

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent pathological liver condition in developed countries. NAFLD results in severe alterations in liver function, including xenobiotic metabolism. Perchloroethylene (PERC) is a ubiquitous environmental pollutant, a known hepatotoxicant in rodents, and a probable human carcinogen. It is known that PERC disposition and metabolism are affected by NAFLD in mice; here, we examined how NAFLD changes PERC-associated liver effects. Male C57Bl6/J mice were fed a low-fat diet (LFD), high-fat diet (HFD), or methionine/folate/choline-deficient diet (MCD) to model a healthy liver, or mild and severe forms of NAFLD, respectively. After 8 weeks on diets, mice were orally administered PERC (300 mg/kg/day) or vehicle (5% aqueous Alkamuls-EL620) for 5 days. PERC-induced liver effects were exacerbated in both NAFLD groups. PERC exposure was associated with up-regulation of genes involved in xenobiotic, lipid, and glutathione metabolism, and down-regulation of the complement and coagulation cascades, regardless of the diet. Interestingly, HFD-fed mice, not MCD-fed mice, were generally more sensitive to PERC-induced liver effects. This was indicated by histopathology and transcriptional responses, where induction of genes associated with cell cycle and inflammation were prominent. Liver effects positively correlated with diet-specific differences in liver concentrations of PERC. We conclude that NAFLD alters the toxicodynamics of PERC and that NAFLD is a susceptibility factor that should be considered in future risk management decisions for PERC and other chlorinated solvents.
Keywords: toxicokinetics, toxicogenomics, mechanisms, liver, xenobiotic, steatosis, steatohepatitis
Inter-individual variability in susceptibility to toxicity associated with exposure to xenobiotics can be a consequence of multiple factors, such as genetics, age, sex, and pre-existing disease states. Nonalcoholic fatty liver disease (NAFLD) is estimated to affect nearly one-quarter of the global population (Younossi etal., 2016b) and its associated health care costs are a considerable financial burden (Younossi etal., 2016a). NAFLD is a spectrum of adverse conditions, spanning from simple steatosis (accumulation of lipid in the liver) to nonalcoholic steatohepatitis (NASH). Unresolved NASH can lead to cirrhosis, hepatocellular carcinoma, and liver-related mortality that may necessitate liver transplantation (Musso etal., 2016).
It is well established that NAFLD profoundly affects liver functions, including hepatic xenobiotic metabolizing capacity (Clarke and Cherrington, 2015). Many chemicals, especially environmental contaminants, undergo metabolism in the liver, which may result in the generation of reactive moieties that can cause tissue injury. While it is presumed that modification of hepatic metabolism of chemicals by NAFLD can potentially exacerbate chemical-induced organ toxicity, no study has directly addressed the effect of NAFLD on the relationship between tissue-specific toxicokinetics and toxicodynamics. As NAFLD prevalence is increasing, its contribution to inter-individual variability in xenobiotic-induced toxicity is also potentially growing.
Perchloroethylene (tetrachloroethylene; PERC) is an excellent case study chemical to examine inter-individual variability in toxicokinetics and toxicodynamics. PERC is an environmental pollutant that is associated with non-cancer effects in humans and rodents, and is a known rodent carcinogen and a probable human carcinogen (Cichocki etal., 2016). Historically, PERC has been a chemical of concern because of its wide use in dry cleaning, although its use in the United States in this industry is being phased out due to technology-based national emission standards imposed by the United States Environmental Protection Agency (U.S. EPA) that went into effect in 2006. However, PERC remains to be a ubiquitous environmental contaminant and exposure in humans is common because PERC is widely present in air, soil, and ground and drinking water (IARC, 2014). Furthermore, most hazardous waste sites contain detectable levels of PERC (National Research Council, 2010). Therefore, health hazards of PERC have been subject to recent international and national human health assessments (IARC, 2014; U.S. EPA, 2011).
Tissue-specific toxicity of PERC is thought to occur via metabolic activation to reactive metabolites (U.S. EPA, 2011). In the liver, oxidation of PERC to trichloroacetate (TCA) is associated with liver effects, while in the kidney, glutathione (GSH) metabolites of PERC are further metabolized to potent nephrotoxicants (Lash and Parker, 2001). In both rodents (Cichocki etal., 2017a) and humans (Birner etal., 1996), levels of TCA excreted in the urine are 3–5 orders of magnitude greater than urinary levels of S-(1,2,2-trichlorovinyl) glutathione, S-(1,2,2-trichlorovinyl)-l-cysteine, and N-acetyl-S-(1,2,2-trichlorovinyl)-l-cysteine.
Recently, we characterized the role of pre-existing NAFLD on the toxicokinetics of PERC in the mouse (Cichocki etal., 2017a). We observed that PERC preferentially adsorbed to fatty liver tissue compared to healthy liver tissue. In addition, hepatic clearance of the major oxidative PERC metabolite TCA was considerably reduced in mice with NAFLD, leading to accumulation of both PERC and TCA in the livers of mice with NAFLD. However, whether or not underlying NAFLD influences the toxicodynamics of PERC has yet to be determined. Thus, the objective of this study was to evaluate the effect of pre-existing experimental NAFLD in the mouse on the liver effects of PERC. We hypothesized that individuals with NALFD would be more susceptible to PERC-associated liver effects specifically through an increase in hepatic exposure to PERC and its hepatotoxic metabolite TCA.
MATERIALS AND METHODS
Chemicals
PERC (CAS 127-18-4) was purchased from Sigma Aldrich (Cat No. 270393, Batch No. SHBD9374V, purity 99.93%; St. Louis, Missouri). S-(1,2,2-trichlorvinyl) glutathione (TCVG; purity: 98.9%), S-(1,2,2-trichlorovinyl)-l-cysteine (TCVC; purity: 98.4%), N-acetyl-S-(1,2,2-trichlorovinyl)-l-cysteine (NAcTCVC; purity 99.7%), 2-amino-5-[(1-{[(13C)carboxy(13C)methyl](15N)amino}-1-oxo-3-[(trichloroethenyl)sulfanyl]propan-2-yl)amino]-5-oxopentanoic acid (TCVG*; purity 90.4%), 2-(15N)amino-3-[(trichloroethenyl)sulfanyl](13C3)propanoic acid (TCVC*; purity 97.5%), and 2-[acetyl(15N)amino]-3-[(trichloroethenyl)sulfanyl](13C3)propanoic acid (NAcTCVC*; purity 99.0%) were synthesized and graciously provided by Dr. Avram Gold at the University of North Carolina, Chapel Hill, North Carolina. Purity was determined via HPLC-UV/Vis. All other chemicals were acquired from local suppliers and were of the highest quality available.
Animals
The animals in this study were maintained on diets and exposed to chemicals as detailed in (Cichocki etal., 2017a). A total of 48 five-week old male C57Bl/6J mice were obtained from the Jackson Laboratory (Bar Harbor, Maine) and housed in polycarbonate cages with Sanichip hardwood chip bedding (P.J. Murphy Forest Products Corp., Montville, New Jersey). Mice were acclimatized for at least 1 week on standard chow (4% calories from fat low-fat diet; LFD; Teklad Rodent Diet #8604; Harlan, Madison, Wisconsin). Mice were randomly assigned into groups (4 mice per cage) fed LFD, a high-fat diet (31% calories from fat, HFD; Diet #519567; Dyets Inc., Bethlehem, Pennsylvania), or a diet with low methionine and devoid of choline and folate (31% calories from fat, MCD, Diet #519541; Dyets). All experimental protocols were approved by the Institutional Animal Care and Use Committee at Texas A&M University and were in accordance with the criteria of the National Institutes of Health (NIH publication # 86-23. Revised 1985).
