

Abstract
Aims
Lp(a) concentrations represent a major cardiovascular risk factor and are almost entirely controlled by one single locus (LPA). However, many genetic factors in LPA governing the enormous variance of Lp(a) levels are still unknown. Since up to 70% of the LPA coding sequence are located in a difficult to access hypervariable copy number variation named KIV-2, we hypothesized that it may contain novel functional variants with pronounced effects on Lp(a) concentrations. We performed a large scale mutation analysis in the KIV-2 using an extreme phenotype approach.
Methods and Results
We compiled an discovery set of 123 samples showing discordance between LPA isoform phenotype and Lp(a) concentrations and controls. Using ultra-deep sequencing, we identified a splice site variant (G4925A) in preferential association with the smaller LPA isoforms. Follow-up in a European general population (n = 2892) revealed an exceptionally high carrier frequency of 22.1% in the general population. The variant explains 20.6% of the Lp(a) variance in carriers of low molecular weight (LMW) apo(a) isoforms (P = 5.75e-38) and reduces Lp(a) concentrations by 31.3 mg/dL. Accordingly the odds ratio for cardiovascular disease was reduced from 1.39 [95% confidence interval (CI): 1.17–1.66, P = 1.89e-04] for wildtype LMW individuals to 1.19 [95%CI: 0.92; 1.56, P = 0.19] in LMW individuals who were additionally positive for G4925A. Functional studies point towards a reduction of splicing efficiency by this novel variant.
Conclusion
A highly frequent but until now undetected variant in the LPA KIV-2 region is strongly associated with reduced Lp(a) concentrations and reduced cardiovascular risk in LMW individuals.
Keywords: Lipoprotein(a), LPA, CVD risk, Kringle IV-type 2, Copy number variation
Translational perspective
A novel frequent variant in LPA specifically reduces Lp(a) in carriers of low molecular weight (LMW) Lp(a) isoforms by 31.3 mg/dL. LMW individuals commonly present an increased cardiovascular risk, but positive carrier status for the variant abolishes this increased risk. This has implications for clinical practice, patient stratification, and treatment development. The selective reduction of Lp(a) in LMW carriers and the resulting reduction in cardiovascular risk significantly adds to previous studies indicating that Lp(a) lowering can be beneficial. Moreover, our data indicate splicing modulation as a potential novel mechanisms for Lp(a) lowering.
Introduction
Increased Lipoprotein(a) [Lp(a)] levels are present in 15–25% of the population1 and confer an up to 2.5-fold increased cardiovascular disease (CVD) risk.2 Since Lp(a) levels are almost entirely controlled by one single gene locus, the LPA gene, they represent one of the most important genetically determined CVD risk factors. LPA encodes the major structural protein of Lp(a),3 apolipoprotein(a) [apo(a)], and accounts for >90% of Lp(a) variance.3
The LPA gene presents a peculiar gene structure, consisting of several, highly homologous ‘kringle-domains’ (K) named KIV type 1 to 10 (KIV-1 to KIV-10). Of note, the KIV-2 domain is encoded by an hypervariable, 5.6 kb sized, coding copy number variation (CNV), that encompasses two exons and is present in up to >40 copies per allele causing a heterozygosity level in the population of >95%.3 It thus generates >40 protein size isoforms in the population. Individuals with at least one low molecular weight allele (LMW; <14 KIV-2 repeats which corresponds to <23 total KIV domains) present 5–10-fold higher median Lp(a) plasma levels (≈50 mg/dL) than individuals with high molecular weight isoforms (HMW; ≥23 KIV; medians ≈5–10 mg/dL).1 Therefore in heterozygous individuals, the shorter allele determines assignment to LMW or HMW group and subsequently the CVD risk.4 Of note, about 30% of the population miss one isoform in plasma despite being heterozygous on DNA level. The non-secreted allele has been termed as ‘operational null allele’5 and in Caucasians, HMW alleles are more likely to be non-expressed than LMW alleles.
