

The kidneys have long been known to play an important role in the metabolism of carbohydrates, proteins, lipids and other nutrients [1–3]. Proximal tubular cells generate glucose through gluconeogenesis, clear insulin from the circulation, perform critical steps of the urea cycle and regulate the metabolism of fat-soluble vitamins such as A and D. In chronic kidney disease (CKD), disruption of these functions may contribute to hypoglycemia, cachexia and cardiovascular diseases.
Insulin resistance, i.e. reduced sensitivity to the actions of insulin, has been extensively investigated as a mechanism through which CKD disrupts homeostasis and increases risks of morbidity and mortality [4]. Total body insulin-mediated glucose disposal measured using hyperinsulinemic-euglycemic clamp techniques, the gold standard measure of insulin sensitivity, is reduced in people with CKD compared with those with normal glomerular filtration rate (GFR) [5–8]. This insulin resistance is driven at least in part by impaired intracellular signaling after binding of insulin to its receptor in skeletal muscle, and obesity and physical inactivity each can contribute to this problem [8, 9]. Insulin resistance may promote cardiovascular diseases directly or through closely related processes such as endothelial dysfunction.
In health, pancreatic beta cells compensate for insulin resistance by secreting more insulin, thereby helping to maintain euglycemia. Insulin secretion is most accurately measured after administration of intravenous or oral glucose using plasma concentrations of C-peptide, which is co-secreted with insulin, but it is more commonly estimated as the plasma insulin response to glucose. Among people with normal glucose tolerance, there is a hyperbolic relationship between insulin sensitivity and the early insulin response to glucose, such that the insulin response increases as insulin sensitivity decreases due to adiposity, physical inactivity or other factors (Figure 1) [10]. Based on this relationship, the product of insulin sensitivity and the early insulin response has been utilized as a measure of beta-cell function and is termed the disposition index [11, 12]. Decreased insulin clearance commonly accompanies insulin resistance and can also increase circulating insulin concentrations, which may help compensate for insulin resistance [13]. When compensation for insulin resistance fails, glucose intolerance and ultimately type 2 diabetes mellitus ensues.
FIGURE 1.
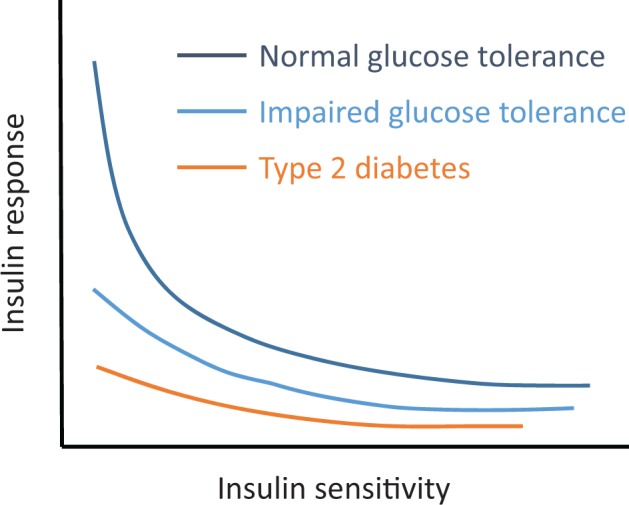
Hyperbolic relationship between beta-cell insulin response and insulin sensitivity. Among people with normal glucose tolerance, decreased insulin sensitivity (i.e. insulin resistance) is accompanied by an augmented insulin response that serves to maintain euglycemia. When insulin response is insufficient to compensate for prevailing insulin resistance, glucose tolerance and ultimately type 2 diabetes ensue. Adapted from Kahn et al. [10].
Is pancreatic beta-cell function affected by CKD? Certainly, the functions of many organs and endocrine systems are impaired in CKD. Moreover, recent work using the 5/6 nephrectomy mouse model and isolated pancreatic islet cells demonstrated that urea reduces the insulin response to glucose by increasing islet cell oxidative stress, O-GlcNAcylation and suppressing phosphofructokinase 1 [14]. The question is, therefore, plausible, and the answer may have important implications for the understanding of impaired systemic metabolism in CKD.
In this context, Guthoff et al. investigated the insulin response to glucose among 107 nondiabetic dialysis patients on a German kidney transplant waiting list, compared with control participants from the Tuebingen Family Study of people without diabetes but at increased risk of developing type 2 diabetes [15]. Matching control participants on age, gender and body mass index (BMI), insulin sensitivity measures were significantly lower among dialysis patients, as expected. Evaluating insulin response, the homeostasis model of assessment beta-cell score (HOMA-B, which is calculated from fasting glucose and insulin concentrations) was higher among dialysis patients (suggesting increased insulin response), and circulating concentrations of C-peptide were higher during an oral glucose tolerance test (OGTT). The insulinogenic index, a more accurate measure of the early insulin response to glucose obtained during the OGTT, was increased, though not significantly. Evaluating insulin response in the context of prevailing insulin sensitivity, the disposition index did not significantly differ.