Study design
Following 8 weeks of feeding with abovementioned diets, unfasted mice were exposed to a single dose of PERC (300 mg/kg/day in 5% Alkamuls-EL620 in saline, 5 ml/kg) or vehicle for 5 consecutive days (n = 8/diet/treatment) via oral gavage. This dose was selected based on our previous study (Cichocki etal., 2017a), which used a single dose of 300 mg/kg PERC (same vehicle) in mice on the same diets. Further, the concentration-time profiles of PERC and its oxidative metabolite TCA in male mice following exposure to 150–1000 mg/kg PERC was previously established in liver and blood (Philip etal., 2007) and did not exceed the maximum tolerated dose in acute (Philip etal., 2007) or sub-chronic studies (National Toxicology Program, 1977). All exposures took place between 07:00 and 10:00 hours. Three days prior to necropsy, animals were administered 5-bromo-2′-deoxyuridine in drinking water (0.02%, w/v). Mice were euthanized 4 h after the final gavage. Animals were deeply anesthetized with pentobarbital (50 mg/kg, i.p.) and exsanguinated through the vena cava, which was the site of serum collection (Z gel tubes; Sarstedt, Nümbrecht, Germany). Tissues were excised, rinsed in saline, blotted dry, weighed, and snap-frozen in liquid nitrogen. The liver left lobe was separated from the rest of the liver prior to freezing. The contents of the cecum were collected and frozen for further analysis. Small sections of the left liver lobe, median liver lobe, and duodenum were fixed in formalin for histological examination. An additional small section of the median liver lobe was embedded into Optimal Cutting Temperature (OCT) compound on dry ice for subsequent frozen sectioning.
Serum clinical chemistry
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured with commercially available kits (Sigma Aldrich, St. Louis, Missouri) according to manufacturer’s instructions.
Triglyceride measurement
Serum and liver triglycerides were measured using a commercially available kits (Wako, Richmond, Virginia).
Histopathology
Tissues were embedded in paraffin, sectioned at 5 µm, and stained with hematoxylin and eosin (H&E) or Sirius Red according to standard protocols. Frozen sections in OCT (5 µm) were used for Oil Red O staining. H&E-stained slides were evaluated by a trained veterinary pathologist who was blinded to study design and animal identification numbers. Sections were scored according to (Kleiner etal., 2005). The scoring system for steatosis ranged from 0 to 7, while the inflammation score ranged from 0 to 6. Immunohistochemical detection of BrdU-positive cells was performed on paraffin-embedded sections using a commercially available kit (Invitrogen, Carlsbad, California). Each slide contained a section of duodenum for a positive control of BrdU incorporation. Five random fields (200×), or at least 500 hepatocytes, were counted for each animal. The ratio of positively stained hepatocytes to total hepatocytes was determined for each animal. Results were verified by another laboratory member who was blinded to study design and animal identification numbers.
mRNA sequencing
mRNA sequencing was performed as previously described (Cichocki etal., 2017a). Differential gene expression was determined with the R package DESeq2 (v 1.12.3) (Love etal., 2014) on the complete list of 18 239 transcripts after removing low count genes. A log2 fold-change (compared with diet-matched vehicle-treated samples) and false discovery rate (FDR)-adjusted P-value cut-off of 0.58 and 0.1, respectively, were necessary for a gene to be deemed as differentially expressed. A cut-off for log2 of 0.58 is approximately a 1.5-fold change in expression compared with the diet-matched vehicle-treated reference group. Default settings in DESeq2 were used for FDR adjustment (Benjamini and Hochberg, 1995). The resulting gene expression data were used for biological pathway analysis using the piano (Varemo etal., 2013) package in R (v. 1.12.0) using the “Mouse Reactome” gene set (www.baderlab.org). For Gene Ontology (GO) pathway analysis, gene lists were uploaded to the Database for Annotation, Visualization, and Integrated Discovery (DAVID, v 6.7) (Huang etal., 2009). Visuals of differentially expressed genes and enriched pathways were generated in R using the VennDiagram (v 1.6.17), gplots (v 3.0.1), and ggplot2 (v 2.1.0) packages.
Quantitative reverse transcriptase real-time polymerase chain reaction (qRT-PCR)
qRT-PCR was performed as previously described (Cichocki etal., 2017a). In brief, 40 ng of liver cDNA was used in a 20-µl reaction with Taqman Probes (Ehhadh, Mm00619685_m1; Acot1, Mm01622471_s1; Acox1, Mm01246834_m1; Gusb, Mm01197698_m1). Expression of each target gene was normalized to Gusb using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Gas chromatography/mass spectrometry (GC/MS) analysis of PERC and TCA
PERC and TCA were measured by GC/MS as previously described (Cichocki etal., 2017a, b). In brief, PERC was measured in methanolic liver or fecal homogenates using dynamic headspace GC/MS. The analysis of TCA in tissues was conducted by a method modified from US EPA method (EPA 815-B-03-002). In brief, liver homogenates were derivatized with a methanolic esterifying reagent to generate TCA methyl ester. After liquid-liquid extraction, the derivatives were analyzed via GC/MS.
Liquid chromatography/mass spectrometry (LC/MS-MS) analysis of TCVG, TCVC, and NAcTCVC
The PERC conjugative metabolites TCVG, TCVC, and NAcTCVC were measured in liver homogenates as detailed elsewhere (Luo etal., 2017). In brief, aqueous tissue homogenates were subjected to solid-phase extraction followed by analysis via LC/MS-MS using positive electrospray ionization and multiple reaction monitoring.
Statistical analyses
GraphPad Prism (v 5.0) was used for statistical analysis of the biochemical and histological data. For comparisons between different exposure groups and disease states, ANOVA followed by Newman-Keuls post-hoc test was employed. For statistical analysis of histopathological scores using the NAFLD activity score (Kleiner etal., 2005), the Kruskal-Wallis test followed by Dunns post-hoc analysis was used. For all tests, a P-value <.05 was deemed statistically significant. For analysis of correlations between transcript abundance and PERC or TCA levels, Pearson correlation coefficients were determined and associated P-values were corrected for multiple comparisons (Benjamini and Hochberg, 1995) to derive q-values using the rcorr function of the Hmisc R package (v. 3.17-4) (Harrell, 2016). To account for diet-dependent effects on transcript levels, all raw counts were expressed as percent of the mean of the diet-matched vehicle-treated control values prior to running correlation analyses.
Data availability
RNA sequencing data are available from GEO. All phenotypes (Supplementary Table 1), correlation matrices among the phenotypes (Supplementary Table 2), differentially expressed genes and enriched pathways (Supplementary Tables S3–S7) are provided as supplementary materials.
RESULTS
Liver Effects
To determine whether underlying NAFLD would affect PERC effects in the liver, mice fed an LFD, HFD, or MCD diets for 8 weeks were exposed to 5 consecutive daily doses of PERC (300 mg/kg/day). Mice fed an HFD or MCD gained significantly more weight compared with LFD-fed mice (Supplementary Figure 1). PERC exposure was associated with a reduction in body weight (∼12%), an effect which was consistent in all groups.
Biochemical analyses were performed to phenotype liver effects of the diets and PERC (Table 1, Supplementary Figure 2). Relative liver weights were significantly greater in MCD-fed mice as compared to LFD-fed mice. In HFD- and MCD-fed mice, PERC exposure resulted in a further significant increase in relative liver weights, with MCD-fed mice being the most sensitive. Compared with diet-matched vehicle-treated controls, serum ALT levels were significantly elevated only in PERC-exposed HFD- and MCD-fed groups, with MCD-fed mice being the most sensitive. Serum AST was increased only in MCD-fed PERC-exposed mice relative to vehicle-treated MCD-fed mice.
Table 1.