The number of KIV-2 repeats explains 40–70% of Lp(a) variance,3 but Lp(a) levels can vary by 200-fold even for alleles with the same size.6,7 This suggests the existence of still unknown modifying genetic variants. Genome-wide association studies indeed reported an impressively large number of independent variants in LPA,8 but causal effects on Lp(a) levels have been shown only for few.9
Depending on the isoform size, up to 70% of the protein coding sequence is derived from the KIV-2 CNV (see Supplementary material online, Figures S1 and S2), but this region remained inaccessible to large-scale mutation analysis until recently, since sensitivity of Sanger sequencing was insufficient to detect one variant in up to 80 KIV-2 copies.10 This resembles observations in other genes with multi-copy coding regions, where novel disease-causing variants were found in similar ‘blind spots’ of the genome (e.g. MUC1, MUC5AC, Kir2.6).11 The KIV-2 region thus represents a natural candidate region for new causal variants.
The ‘extreme-phenotype approach’12 is a powerful instrument to enrich for functional mutations. The association between LMW apo(a) isoforms and high Lp(a) concentrations is well established.3 We therefore reasoned that samples deviating clearly from this pattern can be seen as ‘extreme phenotype’-samples and possibly carry novel causal variants. We therefore compiled 123 samples for sequencing. This set comprised samples showing discordance between Lp(a) isoform phenotypes and Lp(a) concentrations as well as control samples. We sequenced the KIV-2 region in all samples, evaluated the effects of a promising KIV-2 splice site variant on Lp(a) concentrations in an European general population sample (n = 2892) and determined its impact on CVD risk in a high risk population. Subsequently, we confirmed its allelic localization using pulsed-field gel electrophoresis and investigated its effects on invitro splicing assays and human liver mRNA.
Methods
Study design
Lp(a) levels and apo(a) isoforms were determined in three populations (ntotal = 9581): SAPHIR (healthy working population, Austria, n = 1519), GCKD (German Chronic Kidney Disease study, n = 5170) and KORA F4 (population-based study, Southern Germany, n = 2892). Details are given in Supplementary material online, Methods and Table S1. A ‘discovery sample’ of 123 individuals for sequencing was drawn from SAPHIR and GCKD by belonging to either an extreme phenotype (low Lp(a) levels despite LMW isoform or high Lp(a) values despite HMW isoform) or a control group with expected Lp(a) levels (Table 1, see Supplementary material online, Table S2 and Methods). A splice site variant and/or its corresponding proxy single nucleotide polymorphism (SNP) was followed up in the entire KORA F4 and GCKD studies.
Table 1.
Phenotypes of the discovery set
| Phenotype | Group | n | Lp(a) mg/dL [median (IQR)] | Total KIV-2 repeats (qPCR) [median (IQR)] | Short isoform according to Western blot [median (IQR)] | G4925A carrier, n (%) |
|---|---|---|---|---|---|---|
| HMW isoform, low Lp(a) | Controls | 28 | 3.1 (1.4–5.5) | 47 (43–50) | 33 (30–36) | 4 (14.3) |
| LMW isoform, high Lp(a) | Controls | 15 | 55.7 (51.8–83.9) | 32 (30–36) | 15 (15–20) | 1 (6.7) |
| High Lp(a) despite HMW isoform | Extreme phenotype | 33 | 66.0 (55.4–73.9) | 43 (40–47) | 30 (29–33) | 1 (3.0) |
| Low Lp(a) despite LMW isoform | Extreme phenotype | 31 | 4.2 (3.0–6.9) | 39 (36–44) | 20 (19–22) | 22 (71.0) |
| No isoforms detectable in Western blot | Extreme phenotype | 14 | 0.6 (0.3–0.9) | 56 (47–59) | n.a. | 2 (14.3) |
| Othera | Extreme phenotype | 2 | n.a. | n.a. | n.a. | 2 (100) |
| Total | 123 |
HMW, high molecular weight; IQR, interquartile range; LMW, low molecular weight; n.a., not applicable.
1× multiple bands in the Western blot, 1× very faint 20 repeats isoform in the Western blot.
Lp(a) phenotyping and genotyping
All Lp(a) phenotyping was performed centrally at the Division of Genetic Epidemiology, Innsbruck, Austria and evaluated by the same experienced researcher, who was not involved in sequencing. Lp(a) concentration was determined by ELISA and apo(a) isoform phenotyping was done by Western blotting (see Supplementary material online, Methods). Peculiar Western blot phenotypes were confirmed by repeated measurements. For semi-quantitative determination of the relative contribution of both alleles to the total Lp(a) amount in plasma in heterozygous individuals, the sum of both bands in Western Blot was assumed to be 100% and the relative contribution of the smaller isoform was estimated.
