Elsharkawy A. M., Karin M. (2007). Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317, 121–124. [DOI] [PubMed] [Google Scholar]
- Nelson D. R., Zeldin D. C., Hoffman S. M., Maltais L. J., Wain H. M., Nebert D. W. (2004). Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14, 1–18. [DOI] [PubMed] [Google Scholar]
- Nohmi T., Katoh M., Suzuki H., Matsui M., Yamada M., Watanabe M., Suzuki M., Horiya N., Ueda O., Shibuya T., et al. (1996). A new transgenic mouse mutagenesis test system using Spi- and 6-thioguanine selections. Environ. Mol. Mutagen. 28, 465–470. [DOI] [PubMed] [Google Scholar]
- Pfaffl M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian G. S., Ross R. K., Yu M. C., Yuan J. M., Gao Y. T., Henderson B. E., Wogan G. N., Groopman J. D. (1994). A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People’s Republic of China. Cancer Epidemiol. Biomarkers. Prev. 3, 3–10. [PubMed] [Google Scholar]
- Sakuma T., Kitajima K., Nishiyama M., Endo Y., Miyauchi K., Jarukamjorn K., Nemoto N. (2004). Collaborated regulation of female-specific murine Cyp3A41 gene expression by growth and glucocorticoid hormones. Biochem. Biophys. Res. Commun. 314, 495–500. [DOI] [PubMed] [Google Scholar]
- Selevan S. G., Kimmel C. A., Mendola P. (2000). Identifying critical windows of exposure for children’s health. Environ. Health Perspect. 108(Suppl. 3), 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupe T., Sell S. (2004). Low hepatic glutathione S-transferase and increased hepatic DNA adduction contribute to increased tumorigenicity of aflatoxin B1 in newborn and partially hepatectomized mice. Toxicol. Lett. 148, 1–9. [DOI] [PubMed] [Google Scholar]
- Spandidos A., Wang X., Wang H., Seed B. (2010). PrimerBank: A resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 38(Database issue), D792–D799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strosnider H., Azziz-Baumgartner E., Banziger M., Bhat R. V., Breiman R., Brune M. N., DeCock K., Dilley A., Groopman J. D., Hell K., et al. (2006). Workgroup report: Public health strategies for reducing aflatoxin exposure in developing countries. Environ. Health Perspect. 114, 1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner P. C., Collinson A. C., Cheung Y. B., Gong Y., Hall A. J., Prentice A. M., Wild C. P. (2007). Aflatoxin exposure in utero causes growth faltering in Gambian infants. Int. J. Epidemiol. 36, 1119–1125. [DOI] [PubMed] [Google Scholar]
- Van Ness K. P., McHugh T. E., Bammler T. K., Eaton D. L. (1998). Identification of amino acid residues essential for high aflatoxin B1-8,9-epoxide conjugation activity in alpha class glutathione S-transferases through site-directed mutagenesis. Toxicol. Appl. Pharmacol. 152, 166–174. [DOI] [PubMed] [Google Scholar]
- Vesselinovitch S., Mihailovich N., Wogan G. N., Lombard L., Rao K. (1972). Aflatoxin B1, a hepatocarcinogen in the infant mouse. Cancer Res. 32, 2289–2291. [PubMed] [Google Scholar]
- Wattanawaraporn R., Woo L. L., Belanger C., Chang S. C., Adams J. E., Trudel L. J., Bouhenguel J. T., Egner P. A., Groopman J. D., Croy R. G., et al. (2012). A single neonatal exposure to aflatoxin B1 induces prolonged genetic damage in two loci of mouse liver. Toxicol. Sci. 128, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X., Donepudi A. C., Thomas P. E., Slitt A. L., King R. S., Aleksunes L. M. (2013). Regulation of hepatic phase II metabolism in pregnant mice. J. Pharmacol. Exp. Ther. 344, 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Mortality Database. http://www.who.int/healthinfo/mortality_data/en/. Last accessed December 30, 2015. [Google Scholar]
- Wild C. P., Gong Y. Y. (2010). Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 31, 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wogan G. N. (1969). Metabolism and biochemical effects of aflatoxins In Aflatoxin - Scientific Background Control and Implications, pp. 151–186. Academic Press, New York. [Google Scholar]
- Wogan G. N., Edwards G. S., Newberne P. M. (1971). Structure-activity relationships in toxicity and carcinogenicity of aflatoxins and analogs. Cancer Res. 31, 1936–1942. [PubMed] [Google Scholar]
- Woo L. L., Egner P. A., Belanger C. L., Wattanawaraporn R., Trudel L. J., Croy R. G., Groopman J. D., Essigmann J. M., Wogan G. N., Bouhenguel J. T. (2011). Aflatoxin B1-DNA adduct formation and mutagenicity in livers of neonatal male and female B6C3F1 mice. Toxicol. Sci. 122, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wu X., Wang H., Mikheev A. M., Mao Q., Unadkat J. D. (2008). Effect of pregnancy on cytochrome P450 3a and P-glycoprotein expression and activity in the mouse: Mechanisms, tissue specificity, and time course. Mol. Pharmacol. 74, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.