Summary of Physiological/Biochemical Phenotype Data
| Diet | Exposure | Liver weight (g) | LBW (%) | Serum ALT (U/L) | Serum AST (U/L) | Liver TG (mg/g) | Serum TG (mg/dL) | Total histology score |
|---|---|---|---|---|---|---|---|---|
| LFD | Vehicle | 1.15±0.05 | 4.4±0.1 | 7.8±0.8 | 14.3±1.0 | 20.3±4.0 | 32.5±4.6 | 0 (0–0) |
| PERC | 1.21±0.09 | 4.6±0.2 | 9.3±0.6 | 23.9±2.1 | 30.4±3.5 | 18.3±1.9A | 0 (0–0) | |
| HFD | Vehicle | 1.65±0.11A | 4.8±0.2 | 14.6±2.1 | 15.9±1.2 | 44.8±3.8A | 28.4±2.7 | 3.5 (2.0–6.0)A |
| PERC | 1.75±0.05B | 5.6±0.2B,C | 38.1±8.9B,C | 23.7±3.3 | 55.1±2.6B | 17.5±1.1C | 6.0 (3.0–8.0)B,C | |
| MCD | Vehicle | 1.72±0.08A | 5.9±0.2A,C | 27.6±5.5 | 18.6±1.6 | 48.3±4.1A | 31.8±3.1 | 7.5 (5.0–10.0)A,C |
| PERC | 1.89±0.05B | 6.5±0.1B,D | 64.2±8.2B,D,E | 28.7±3.5E | 53.5±2.6B | 23.0±1.9 | 8.0 (6.0–9.0)B,D |
Values represent mean ± SE (n = 8/group), except for histopathological scoring, where values represent median (range). Two-way ANOVA (Tukey’s range post hoc test) was used for testing statistical significance of the effects of diet (irrespective of treatment) and treatment (irrespective of diet). Significant differences (P < .05) are denoted as compared to: A, vehicle (LFD); B, PERC (LFD); C, Vehicle (HFD); D, PERC (HFD); E, Vehicle (MCD). Abbreviations used in the table: LBW, liver-to-bodyweight ratio; ALT, serum alanine aminotransferase activity; AST, serum aspartate aminotransferase activity; Liver TG, liver triglycerides; Serum TG, serum triglycerides.
PERC exposure has been associated with lipid accumulation in the mouse liver (Buben and O’Flaherty, 1985; Philip etal., 2007); therefore, we measured liver and serum triglyceride levels. Liver triglyceride levels were increased in HFD- and MCD-fed mice compared with LFD-fed mice (Table 1). PERC exposure had no statistically significant effect on liver triglyceride levels compared with diet-matched vehicle-treated controls. Serum triglyceride levels were significantly reduced in PERC-exposed LFD- and HFD-fed mice compared with diet-matched vehicle-treated controls.
As expected, 8 weeks of HFD or MCD administration was associated with development of a NAFLD phenotype, as indicated by histopathology (Table 1, Supplementary Figure 3). PERC exposure influenced steatosis scores in HFD-fed mice only. PERC exposure did not have an effect on inflammation as indicated by histopathological scoring of liver tissue, according to the result of two-way ANOVA using the 3 diet and 2 treatment groups (Table 1). When the group means were normalized to diet-matched vehicle-treated control values, the means between the 3 diet groups were significantly different, with HFD-fed mice being the most sensitive to PERC-induced histopathological changes (Supplementary Figure 2). No steatosis was noted in H&E-stained slides from LFD-fed PERC-exposed mice even though liver triglyceride levels were elevated (although not statistically-significant; see above). To confirm the biochemical measurements, Oil Red O staining was employed. Red-stained neutral lipids were apparent in liver sections of PERC-exposed LFD-fed animals, albeit the degree of lipid accumulation was less pronounced in comparison to liver sections from HFD- or MCD-fed animals exposed to vehicle or PERC.
Diet-Related Transcriptomic Effects
We found that 1129 and 1881 transcripts were differentially expressed in vehicle-treated HFD- and MCD-fed mice, respectively, compared with vehicle-treated LFD-fed mice (Figs. 1A and B). There were 787 differentially expressed genes that were in common between two groups and the magnitude of changes in gene expression was highly correlated between HFD- and MCD-fed vehicle-treated animals (r2 = 0.665, Spearman’s ρ = 0.796). Enriched KEGG pathways in either HFD- and MCD-fed animals included those involved in xenobiotic, retinol, linoleic acid, and nitrogen metabolism, in addition to cytokine-cytokine receptor interactions (Table 2, individual gene level data are provided in Supplementary Tables S3 and S4). From these 6 significantly enriched KEGG pathways, 53 unique genes were identified and extracted for further analyses. The directionality of the change in expression of these 53 genes was similar between HFD- and MCD-fed mice, but diet-specific changes were also evident (Figure 1C). For example, an increased expression of inflammatory genes (e.g., Cx3cr1, Ccl2, and Il1a) was observed in MCD-fed mice compared with HFD-fed mice. Consistent with the previous reports by our group (Cichocki etal., 2017a) and others (as reviewed in Clarke and Cherrington, 2015), mRNA levels of multiple xenobiotic metabolizing enzymes were significantly repressed in HFD- and MCD-fed vehicle-treated mice compared with LFD-fed vehicle-treated mice.
Figure 1.
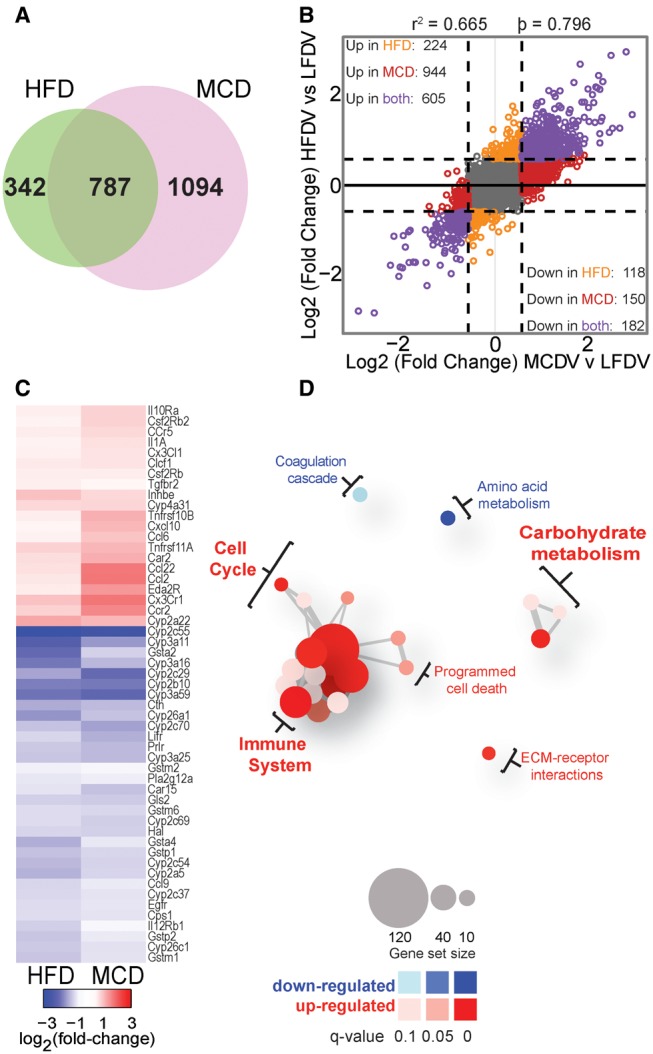
Effect of 8 week HFD or MCD feeding on liver gene expression. All comparisons are made between vehicle-treated LFD-fed mice. A, Venn diagram showing numbers of differentially expressed genes in HFD- or MCD-fed mice compared to LFD-fed mice. B, Plot of fold-changes (log2, compared with LFD) of individual transcripts in HFD- and MCD-fed animals. In order to be deemed differentially expressed, transcripts needed to have an absolute value of log2 fold-change > 0.58 (represented by dotted-lines) and an FDR-adjusted q-value <0.1. Transcripts that did not meet these requirements are colored grey. Differentially expressed genes are color-coded by whether they were changed by HFD (orange), MCD (red), or both (purple) diets. C, Heatmap of log2 fold-change values for differentially expressed genes modified by both HFD and MCD diets. Genes were extracted from the 5 KEGG pathways listed in Table 2 as described in the text. D, Network plots of gene sets enriched specifically in the 1094 genes differentially expressed specifically in MCD-fed mice. Red circles represent pathways that were significantly induced, while blue circles represent repressed pathways (FDR-adjusted q-value <0.1). The size of the circle is proportional to the number of genes that are enriched in that pathway. The thickness of connecting lines between circles is proportional to the number of common genes between those 2 pathways; at least 10 genes needed to be common between pathways in order for a line to be drawn. Color and size keys are provided at the bottom.