KIV-2 batch amplification and next-generation sequencing
The KIV-2 region was amplified using a ‘batch amplification’ approach,10,13 which leverages the homology of KIV-2 repeats to simultaneously amplify all KIV-2 repeats as an amplicon mixture (Figure 1) and subjected to ultra-deep next-generation sequencing (NGS). Like mitochondrial heteroplasmies, variants occurring only in a subset of repeats will be present only in a corresponding subfraction of NGS reads. Data analysis was therefore performed by aligning all reads to one single kringle repeat and low-level variants were called using the highly specific14 mutation detection pipeline ‘mtDNA-Server’14 with slight adaptions (https://mtdna-server.uibk.ac.at/, 25 March 2017) (see Supplementary material online, Methods). The LPA KIV-2 specific variant calling pipeline will be available at lpa-server.i-med.ac.at, 25 March 2017 (manuscript in preparation). Two known splice site defects (rs41272114 and rs14343136815) were determined by Sanger sequencing (see Supplementary material online, Tables S3 and S4). See Supplementary material online, Methods for details, reference sequence selection strategy and analysis of 1000Genomes and GenomeAustria datasets.
Figure 1.

Concept of the batch sequencing approach. All KIV-2 repeats are amplified with the same primer pair by leveraging the high homology. By aligning all repeats to one single copy, a variant occurring in only one repeat will be present only in a subset of amplicons. This resembles mitochondrial heteroplasmies or somatic mutations and can thus be detected using the same approaches.
G4925A genotyping by competitive allele-specific TaqMan PCR
Given the technically demanding necessity to type a variant within up to >70 KIV-2 copies, we used a custom-made commercial competitive allele-specific TaqMan PCR (castPCR)16 assay (competitive allele-specific TaqMan PCR; also known as ASB-PCR) (ThermoFisher Scientific). This allele specific qPCR assay allows detection of somatic mutations down to at least 0.1% level16 and thus provides sufficient sensitivity for determining SNPs present in one of >70 KIV-2 repeats. For technical details, quality figures from assay validation and quality control measures see Supplementary material online, Methods.
Minigene assays and liver mRNA analyses
The G4925A variant is located at the donor splice site of the second KIV-2 exon, which is spliced to the first exon of the next KIV-2. Therefore a PCR fragment spanning the short intron between two KIV-2 repeats as well as the two boundary exons was subcloned into a pSPL317 minigene plasmid by standard PCR subcloning techniques (see Supplementary material online, Figures S3 and S4). Invitro splicing assays were performed in HepG2. Details are given in Supplementary material online, Tables S5 and S6. In order to study mRNA from G4925A carriers, we obtained liver biopsy samples from a G4925A carrier and three G4925A-negative controls, amplified the G4925A context from both mRNA and pre-mRNA and analysed these by NGS and castPCR. Additionally, the presence of exon skipping was investigated (see Supplementary material online, Methods).
Pulsed field gel electrophoresis
Apo(a) isoforms in plasma and G4925A carrier status were determined by Western blotting and castPCR in 40 fresh blood samples from anonymous donors (Central Institute for Blood Transfusion, State Hospital of Innsbruck, Austria). Seven out of 11 G4925A carriers presented both apo(a) alleles in plasma and 5 of these were subjected to KIV-2 genotyping by pulsed field gel electrophoresis (PFGE) together with six G4925A-negative controls. PFGE allows the determination of the number of both KIV-2 present on genomic DNA,10 independently from the occurrence of operational null alleles (see Supplementary material online, Methods).
Statistical methods
Prevalence of G4925A in the discovery set phenotypic groups was compared by Fisher exact test (two-sided) and relative contribution of the smaller band in the Western blot to the total signal in Western blot was compared by Wilcoxon test. The association of G4925A with Lp(a) was tested in the entire KORA F4 study and in LMW/HMW subgroups by linear regression analysis. In KORA F4 and GCKD, regression analysis on Lp(a) was also performed for the SNP rs75692336, which tags G4925A. Finally, effects of the tagging SNP rs75692336, which serves as a proxy for G4925A, on CVD prevalence in the GCKD study were tested by a logistic regression. For details on statistical methods and the tagging SNP approach see the Supplementary material online, Methods.