In a second analysis, control subjects were matched to the dialysis patients on plasma glucose obtained 2h after a 75-g glucose load (a measure of glucose tolerance obtained during an OGTT), in addition to age, gender and BMI. Because insulin sensitivity and insulin response are the determinants of glucose tolerance, it is a truism that populations with reduced insulin sensitivity must have increased insulin response when matched on glucose tolerance. As expected, therefore, the insulin resistant dialysis patients had greater insulin response to glucose in this analysis.
Guthoff et al. conclude that insulin secretion is appropriately increased in end-stage renal disease (ESRD) to compensate for insulin resistance. This conclusion is supported by the higher insulin and C-peptide concentrations and the similar disposition index observed when comparing dialysis patients with controls matched on age, gender and BMI. However, there are important caveats to this conclusion.
First, the kidneys are important in the clearance of both insulin and C-peptide, making interpretations of insulin secretion based on measured circulating concentrations difficult. Insulin secretion is generally calculated using deconvolution of C-peptide kinetics [16]. This process relies on known values for renal clearance of C-peptide in normal individuals, which will be markedly diminished in the setting of ESRD. Thus, while conclusions regarding insulin responses that are based solely on circulating insulin concentrations may be valid, conclusions regarding insulin secretion that rely on C-peptide levels are problematic in this patient population. Second, HOMA-B is a poor measure of insulin secretion because it is obtained in the fasting state, in which it is difficult to disentangle insulin secretion and insulin resistance. Estimates of insulin resistance based on fasting measurement are biased in CKD, perhaps due in part to differences in insulin clearance [17], and this bias is likely to extend to HOMA-B. The OGTT-based insulinogenic index is a much better test of beta-cell function and was non-significantly higher in ESRD.
Third, the analysis matching participants on 2-h OGTT glucose cannot be used to assess insulin response in the ESRD population, as it simply demonstrates the known paradigm that insulin secretion and insulin sensitivity together determine glucose tolerance. Most importantly, the populations studied may have introduced a selection bias. Dialysis patients on the kidney transplant waiting list, studied here, are well known to be healthier than those who are not listed for transplant. On the other hand, control subjects were drawn from a population selected for increased risk of type 2 diabetes, a disease defined by impaired beta-cell function. The comparison of dialysis patients who were healthier than average with control participants who may have had an increased prevalence of beta-cell dysfunction may have obscured defects in insulin secretion in ESRD.
The study by Guthoff et al. is notable in part because there is currently a scarcity of data addressing insulin secretion in CKD. In early studies of ESRD patients, DeFronzo et al. found a large range of insulin response in response to insulin resistance [18]. In a study comparing participants with Stages 3–5 predialysis CKD versus controls with normal estimated GFR (eGFR), we found that insulin response to intravenous or oral glucose was not reduced with CKD, but it also did not significantly increase to compensate for insulin resistance; as a result, glucose intolerance was common in the CKD participants, albeit not significantly more so than the control subjects [8]. Like the study by Guthoff et al., our study was inconclusive as to the association of CKD with insulin secretion.
Beta-cell failure is the proximate cause of type 2 diabetes mellitus, and one would expect that populations with impaired beta-cell function would go on to develop diabetes over time. At least three community-based cohort studies have tested the hypothesis that CKD is a risk factor for incident diabetes. In two cohort studies, lower eGFR or higher serum cystatin C concentration were associated with increased risk of developing type 2 diabetes [19, 20]. In the third study, eGFR <60 mL/min/1.73 m2 was associated with insulin resistance but also increased insulin response (measured by OGTT), and no difference in risk of incident diabetes [21].
In summary, the published literature available today does not provide a clear picture of pancreatic function in CKD. Additional well-designed human studies could determine whether there are clinically important defects in beta-cell function that could be targeted to improve glucose homeostasis in CKD. In addition to beta-cell function, little is known about pancreatic alpha-cell function in CKD. Moreover, insulin secretion has implications for other metabolic pathways, e.g. protein and lipid metabolism. Further investigation in this area is likely to help understanding of the complex metabolic abnormalities present in CKD and perhaps offer new strategies for reducing morbidity and mortality in the high-risk CKD population.
ACKNOWLEDGEMENTS
I.H.d.B. is supported by grants R01DK088762 and R01DK099199 from the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK), grant R01HL096875 from the National Heart Lung and Blood Institute (NHLBI), grant 4-15-CKD-20 from the American Diabetes Association, an unrestricted fund from Northwest Kidney Centers and the Veteran Affairs Administration. K.M.U. is supported by the Veteran Affairs Administration and NIH grants R01DK092568-01, U01DK098246, P30DK017047 and U01DK094406.