Table 2.
Pathways Enriched in Both HFD- and MCD-Fed Vehicle-Treated Micea
| KEGG Pathway IDb | No. of genes | Fold enrichment | Benjamini-corrected q-value |
|---|---|---|---|
| mmu00980:Metabolism of xenobiotics by cytochrome P450 | 16 | 5.21 | 1.73E−05 |
| mmu00982:Drug metabolism | 18 | 5.16 | 5.46E−06 |
| mmu00830:Retinol metabolism | 15 | 4.74 | 1.06E−04 |
| mmu00591:Linoleic acid metabolism | 10 | 4.67 | 8.57E−03 |
| mmu04060:Cytokine-cytokine receptor interaction | 23 | 2.03 | 6.15E−02 |
As compared with LFD-fed vehicle-treated animals
KEGG pathways were retrieved from DAVID v 6.7, as described in the Materials and Methods section.
We next used network plots to visualize which Reactome pathways were enriched in the gene expression data from HFD- or MCD-fed mice (Figure 1D). Of the 1094 differentially expressed genes specific to MCD-fed mice, the immune system, cell cycle, apoptosis, and extracellular matrix pathways were found to be significantly up regulated, while amino acid metabolism and fibrin clot formation pathways were significantly repressed specifically in the MCD-fed group. No pathways were found to be enriched (at the stringent statistical significance thresholds applied herein) for the 342 differentially expressed genes specific to HFD-fed animals.
Effect of NAFLD on PERC Metabolism
Characterization of PERC metabolism is critical to understanding its toxicity (Cichocki etal., 2016). We have previously showed that NAFLD has a profound effect on PERC toxicokinetics in a study of a single oral dose of PERC (Cichocki etal., 2017a). To assess whether the same effects are observed following repeat exposures, we measured PERC, TCA, TCVG, TCVC, and NAcTCVC levels in liver tissue, as well as PERC level in feces (Figure 2). We observed that levels of PERC and its primary oxidative metabolite TCA were increased in HFD- and MCD-fed mice, compared with LFD-fed mice. Specifically, levels of PERC were highest in MCD-, followed by HFD-, then LFD-fed mice. Levels of TCA were highest in HFD-, followed by MCD-, then LFD-fed mice. The ratio of PERC to TCA was highest in MCD-fed mice compared with the other groups. Levels of glutathione conjugation metabolites TCVG, TCVC, and NAcTCVC were decreased in HFD- and MCD-fed mice, compared with LFD-fed mice. PERC levels in the cecum contents showed that although the LFD group had higher concentrations of PERC as compared with the HFD or MCD groups, in all cases, the amount in feces constituted a very small percentage of the administered PERC dose. Correlation among PERC and its metabolite levels and liver phenotypes was evaluated (Figure 3). Strong positive correlations between hepatic levels of PERC and markers of liver effects were observed. Interestingly, the glutathione conjugation metabolites TCVG and TCVC were negatively correlated with the same phenotypes.
Figure 2.

Concentrations of PERC and its metabolites. Data expressed as mean ± SE, n = 8. *P < .05 compared with LFD-fed mice via ANOVA with Newman-Keuls post-hoc test.
Figure 3.
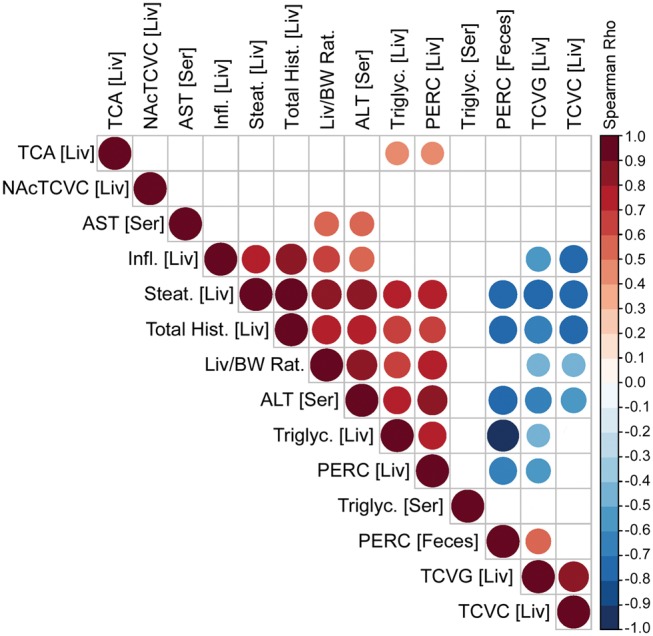
Correlation analysis of PERC and its liver metabolites and toxicity phenotypes. Spearman rank-based correlation analysis is shown for pair-wise comparisons among PERC in liver, NAcTCVC in liver, serum AST, liver inflammation, steatosis and total histopathology scores, liver-to-body weight ratios, serum ALT, liver triglycerides, liver PERC, serum triglycerides, PERC in feces, and liver TCVG and TCVC. Only correlations that were significant (Bonferroni-corrected P-value ≤.05) are shown and the size and color of each bubble is proportional to the correlation value as shown in the color bar.
Effect of NAFLD on PERC-Induced Modulation of Liver Gene Expression
When compared with diet-matched vehicle-treated mice, PERC exposure was associated with differential expression of 851, 1762, and 1041 genes in LFD-, HFD-, and MCD-fed mice, respectively. Of these differentially expressed genes, 328, 914, and 247 were specific to LFD-, HFD-, and MCD-fed animals, respectively (Figure 4A).
Figure 4.

Diet-specific effects of PERC exposure on liver gene expression. Mice were fed a LFD, HFD, or MCD for 8 weeks prior to oral exposure to PERC (300 mg/kg/day; 5 days). All comparisons are made between vehicle-treated diet-matched control mice. A, Venn diagram showing numbers of differentially expressed genes in PERC-exposed mice. B–C, Network plots of gene sets enriched specifically in the 914 and 247 genes differentially expressed specifically in HFD-fed (B) and MCD-fed mice (C), respectively. Red circles represent pathways that were significantly induced, while blue circles represent repressed pathways (FDR-adjusted q-value <0.05). The size of the circle is proportional to the number of genes that are enriched in that pathway. The thickness of connecting lines between circles is proportional to the number of common genes between those 2 pathways. At least 10 genes needed to be common between pathways in order for a line to be drawn. Color and size keys are provided on the right.
For the 914 genes specific to HFD-fed PERC-treated mice, a total of 46 Reactome pathways were significantly enriched, with all of them being significantly up-regulated (Figure 4B). The pathways with the most differentially expressed genes were the cell cycle (43 genes), metabolism of proteins (42 genes), gene expression (41 genes), and adaptive immune system (32 genes). With a Benjamini-adjusted P-value cut-off of .05, no gene sets were enriched for the 328 genes that were specific to LFD-fed PERC-treated mice. A total of 5 pathways were significantly up-regulated specifically in MCD-fed PERC-treated animals from 247 selected genes. These pathways were involved in lipid and fatty acid synthesis and metabolism (Figure 4C).