Results
Mutation analysis
We ultra-deep sequenced the KIV-2 repeat in 123 individuals [drawn from the 2 populations (SAPHIR and GCKD, ntotal = 6689)] either showing discordance between Lp(a) isoform phenotype and Lp(a) concentrations: (i) low Lp(a) despite LMW isoform, (ii) high Lp(a) despite HMW isoform, (iii) no detectable isoform in blood or to a control group, (iv) LMW with high Lp(a) concentrations, and (v) HMW with expected Lp(a) concentrations (Table 1, see Supplementary material online, Table S2).
The two known functional SNPs rs41272114 and rs14343136815 did not explain the observed extreme phenotypes (see Supplementary material online, Table S7). Ultra-deep NGS (average depth: 24 418×) and dedicated low-level mutation detection of ‘batch amplification’13 products (Figure 1) revealed, however, a novel variant in the exonic donor splice site of the second KIV-2 exon, which was strongly over-represented in the phenotype ‘low Lp(a) levels despite LMW isoform’ (71% carriers compared to 11% in all other groups combined, PFisher = 4.47e-10; Table 1). Since unique position annotation is not possible, we use our working name G4925A throughout the manuscript [Human Genome Variation Society (HGVS) annotation provided in Supplementary material online, Methods]. By dividing the NGS mutation level by the total number of KIV-2 repeats estimated by KIV-2 qPCR, we estimate that the variant occurs mostly in only one or two of all KIV-2 repeats present in an individual (see Supplementary material online, Figure S5). Despite its seemingly high frequency and its promising location, the variant has thus likely escaped detection in earlier studies10,13 due to insufficient sensitivity of Sanger sequencing.
Next-generation sequencing validation and population data
We analysed three public datasets [1000G Exome samples with >1000× coverage at the variant position (n = 1589, 26 populations), 1000G high-coverage whole-genome samples (n = 30), GenomeAustria whole-genome samples (n = 8)] to exclude technical artefacts in our sequencing procedure and investigate the frequency of G4925A in different populations on the other hand (see Supplementary material online, Methods and Tables S4–S7). Despite different sequencing technologies, G4925A was observed in all three control datasets. Largely varying frequencies were observed in the 1000Genomes populations (Figure 2, see Supplementary material online, Tables S8–S11).
Figure 2.

G4925A carrier frequency in the 1000 Genomes populations. Only exome data with >1000× coverage from unrelated samples were used (n = 1589). The total number of individuals passing the coverage requirements and the G4925A overall frequency (F) in each super population is reported on the x-axis. Details and population code are given in Supplementary material online, Tables S8 and S9. Admixed African populations (ACB and ASW) were excluded from this figure, but are reported in Supplementary material online, Table S8. In Chinese Dai in Xishuangbanna (CDX), only five individuals passed the coverage requirements.
Association study in the general population
To assess the impact of G4925A in the general population, we determined the G4925A carrier status in the population-based study KORA F4 (n = 2892; Southern Germany) using highly sensitive castPCR (validation results shown in Supplementary material online, Table S12). The frequency of G4925A carriers in KORA F4 was 22.1% and carriers preferentially presented a smaller apo(a) isoform with 19-25 KIV (Figure 3A). 56.4% of all carriers presented a smaller apo(a) isoform with 23 or 24 KIV. These isoforms represent a threshold at which the median Lp(a) concentrations suddenly decrease from ≈45–50 mg/dL in carriers of isoforms with ≤22 KIV to ≈10 mg/dL in carriers of ≥23 KIV isoforms1 (see Supplementary material online, Figure S6). This sudden decrease is smoothened when accounting for G4925A carrier status (Supplementary Figure S6), thus indicating that G4925A might substantially account for this observation.
Figure 3.