CONFLICT OF INTEREST STATEMENT
None declared.
(See related article by Guthoff et al. Impact of end-stage renal disease on glucose metabolism—a matched cohort analysis. Nephrol Dial Transplant 2017; 32: 670--676)
REFERENCES
- 1. Zubrod CG, Eversole SL, Dana GW.. Amelioration of diabetes and striking rarity of acidosis in patients with Kimmelstiel-Wilson lesions. N Engl J Med 1951; 245: 518–528 [DOI] [PubMed] [Google Scholar]
- 2. Hutchings R, Hegstrom R, Scribner BH.. Glucose intolerance in patients on long-term intermittent dialysis. Ann Intern Med 1966; 65: 275–285 [Google Scholar]
- 3. Joven J, Vilella E, Ahmad S. et al. Lipoprotein heterogeneity in end-stage renal disease. Kidney Int 1993; 43: 410–418 [DOI] [PubMed] [Google Scholar]
- 4. Spoto B, Pisano A, Zoccali C.. Insulin resistance in chronic kidney disease: a systematic review. Am J Physiol Renal Physiol 2016; 311: F1087–F1108 [DOI] [PubMed] [Google Scholar]
- 5. DeFronzo RA, Alvestrand A, Smith D. et al. Insulin resistance in uremia. J Clin Invest 1981; 67: 563–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fliser D, Pacini G, Engelleiter R. et al. Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int 1998; 53: 1343–1347 [DOI] [PubMed] [Google Scholar]
- 7. Pham H, Utzschneider KM, de Boer IH.. Measurement of insulin resistance in chronic kidney disease. Curr Opin Nephrol Hypertens 2011; 20: 640–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Boer IH, Zelnick L, Afkarian M. et al. Impaired glucose and insulin homeostasis in moderate-severe CKD. J Am Soc Nephrol 2016; 27: 2861–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bowlby W, Zelnick LR, Henry C. et al. Physical activity and metabolic health in chronic kidney disease: a cross-sectional study. BMC Nephrol 2016; 17: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kahn SE, Prigeon RL, McCulloch DK. et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 1993; 42: 1663–1672 [DOI] [PubMed] [Google Scholar]
- 11. Utzschneider KM, Prigeon RL, Carr DB. et al. Impact of differences in fasting glucose and glucose tolerance on the hyperbolic relationship between insulin sensitivity and insulin responses. Diabetes Care 2006; 29: 356–362 [DOI] [PubMed] [Google Scholar]
- 12. Utzschneider KM, Prigeon RL, Faulenbach MV. et al. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 2009; 32: 335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haffner SM, Stern MP, Watanabe RM. et al. Relationship of insulin clearance and secretion to insulin sensitivity in non-diabetic Mexican Americans. Eur J Clin Invest 1992; 22: 147–153 [DOI] [PubMed] [Google Scholar]
- 14. Koppe L, Nyam E, Vivot K. et al. Urea impairs beta cell glycolysis and insulin secretion in chronic kidney disease. J Clin Invest 2016; 126: 3598–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guthoff M, Wagner R, Vossler D. et al. Impact of end-stage renal disease on glucose metabolism—a matched cohort analysis. Nephrol Dial Transplant 2017; 32: 670--676 [DOI] [PubMed] [Google Scholar]
- 16. Van Cauter E, Mestrez F, Sturis J. et al. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992; 41: 368–377 [DOI] [PubMed] [Google Scholar]
- 17. Ahmad I, Zelnick LR, Robinson NR. et al. Chronic kidney disease and obesity bias surrogate estimates of insulin sensitivity compared to the hyperinsulinemic-euglycemic clamp. Am J Physiol Endocrinol Metab 2017; doi:10.1152/ajpendo.00394.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeFronzo RA, Tobin JD, Rowe JW. et al. Glucose intolerance in uremia. Quantification of pancreatic beta cell sensitivity to glucose and tissue sensitivity to insulin. J Clin Invest 1978; 62: 425–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lorenzo C, Nath SD, Hanley AJ. et al. Risk of type 2 diabetes among individuals with high and low glomerular filtration rates. Diabetologia 2009; 52: 1290–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sahakyan K, Lee KE, Shankar A. et al. Serum cystatin C and the incidence of type 2 diabetes mellitus. Diabetologia 2011; 54: 1335–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pham H, Robinson-Cohen C, Biggs ML. et al. Chronic kidney disease, insulin resistance, and incident diabetes in older adults. Clin J Am Soc Nephrol 2012; 7: 588–594 [DOI] [PMC free article] [PubMed] [Google Scholar]