Of particular interest was the finding that the cell cycle was significantly enriched only in HFD-fed mice. It has previously been shown that PERC exposure is associated with hepatocyte proliferation (Philip etal., 2007) and with liver tumors in rodents (Japanese Industrial Safety Association, 1993; National Toxicology Program, 1977, 1986). To this end, we further evaluated the effect of PERC on hepatocyte proliferation (Supplementary Figure 4). In vehicle-treated HFD- and MCD-fed mice, an increase in BrdU staining of hepatocyte nuclei was observed compared with LFD-fed vehicle-treated controls. PERC exposure had no significant effect on the percentage of BrdU-positive nuclei. In addition, we investigated hepatic expression levels of differentially expressed genes (56 genes) classified in the GO Biological Process pathway as part of “cell cycle process.” Clear clustering of vehicle-and PERC-treated animals, with the exception of 3 MCD-fed vehicle-treated mice, was observed. LFD-fed PERC-treated animals were separated from HFD- and MCD-fed PERC-treated animals; however, there was no clear clustering between these latter two groups.
Common Effects of PERC Exposure in Both HFD- and MCD-Fed Mice
A total of 386 genes overlapped between HFD- and MCD-fed animals exposed to PERC (Figure 4A); thus, these genes were differentially expressed in PERC-exposed mice in either model of NAFLD. GO pathway analysis of these 386 genes implicated the mitochondria as a potential molecular target of PERC in HFD- and MCD-fed mice (Supplementary Table 5, individual gene level data are provided in Supplementary Tables S6 and S7). Further, the magnitude of change in global hepatic gene expression between HFD- and MCD-fed PERC-treated mice was positively correlated (Pearson’s r2 = 0.565) (Supplementary Figure 5).
Common Effects of PERC Exposure in LFD-, HFD-, and MCD-Fed Mice
We identified 347 differentially expressed genes which overlapped among LFD-, HFD-, and MCD-fed mice treated with PERC (Figure 5A), suggesting that these genes were differentially expressed in a diet-independent manner. Pathways related to cellular metabolism, including xenobiotic and fatty acid metabolism, and the complement and coagulation cascade pathway were significantly enriched in PERC-exposed mouse liver (Table 3, individual gene level data are provided in Supplementary Tables S6–S8). Unsupervised hierarchical clustering of the gene expression of these 347 genes (Figure 5A) revealed distinct clustering of LFD-fed PERC-treated animals, while PERC-treated HFD- and MCD-fed animals were interspersed in a separate cluster. Expression of 3 of these genes was measured by qRT-PCR (Figure 5B). These genes were found to be significantly differentially expressed following PERC exposure in all 3 diet groups, but the effect in mice with NAFLD, particularly HFD-fed mice, was generally more pronounced.
Figure 5.
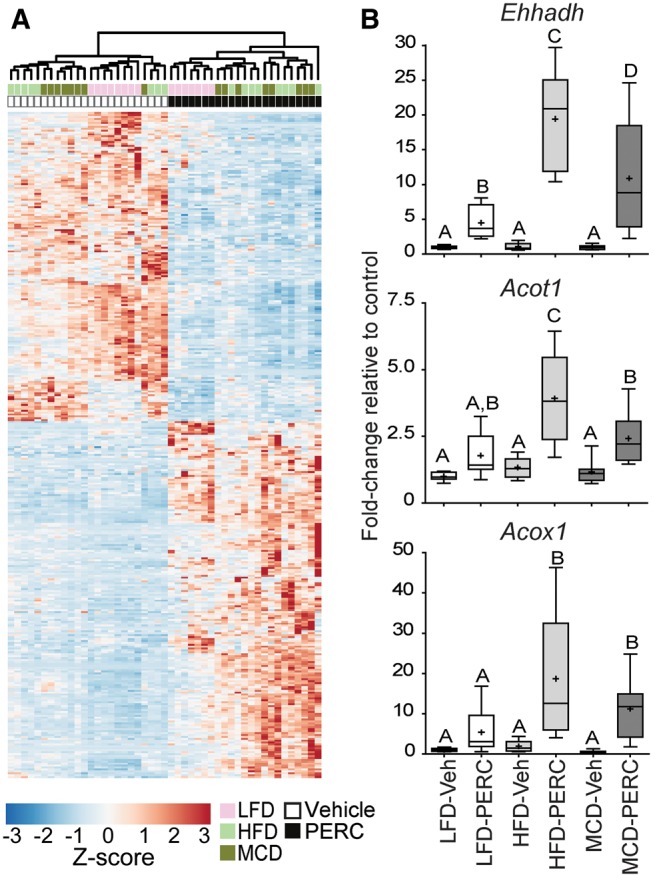
Diet-independent effects of PERC exposure on liver gene expression. Mice were fed an LFD, HFD, or MCD for 8 weeks prior to oral exposure to PERC (300 mg/kg/day; 5 days). A, Heatmap and dendrogram for unsupervised hierarchical clustering of row Z-scores of the normalized count data for 347 genes that were commonly differentially expressed among all 3 groups exposed to PERC (see Venn diagram, Figure 2). B, qRT-PCR analysis of selected genes which were differentially-expressed in all 3 groups. Expression was normalized to Gusb expression using the 2−ΔΔCT method. Different letters represent different statistical groups (P < .05, ANOVA with Newman-Keuls post hoc test). Whiskers, min and max; +, group mean; line, median; box, interquartile range.
Table 3.
Pathways Enriched in All PERC-treated Groupsa
| KEGG Pathway IDb | No. of genes | Fold enrichment | Benjamini-corrected q-value |
|---|---|---|---|
| mmu00053:Ascorbate and aldarate metabolism | 4 | 13 | 3.61E−02 |
| mmu00980:Metabolism of xenobiotics by cytochrome P450 | 15 | 11 | 1.99E−09 |
| mmu01040:Biosynthesis of unsaturated fatty acids | 6 | 11 | 2.70E−03 |
| mmu00982:Drug metabolism | 16 | 10 | 1.87E−09 |
| mmu00071:Fatty acid metabolism | 9 | 9.6 | 6.56E−05 |
| mmu00830:Retinol metabolism | 13 | 9.2 | 3.42E−07 |
| mmu00590:Arachidonic acid metabolism | 13 | 7.6 | 2.63E−06 |
| mmu00480:Glutathione metabolism | 8 | 7.4 | 1.32E−03 |
| mmu03320:PPAR signaling pathway | 11 | 6.7 | 7.65E−05 |
| mmu00140:Steroid hormone biosynthesis | 6 | 6.4 | 2.68E−02 |
| mmu04610:Complement and coagulation cascades | 7 | 4.5 | 4.29E−02 |
As compared with diet-matched vehicle-treated animals.
KEGG pathways were retrieved from DAVID v 6.7, as described in the Materials and Methods section.
Relationship Between PERC and TCA Levels and Liver Gene Expression
To explore concentration-response relationships we performed correlation analyses between hepatic gene transcript abundance and liver PERC or TCA concentrations. We used the 347 genes that were common among all PERC-treated animals for these analyses. A total of 49 genes were identified that correlated with PERC and/or TCA levels in the liver (Figure 6, Supplementary Table 9) with cut-offs of 0.6 and 0.05 for Pearson’s r and FDR-adjusted q-value, respectively. Hepatic expression of 45 of these genes correlated strongly with TCA levels, these were highly enriched (q-value = 0.016) for GO pathway “oxidation reduction.” Transcript abundances of 11 genes were correlated with hepatic levels of PERC, of which 7 of these were also correlated with TCA levels. Select genes that were highly correlated with PERC or TCA are shown as correlation plots.
Figure 6.

Correlation of hepatic concentrations of PERC and TCA with liver gene expression changes. A,. Heatmap showing the Pearson’s r correlation coefficient for genes significantly correlated with hepatic concentrations of PERC (left) or TCA (right). All genes shown had a Benjamini-Hochberg adjusted FDR q-value < 0.05. Pearson’s r values are shown within each cell. Red cells = positively correlated (r ≥ 0.6); Blue cells = negatively correlated (r ≤ 0.6; Gray cells = not correlated (−0.6 < r < 0.6). B, Select genes from (A) are presented along as linear regression of gene expression to hepatic PERC (top) or TCA (bottom) concentrations. Each dot represents an individual animal and the red or blue lines represent the linear regression lines. To eliminate the effect of diet alone, individual gene raw count data were normalized to the percentage of the average diet-matched vehicle-treated control prior to running correlation analyses.