Distribution and effects of G4925A. (A) Carrier frequency in the single isoform groups (based on the smaller isoform). The values report the percentage of wild type (wt), respectively G4925A carriers that present the mentioned isoform. The variant clusters in isoforms ≈19–25, peaking at 23 and 24 (32.7% and 23.7% of all carriers). (B) Median Lp(a) concentrations depending on isoform and carrier status (solid line). The variant is associated with lower Lp(a) concentrations throughout the whole isoforms range with the most pronounced effect in LMW carriers (defined ≤22 KIV repeats). Dotted lines: 1st and 3rd quartile. Isoform was binned to encompass ≥10 carrier per group (except for isoforms >34, n = 9). WB: Western blot.
G4925A carrier status was highly significantly associated with inverse-normal transformed Lp(a) concentrations in the clinically relevant LMW group (encompassing 24.6% of the population): positive G4925A carrier status was associated with Lp(a) reduction by 31.3 mg/dL, explaining 20.6% of Lp(a) variance (P = 5.75e-38) (Figure 3B). After adjustment for KIV number, the association became highly significant also in the total population (P = 1.05e-55) and in the HMW group (KORA F4 HMW: P = 1.69e-10). In contrast, the association in the LMW group was virtually unaffected by isoform adjustment (P = 3.0e-36). Full results are reported in Table 2.
Table 2.
Results of association study of G4925A on Lp(a) concentrations
| Group | βinverse norm | βoriginal | P-value | Variance explained (%)a |
|---|---|---|---|---|
| Unadjusted model | ||||
| KORA F4 all (n = 2892) | −0.122 | −8.1 | 0.006 | 0.2 |
| KORA F4 LMW only (n = 716) | −0.891 | −31.3 | 5.57e-38 | 20.6 |
| KORA F4 HMW only (n = 2176) | 0.054 | −2.5 | 0.231 | 0.02 |
| Adjusted model (isoform-adjusted) | ||||
| KORA F4 all (n = 2864) | −0.618 | −21.3 | 1.05e-55 | 6.1 |
| KORA F4 LMW only (n = 715) | −0.868 | −30.6 | 3.0e-36 | 19.3 |
| KORA F4 HMW only (n = 2149) | −0.310 | −10.0 | 1.69e-10 | 1.6 |
P-value, βinverse norm, and explained variance (R2) are based on linear regression from G4925A carrier status on inverse normal-transformed Lp(a) concentrations. Additional effect estimates were obtained from linear regression on the original Lp(a) scale in mg/dL (βoriginal).
For the adjusted model, this represents the variance explained by G4925A additionally to apo(a) isoforms.
Association with cardiovascular disease in the GCKD study group
We reasoned that the strong Lp(a) reduction in LMW individuals with positive G4925A carrier status may alleviate LMW-mediated CVD risk. A direct evaluation of the effect of G4925A on CVD is hampered by the fact that variants in KIV-2 are not contained in the imputed whole-genome SNP datasets commonly available in large, genetic-epidemiological studies on CVDs. By combining 1000G-imputed SNP data and G4925A data in KORA F4 (see Supplementary material online, Methods), we identified rs75692336 as proxy for G4925A (r2 = 0.82, D’ = 0.99) (see Supplementary material online, Table S13). The minor allele of rs75692336 was associated with a similar, but slightly smaller Lp(a) reduction than G4925A (see Supplementary material online, Table S14).
This proxy SNP was then used to assess the effect of G4925A on CVD in the high risk GCKD population. rs75692336 was associated with lower odds ratio (OR) for CVD following a dominant model (see the Supplementary material online, Methods for the rationale of the model selection). When comparing LMW individuals of GCKD to HMW individuals of GCKD, the LMW individuals present an increased risk for CVD, as would be expected4 (OR = 1.34, P = 6.29e-05) (Table 3). However, when the population is stratified according to G4925A carrier status, this risk elevation in LMW individuals carrying also a minor allele of rs75692336 is no longer significant (OR = 1.19, P = 0.19) (Table 3). Conversely, in LMW individuals not carrying G4925A the CVD risk was even slightly higher than in the whole population (OR = 1.39, P = 1.89e-04). The results from the extended adjustment model remained very consistent to the unadjusted model (Table 3).
Table 3.
Association of LMW apo(a) isoforms with cardiovascular disease risk in the GCKD study (compared to high molecular weight isoform), for all participants and also stratified by the carrier status for at least one rare allele of rs75692336 (proxy to G4925A)
| Number (%) of CVD events in |
Unadjusted model |
Adjusted modela |
||||
|---|---|---|---|---|---|---|
| Group | LMW group | HMW group | OR [95% CI] (LMW vs. HMW carriers) | P-value | OR [95% CI] (LMW vs. HMW carriers) | P-value |
| All (n = 4883) | 368 of 1208 (30.5%) | 905 of 3675 (24.6%) | 1.34 [1.16–1.54] | 6.29e-05 | 1.35 [1.16;1.58] | 1.49e-04 |
| rs75692336 carrier (CA/AA) (n = 1151) | 127 of 425 (29.9%) | 191 of 726 (26.3%) | 1.19 [0.92–1.56] | 0.19 | 1.21 [0.90;1.63] | 0.20 |
| rs75692336 non-carrier (CC) (n = 3732) | 241 of 783 (30.8%) | 714 of 2949 (24.2%) | 1.39 [1.17–1.66] | 1.89e-04 | 1.40 [1.16;1.70] | 4.41e-04 |
HMW, high molecular weight; LMW, low molecular weight; CVD, cardiovascular disease; CI, confidence interval; OR, odds ratio.
Adjusted for age, gender, type 2 diabetes, LDL-cholesterol corrected for lipid-lowering medications and Lp(a), log(triglycerides), eGFR and log(UACR).
Molecular analysis of G4925A localization
Figure 3A suggests that the G4925A is located on smaller LPA alleles and Figure 4 shows that in KORA F4 samples presenting both isoforms in plasma the contribution of the small allele to the total Western blot signal was significantly lower in G4925A carriers than in wild-type samples [PWilcoxon = 0.0041 (all samples) and PWilcoxon = 2.42e-09 (LMW samples only); Figure 4]. This indicates a reduced protein amount originating from this allele. To investigate whether the variant is truly located on the short allele, we selected 11 healthy volunteers (five G4925A carriers), who presented both protein isoforms in plasma, separated genomic LPA alleles by PFGE, isolated the 2 alleles from the gel as previously described10 and tested the separated alleles by castPCR. In all carriers, the G4925A variant was indeed localized of on the Lp(a) level-determining short LPA allele. This provides an important prerequisite for a true functional impact of G4925A on Lp(a) levels.
Figure 4.
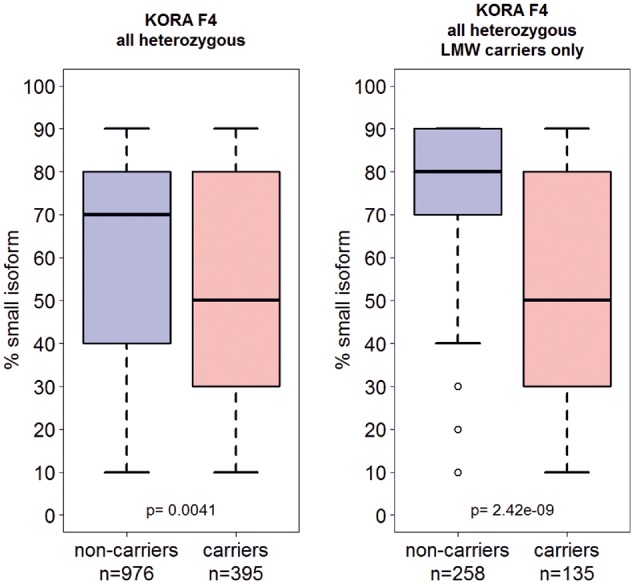
Relative contribution of the smaller apo(a). Western blot band to the total Western blot signal. Assuming the summed Western blot signal of both alleles being 100%, we estimated the contribution of the smaller allele to the total (see Methods). The contribution of the smaller allele to the circulating Lp(a) was significantly lower in carriers of G4925A (Wilcoxon test).