DISCUSSION
We previously reported that NAFLD has a profound effect on PERC toxicokinetics in mice because of (i) increased affinity of PERC, a lipophilic chemical, for fatty liver tissue compared with lean liver tissue, and (ii) decreased hepatic elimination of TCA in HFD- or MCD-fed mice compared with LFD-fed mice (Cichocki etal., 2017a). In this study, we tested the hypothesis that mice with NAFLD would be more susceptible to PERC-induced liver effects, specifically through increased hepatic exposure to PERC and TCA. This study was needed to address a critical gap in our existing knowledge of how common conditions such as NAFLD, which affects nearly 25% of adults globally, modulates the metabolism of and the response to PERC, a ubiquitous environmental contaminant. Our principal finding is that mice with NAFLD are susceptible to the liver effects of PERC.
Despite being a high-volume production chemical to which humans are commonly exposed, surprisingly little is known about the mechanism of PERC-induced hepatotoxicity (Cichocki etal., 2016). Therefore, in order to determine the effect of NAFLD on the toxicology of PERC, we first examined diet-independent effects of PERC. In doing so, we report for the first time the effects of PERC exposure on the mouse liver transcriptome. By analyzing the genes which were modulated independently by diet, we show that PERC exposure resulted in enrichment of pathways involved in xenobiotic, glutathione, and lipid metabolism, PPARα signaling, and complement and coagulation cascades.
Exposure to PERC has been shown to be associated with altered hepatic lipid homeostasis (Buben and O’Flaherty, 1985; Cichocki etal., 2017b; Philip etal., 2007), peroxisome proliferation, and activation of PPARα (Odum etal., 1988; Philip etal., 2007). While the overall database on the contribution of PPARα to the hepatotoxic effects of PERC is limited (Cichocki etal., 2016; IARC, 2014; U.S. EPA, 2011), activation of this nuclear receptor has been implicated in chemical-induced hepatocarcinogenesis in the mouse (Bull, 2000; Corton etal., 2000). While PERC exposure did not have an effect on hepatic glutathione levels (Philip etal., 2007), PERC is conjugated with glutathione in the liver, therefore it is not surprising that exposure to PERC would modify genes involved in glutathione synthesis or transferase activity. Interestingly, the effect of PERC on the complement and coagulation cascade has not been previously reported and may represent a novel mechanism of PERC-induced hepatotoxicity. Further studies are warranted, especially considering the role of complement activity in liver regeneration, hepatocyte proliferation, and liver fibrosis (Ricklin etal., 2010).
Our main finding is that NAFLD is an important susceptibility factor for PERC-induced liver effects in mice. This study used histopathological, biochemical, and transcriptomic endpoints to evaluate the hepatic response to a toxicant exposure in both HFD and MCD models of NAFLD. One may suspect that MCD-fed mice would have the greatest response to PERC, as they already exhibit marked liver injury prior to exposure. However, 3 times more genes were differentially expressed specifically in HFD-fed animals compared to LFD- or MCD-fed animals following PERC exposure. Specifically, we found that HFD-fed mice exhibited changes in pathways involved in the adaptive immune system, protein translation, cell signaling, and the cell cycle, while MCD-fed mice experienced changes in only a few pathways related to lipid and fatty acid synthesis and metabolism. The differential response among these NAFLD-related diet groups is perhaps not surprising given that HFD-fed mice had the highest hepatic concentrations of TCA following exposure to PERC. Indeed, many of the genes that were differentially expressed as a result of PERC treatment in all 3 diet groups were significantly correlated with hepatic PERC or TCA concentrations. Interestingly, genes involved in “oxidation reduction” were significantly correlated with TCA levels, which may be due to the activation of PPARα by TCA (Maloney and Waxman, 1999), as PPARα has been shown to modulate xenobiotic metabolism (Janssen etal., 2015; Yoo etal., 2015), although other nuclear receptors or transcription factors may be involved. Taken together, our data strongly suggest that the tissue dose of PERC and/or TCA is a strong predictor of the transcriptomic phenotype. However, it is also possible that underlying disease modulated the response to PERC in a toxicokinetic-independent fashion.
Furthermore, we found that vehicle-treated mice with NAFLD exhibited up-regulation of genes involved in cytokine-cytokine receptor interaction; it is well established that pre-treatment of laboratory animals with pro-inflammatory agents (e.g., lipopolysaccharide) can intensify the effects of idiosyncratic hepatotoxicants (Deng etal., 2009; Shaw etal., 2010). It is known that inflammatory cytokines can modulate xenobiotic metabolism, thereby altering toxicity (Woolbright and Jaeschke, 2015). Interestingly, MCD-fed mice were most sensitive to increases in serum transaminase levels following exposure to PERC, while HFD-fed mice were most sensitive to PERC-induced histopathological changes and liver transcriptomic effects. It is possible that histopathological assessment was not sensitive enough to capture the effect of PERC in MCD-fed mice, given that MCD-fed mice had severe underlying injury. It is also plausible that this underlying disease state in MCD-fed mice precluded any hepatic transcriptomic response to PERC exposure. Concomitantly, mild lobular inflammation in HFD-fed mice may have potentiated the effects of PERC treatment. Thus, while delivered dose is obviously important for target organ toxicity of a chemical, there are other factors, such as pre-existing chronic inflammation, which may contribute to liver effects associated with PERC exposure.
One hypothesis to explain greater susceptibility in the HFD and MCD groups is that the diets may affect PERC absorption as compared with the LFD group; we posit this is not the case. First, the LFD is the “normal” diet typically given to laboratory mice; thus, prior toxicokinetic studies can be informative as to the expected degree of absorption of PERC. Multiple radiolabel studies in mice, rats, and dogs have suggested near complete absorption after oral dosing. For instance, Schumann etal. (1980) administered 500 mg/kg of radiolabeled PERC to B6C3F1 mice, and recovered only 1.2% of the radiolabel in feces after 72 h. Thus, the expectation based on prior studies would be that absorption is nearly complete, and any additional absorption in the HFD or MCD groups would have a negligible influence on the total amount absorbed. Second, we found that measured PERC levels in the feces constitute a very small percentage of the administered PERC dose. Specifically, given that mice produce about 1 g of feces per day, the measured fecal concentrations of PERC imply <1% of the administered PERC dose of 300 mg/kg/day being excreted in feces (0.77% for LFD, 0.15% for HFD, and 0.18% for MCD). Therefore, it is unlikely that greater absorption of PERC in the HFD and MCD groups can explain the differences in the observed effects.
The effects of NAFLD on xenobiotic metabolism and toxicity is well-established; both experimental and human NAFLD can affect xenobiotic disposition and metabolism, which is typically associated with the effect of NAFLD on expression of xenobiotic-metabolizing enzymes and xenobiotic transporter, as recently reviewed in Clarke and Cherrington (2015). The relationships between chemical metabolism and experimental NAFLD that render rodents susceptible to the hepatotoxic effects have been shown for Aroclor 1260 (Wahlang etal., 2014), methotrexate (Hardwick etal., 2014), and carbon tetrachloride (Donthamsetty etal., 2007). Conversely, HFD-fed mice were relatively insensitive to thioacetamide- (Shirai etal., 2013) or acetaminophen- (Ito etal., 2006) induced hepatocellular injury. Our study adds to this body of literature by showing that NAFLD results in different delivered doses of PERC and TCA to the liver which in turn influences the hepatic response to PERC.