In vitro analysis of functional effects of G4925A
Although occurring at a splice site, G4925A was not associated with null alleles (see Supplementary material online, Figure S7). Since LPA is exclusively liver-expressed3 and human mRNA is therefore utmost difficult to obtain, we used comprehensive pSPL317 minigenes (see Supplementary material online, Figure S3; assay concept explained in Supplementary material online, Figure S8) to assess splicing patterns in HepG2 cells. In our analyses, the minor allele (A) abolished splice site recognition invitro and activated a cryptic intronic splice site leading to retention of the first 87 intronic bases and resulting in inclusion of a premature stop codon invitro (see Supplementary material online, Figures S4 and S9). The comparison of PFGE and Western blot data from G4925A carriers and controls (see Supplementary material online, Table S15) indicated that invivo the reduced splicing efficiency of the minor allele does not result in a complete protein truncation, but rather in a clear reduction of the abundance of the (variant-carrying) small allele (Figure 4). To investigate this observation, we obtained mRNA from liver biopsies of a G4925A carrier and three controls and analysed a RT-PCR product, which bridged the exon–intron boundary at G4925A (see Supplementary material online, Figure S10) by deep sequencing. The occurrence of the A-allele in the mature mRNA was below detection limit and only the G allele was detected, while a castPCR on an amplicon amplifying the G4925A context sequence from pre-mRNA instead of mature mRNA was still positive for the A allele, suggesting loss of the variant A allele during mRNA maturation (discussed below). No exon skipping was observed (see Supplementary material online, Figure S11).
Discussion
The LPA gene represents a major CVD risk locus and the measurement of Lp(a) is especially recommended in people with high CVD risk or a strong family history of premature atherothrombotic disease.18 Several independent association signals for Lp(a) have been described in LPA region,8 but a consistent portion was inaccessible to mutation screening until recently. Accordingly, factors governing the variance of the Lp(a) besides the KIV-2 repeat are still largely unknown.
In the work at hand, we identified a novel variant that overrides the major effect of LMW isoforms on Lp(a) concentrations and is associated with a pronounced Lp(a) reduction. It exerts its largest effect in the LMW group (Figure 3), which is commonly associated with increased Lp(a) levels and CVD risk.4 Conversely, in the HMW group and in the total population its effect was initially masked by its association with smaller isoforms at 23–25 KIV (explained detail in Supplementary material online, Figure S12). This observation reminds the regulatory variant rs1853021, whose effect is masked by its association with HMW isoforms,19 and exemplifies how failure to account for apo(a) isoforms in association studies can mask the effect of high-impact variants. It suggests a complex interplay of KIV-2 number and SNPs in determining the Lp(a) levels. G4925A seems instrumental both in the large concentration range observed especially in LMW isoforms (Figure 3) as well as in the sudden Lp(a) decrease at 23-24 KIV (see Supplementary material online, Figure S6).
Using a proxy SNP present in common imputed data sets we finally show that the G4925A carrier status modifies the genetic CVD risk of LMW individuals (which would otherwise be at high genetic CVD risk). This confirms recent studies on other, rarer variants associated with low Lp(a),15,20 which reported similar CVD risk reductions. Given the high frequency in the population, we anticipate, that inclusion of G4925A status in studies assessing the impact of Lp(a) isoforms may increase discrimination.
The mechanism behind G4925A-mediated Lp(a) lowering is intriguing and warrants further research. We show that G4925A affects splice site recognition invitro. By combining Western blot and castPCR on PFGE-separated alleles, we also show that G4925A is indeed located on the Lp(a) level-determining small allele and is associated with a reduced amount of protein originating from this allele (Figure 4). The detection of a guanosine instead of the expected adenosine in the mature liver mRNA of a mutation carrier would be consistent with an ‘A-to-I RNA editing’21 mechanism, since the expected adenosine was still detected in amplicons on pre-mRNA from of the same sample.
A-to-I RNA editing happens during maturation of pre-mRNA to mRNA where ADAR enzymes desaminate adenosines to inosines that are then interpreted by the cell (and PCR) as guanosines.21 Indeed, in silico mRNA structure modelling predicts extended double-stranded RNA structures at G4925A (see Supplementary material online, Figure S13), which represent canonical RNA editing recognition motifs.21 In addition, both the nucleotide context of G4925A (5’-A-[G4925A]-G-3’)22 and inefficient splicing23 have been shown to strongly promote editing. We hypothesize that G4925A reduces splice site recognition, as shown by our invitro assays, which in turn prolongs nuclear retention and promotes so editing of the A-allele to the wild-type guanosine, explaining so why only guanosines are detected in RT-PCR NGS on mature mRNA. Not much is known about editing of protein-coding genes, but hyperedited Alu repeat mRNA has been shown to undergo mRNA decay.21 Several lines of evidence therefore argue for a combination of reduced splicing efficiency and possibly RNA editing-related processes as mechanistic basis for the observed association. These mechanisms might finally lead to the observed reduced contribution of the short allele to the total apo(a) protein level in plasma. Indeed, Lp(a) levels are known to be mainly determined by synthesis rate9 and are modifiable by LPA expression modulation (reviewed in3). Future, in-depth studies in large collections of native human liver samples will be needed to test this working hypothesis.