There are several important limitations to this study. First, the dose of PERC was high as compared with potential human environmental exposures even though it was in line with the doses used in sub-chronic and cancer studies of PERC. Only one dose level was administered. Thus, additional studies are needed to fully characterize concentration-response relationship of PERC-induced liver effects in the context of NAFLD. While transcriptomic data are important for understanding the molecular pathways that are affected by NAFLD or PERC, future studies may need to utilize proteomic or metabolic approaches to better characterize these molecular networks. Further, only one sex and strain of mouse was used in these studies. It is known that the hepatic responses to high fat/high sucrose (Hui etal., 2015) and MCD (Tryndyak etal., 2012) diets are strain-specific in mice. In humans, NAFLD pathogenesis and progression is thought to be mediated through gene-environment interactions (Anstee etal., 2011). In mice, the relationship between PERC toxicokinetics and toxicodynamics is strain-specific (Cichocki etal., 2017b). The toxicokinetics of PERC is also variable in humans (Chiu etal., 2007; Monster and Houtkooper, 1979; Opdam, 1989; Volkel etal., 1998), an effect which may be mediated by multiple factors, including underlying disease state (National Research Council, 2009, 2010; U.S. EPA, 2011). Future studies are needed to determine the extent of interplay between genetics and disease in response to environmental chemical exposure.
These limitations notwithstanding, this study has a number of strengths and contributes novel findings to the study of liver diseases and toxicological sciences. First, this study is first to examine the effects of PERC exposure on the liver transcriptome. Secondly, we directly compared differences in global liver gene expression in HFD- and MCD-fed mice, which are 2 distinct models of NAFLD (Maher, 2011). Our findings here in HFD- and lipogenic MCD-induced NAFLD models were similar to the microarray results of Teufel etal. (2016), where the authors reported that pathways related to xenobiotic metabolism (ABC transporters, glutathione metabolism), lipid metabolism, ECM-receptor interaction, and inflammation were differentially regulated by a “western-type diet” murine model of NAFLD. Importantly, Teufel etal. (2016) performed pathway analysis and reported that human NAFLD/NASH samples clustered more closely with the “western-type diet” group, compared with other murine models, suggesting that the lipogenic models used in our study are appropriate for modeling human NAFLD.
Lastly, by using two distinct mouse models, we show that experimental NAFLD can modulate the response to PERC, a ubiquitous environmental contaminant; this was accomplished by applying transcriptomics and traditional toxicity phenotyping. Interestingly, we show that MCD-fed mice are most susceptible to PERC-induced increases in serum transaminase levels, while HFD-fed mice are most susceptible to the liver transcriptomic effects of PERC; these findings may be due to increased hepatic levels of PERC in MCD-fed mice, increased TCA in HFD-fed mice, or due to effects not directly related to toxicokinetics.
These data have implications for future risk management decisions for environmental chemicals, particularly because the prevalence of NAFLD is expected to increase in the coming years (Sayiner etal., 2016). Our experimental evidence shows that underlying NAFLD substantially impacts susceptibility to toxicity associated with PERC exposure in mice. Lipophilic molecules, such as PERC, will preferentially partition into fatty liver tissue compared with healthy liver tissue (Cichocki etal., 2017a); it is therefore expected that, given the same exposure conditions, individuals with NAFLD will have increased hepatic concentrations of these agents compared with healthy individuals, which may contribute to chemical-induced toxicity. Incorporation of disease state as a susceptibility factor into public health assessments of environmental chemicals may serve to better characterize potentially susceptible subpopulations, such as individuals with NAFLD.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
J.A.C was a recipient of a National Research Service Award [grant number F32 ES026005] through the National Institutes of Health (NIH). This study was supported, in part, by a cooperative agreement [STAR RD83561202] from US Environmental Protection Agency (EPA) to Texas A&M University. The views expressed in this paper are those of the authors and do not necessarily reflect the views or policies of US Food and Drug Administration, NIH, or EPA.
Author contributions: Participated in study design (JAC, WAC, DWT, IPP, IR); conducted experiments (JAC, SF, YL, YI); provided assistance with bioinformatics (KK); wrote the paper (JAC, IR). All authors read and approved the final version of the manuscript.
Supplementary Material
REFERENCES
- Anstee Q. M., Daly A. K., Day C. P. (2011). Genetic modifiers of non-alcoholic fatty liver disease progression. Biochim. Biophys. Acta 1812, 1557–1566. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 57, 289–300. [Google Scholar]
- Birner G., Rutkowska A., Dekant W. (1996). N-acetyl-S-(1,2,2-trichlorovinyl)-L-cysteine and 2,2,2-trichloroethanol: two novel metabolites of tetrachloroethene in humans after occupational exposure. Drug Metab. Dispos. 24, 41–48. [PubMed] [Google Scholar]
- Buben J. A., O’Flaherty E. J. (1985). Delineation of the role of metabolism in the hepatotoxicity of trichloroethylene and perchloroethylene: a dose-effect study. Toxicol. Appl. Pharmacol. 78, 105–122. [CrossRef [DOI] [PubMed] [Google Scholar]
- Bull R. J. (2000). Mode of action of liver tumor induction by trichloroethylene and its metabolites, trichloroacetate and dichloroacetate. Environ. Health Perspect. 108, 241–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu W. A., Micallef S., Monster A. C., Bois F. Y. (2007). Toxicokinetics of inhaled trichloroethylene and tetrachloroethylene in humans at 1 ppm: empirical results and comparisons with previous studies. Toxicol. Sci. 95, 23–36. [DOI] [PubMed] [Google Scholar]
- Cichocki J. A., Furuya S., Konganti K., Luo Y. S., McDonald T. J., Iwata Y., Chiu W. A., Threadgill D. W., Pogribny I. P., Rusyn I. (2017a). Impact of nonalcoholic fatty liver disease on toxicokinetics of tetrachloroethylene in mice. J. Pharmacol. Exp. Ther. 361, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki J. A., Furuya S., Venkatratnam A., McDonald T. J., Knap A. H., Wade T., Sweet S., Chiu W. A., Threadgill D. W., Rusyn I. (2017b). Characterization of variability in toxicokinetics and toxicodynamics of tetrachloroethylene using the collaborative cross mouse population. Environ Health Perspect. 125, 057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki J. A., Guyton K. Z., Guha N., Chiu W. A., Rusyn I., Lash L. H. (2016). Target organ metabolism, toxicity, and mechanisms of trichloroethylene and perchloroethylene: key similarities, differences, and data gaps. J. Pharmacol. Exp. Ther. 359, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J. D., Cherrington N. J. (2015). Nonalcoholic steatohepatitis in precision medicine: Unraveling the factors that contribute to individual variability. Pharmacol. Ther. 151, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton J. C., Lapinskas P. J., Gonzalez F. J. (2000). Central role of PPARalpha in the mechanism of action of hepatocarcinogenic peroxisome proliferators. Mutat. Res. 448, 139–151. [DOI] [PubMed] [Google Scholar]
- Deng X., Luyendyk J. P., Ganey P. E., Roth R. A. (2009). Inflammatory stress and idiosyncratic hepatotoxicity: hints from animal models. Pharmacol. Rev. 61, 262–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donthamsetty S., Bhave V. S., Mitra M. S., Latendresse J. R., Mehendale H. M. (2007). Nonalcoholic fatty liver sensitizes rats to carbon tetrachloride hepatotoxicity. Hepatology 45, 391–403. [DOI] [PubMed] [Google Scholar]
- Hardwick R. N., Clarke J. D., Lake A. D., Canet M. J., Anumol T., Street S. M., Merrell M. D., Goedken M. J., Snyder S. A., Cherrington N. J. (2014). Increased susceptibility to methotrexate-induced toxicity in nonalcoholic steatohepatitis. Toxicol. Sci. 142, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell F. E. (2016). Hmisc: Harell Miscellaneous. R package version 3.17-4.
- Huang D. W., Sherman B. T., Lempicki R. A. (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S. T., Parks B. W., Org E., Norheim F., Che N., Pan C., Castellani L. W., Charugundla S., Dirks D. L., Psychogios N., et al. (2015). The genetic architecture of NAFLD among inbred strains of mice. Elife 4, e05607.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC (2014). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans (Vol. 106): Trichloroethylene, Tetrachloroethylene and Some Other Chlorinated Agents. [PMC free article] [PubMed]
- Ito Y., Abril E. R., Bethea N. W., McCuskey M. K., McCuskey R. S. (2006). Dietary steatotic liver attenuates acetaminophen hepatotoxicity in mice. Microcirculation 13, 19–27. [DOI] [PubMed] [Google Scholar]
- Janssen A. W., Betzel B., Stoopen G., Berends F. J., Janssen I. M., Peijnenburg A. A., Kersten S. (2015). The impact of PPARalpha activation on whole genome gene expression in human precision cut liver slices. BMC Genomics 16, 760.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Japanese Industrial Safety Association (1993). Carcinogenicity study of tetrachloroethylene by inhalation in rats and mice. J. I. S. Association.
- Kleiner D. E., Brunt E. M., Van Natta M., Behling C., Contos M. J., Cummings O. W., Ferrell L. D., Liu Y. C., Torbenson M. S., Unalp-Arida A., et al. (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Lash L. H., Parker J. C. (2001). Hepatic and renal toxicities associated with perchloroethylene. Pharmacol. Rev. 53, 177–208. [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y. S., Cichocki J. A., McDonald T. J., Rusyn I. (2017). Simultaneous detection of the tetrachloroethylene metabolites S-(1,2,2-trichlorovinyl) glutathione, S-(1,2,2-trichlorovinyl)-L-cysteine, and N-acetyl-S-(1,2,2-trichlorovinyl)-L-cysteine in multiple mouse tissues via ultra-high performance liquid chromatography electrospray ionization tandem mass spectrometry. J. Toxicol. Environ. Health A, in press. [DOI] [PMC free article] [PubMed]
- Maher J. J. (2011). New insights from rodent models of fatty liver disease. Antioxid Redox Signal. 15, 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney E. K., Waxman D. J. (1999). Trans-activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol. Appl. Pharmacol. 161, 209–218. [DOI] [PubMed] [Google Scholar]
- Monster A. C., Houtkooper J. M. (1979). Estimation of individual uptake of trichloroethylene, 1,1,1-trichloroethane and tetrachloroethylene from biological parameters. Int. Arch. Occup. Environ. Health 42, 319–323. [DOI] [PubMed] [Google Scholar]
- Musso G., Cassader M., Gambino R. (2016). Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 15, 249–274. [DOI] [PubMed] [Google Scholar]
- National Research Council (2009). Science and Decisions: Advancing Risk Assessment. National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- National Research Council (2010). Review of the Environmental Protection Agency's Draft IRIS Assessment of Tetrachloroethylene. The National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- National Toxicology Program (1977). Bioassay of tetrachloroethylene for possible carcinogenicity. Natl Cancer Inst. Carcinog Tech. Rep. Ser. 13, 1–83. [PubMed] [Google Scholar]
- National Toxicology Program (1986). NTP toxicology and carcinogenesis studies of tetrachloroethylene (perchloroethylene) (CAS No. 127-18-4) in F344/N rats and B6C3F1 mice (inhalation studies). Natl Toxicol Program Tech Rep Ser 311, 1–197. [PubMed] [Google Scholar]
- Odum J., Green T., Foster J. R., Hext P. M. (1988). The role of trichloracetic acid and peroxisome proliferation in the differences in carcinogenicity of perchloroethylene in the mouse and rat. Toxicol. Appl. Pharmacol. 92, 103–112. [DOI] [PubMed] [Google Scholar]
- Opdam J. J. (1989). Intra and interindividual variability in the kinetics of a poorly and highly metabolising solvent. Br. J. Ind. Med. 46, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip B. K., Mumtaz M. M., Latendresse J. R., Mehendale H. M. (2007). Impact of repeated exposure on toxicity of perchloroethylene in Swiss Webster mice. Toxicology 232, 1–14. [DOI] [PubMed] [Google Scholar]
- Ricklin D., Hajishengallis G., Yang K., Lambris J. D. (2010). Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayiner M., Koenig A., Henry L., Younossi Z. M. (2016). Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the Rest of the World. Clin. Liver Dis. 20, 205–214. [DOI] [PubMed] [Google Scholar]
- Schumann A. M., Quast J. F., Watanabe P. G. (1980). The pharmacokinetics and macromolecular interactions of perchloroethylene in mice and rats as related to oncogenicity. Toxicol. Appl. Pharmacol. 55, 207–219. [DOI] [PubMed] [Google Scholar]
- Shaw P. J., Ganey P. E., Roth R. A. (2010). Idiosyncratic drug-induced liver injury and the role of inflammatory stress with an emphasis on an animal model of trovafloxacin hepatotoxicity. Toxicol. Sci. 118, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai M., Arakawa S., Miida H., Matsuyama T., Kinoshita J., Makino T., Kai K., Teranishi M. (2013). Thioacetamide-induced hepatocellular necrosis is attenuated in diet-induced obese mice. J. Toxicol. Pathol. 26, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel A., Itzel T., Erhart W., Brosch M., Wang X. Y., Kim Y. O., von Schonfels W., Herrmann A., Bruckner S., Stickel F., et al. (2016). Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 151, 513–525. [DOI] [PubMed] [Google Scholar]
- Tryndyak V., de Conti A., Kobets T., Kutanzi K., Koturbash I., Han T., Fuscoe J. C., Latendresse J. R., Melnyk S., Shymonyak S., et al. (2012). Interstrain differences in the severity of liver injury induced by a choline- and folate-deficient diet in mice are associated with dysregulation of genes involved in lipid metabolism. FASEB J. 26, 4592–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA (2011). Toxicological Review of Tetrachloroethylene (CAS No. 127-18-4): In Support of Summary Information on the Integrated Risk Information System (IRIS). U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- Varemo L., Nielsen J., Nookaew I. (2013). Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 41, 4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkel W., Friedewald M., Lederer E., Pahler A., Parker J., Dekant W. (1998). Biotransformation of perchloroethene: dose-dependent excretion of trichloroacetic acid, dichloroacetic acid, and N-acetyl-S-(trichlorovinyl)-L-cysteine in rats and humans after inhalation. Toxicol. Appl. Pharmacol. 153, 20–27. [DOI] [PubMed] [Google Scholar]
- Wahlang B., Song M., Beier J. I., Cameron Falkner K., Al-Eryani L., Clair H. B., Prough R. A., Osborne T. S., Malarkey D. E., Christopher States J., et al. (2014). Evaluation of Aroclor 1260 exposure in a mouse model of diet-induced obesity and non-alcoholic fatty liver disease. Toxicol. Appl. Pharmacol. 279, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright B. L., Jaeschke H. (2015). Xenobiotic and endobiotic mediated interactions between the cytochrome P450 system and the inflammatory response in the liver. Adv. Pharmacol. 74, 131–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H. S., Cichocki J. A., Kim S., Venkatratnam A., Iwata Y., Kosyk O., Bodnar W., Sweet S., Knap A., Wade T., et al. (2015). The contribution of peroxisome proliferator-activated receptor alpha to the relationship between toxicokinetics and toxicodynamics of trichloroethylene. Toxicol. Sci. 147, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi Z. M., Blissett D., Blissett R., Henry L., Stepanova M., Younossi Y., Racila A., Hunt S., Beckerman R. (2016a). The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 64, 1577–1586. [DOI] [PubMed] [Google Scholar]
- Younossi Z. M., Koenig A. B., Abdelatif D., Fazel Y., Henry L., Wymer M. (2016b). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data are available from GEO. All phenotypes (Supplementary Table 1), correlation matrices among the phenotypes (Supplementary Table 2), differentially expressed genes and enriched pathways (Supplementary Tables S3–S7) are provided as supplementary materials.