Our study has strengths and limitations. We describe a novel, highly frequent variant in the highly repetitive KIV-2 with a pronounced effect on Lp(a) concentrations and describe methods to type this variant. The variant was confirmed in 1000G data and in a denovo typed, large Caucasian general population-based sample. The widely varying levels between the different 1000G populations argue against ‘phantom mutation’ at a sequencing error hot spot. We also show that the variant reduces CVD risk in individuals at otherwise high genetic CVD risk. The use of a proxy SNP presents for CVD risk estimation can be seen as a limitation but is supported by the very similar effect size of the proxy on Lp(a) and allows the replication and extension of our findings by others (provided LPA isoform data for adjustment are available). Additional replication in possibly even larger studies on CVD with apo(a) isoform data available will be crucial to establish the role of our findings. Albeit G4925A location is likely to be functional and we provide first mechanistic insights in the Lp(a) lowering mechanism, we clearly cannot completely exclude that a variant in linkage disequilibrium is the real causal one.
In conclusion, our investigation summarized in Figure 5 adds to previous works showing the utility of sequencing under genome-wide association study (GWAS) peaks. We identified a highly frequent splice site variant, which markedly reduces CVD risk in LMW carriers and is instrumental in the strikingly large range of Lp(a) levels observed in LMW carriers.6,7 Much emphasis has been put on the deleterious consequences of high Lp(a) (reviewed in24), but Lp(a) function remains elusive. The investigation of variants causing low Lp(a) might therefore provide novel perspectives (as recently shown15,25) and our observations on G4925A-mediated Lp(a) reduction highlights splicing efficiency modulation by antisense oligos or transsplicing26 as a potential novel Lp(a) lowering approach .
Figure 5.

Summarizing illustration of the key messages of this article.
Supplementary material
Supplementary material is available at European Heart Journal online.
Supplementary Material
Acknowledgements
We would like to express our gratitude to Thomas van Overeem Hansen (Center for Genomic Medicine, Rigshospitalet, University of Copenhagen, Denmark) for kind gift of pSPL3 vector and to Annekatrin Wernstedt, PhD, PD Dr Martina Witsch-Baumgartner and University Professor Dr Johannes Zschocke from the Division of Human Genetics of the Medical University of Innsbruck for assistance with MiSeq sequencing.
Funding
This work was supported by the intramural funding program of the Medical University of Innsbruck for young scientists MUI-START, project 2015-06-007, and the D•A•CH Advancement Award Lipidology 2015 (supported by the Christine Katharine Schmitz Foundation) of the D•A•CH-Society Prevention of Cardiovascular Diseases to S.C., by the Austrian Science Fund (FWF): Project Number P 266600-B13 to C.L. and by the Austrian Genome Project ‘GOLD’ to F.K. The GCKD study is funded by grants from the German Ministry of Education and Research (BMBF) (www.gesundheitsforschung-bmbf.de/de/2101.php, 25 March 2017; grant number 01ER 0804, 01ER 0818, 01ER 0819, 01ER 0820 und 01ER 0821) and the KfH Foundation for Preventive Medicine (http://www.kfh-stiftung-praeventivmedizin.de/content/stiftung, 25 March 2017).
The KORA study was initiated and financed by the Helmholtz Zentrum München—German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ.
Conflict of interest: none declared.
References
- 1. Kronenberg F. Human genetics and the causal role of Lipoprotein(a) for various diseases. Cardiovasc Drugs Ther 2016;30:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG.. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. Jama 2009;301:2331–2339. [DOI] [PubMed] [Google Scholar]
- 3. Kronenberg F, Utermann G.. Lipoprotein(a): resurrected by genetics. J Intern Med 2013;273:6–30. [DOI] [PubMed] [Google Scholar]
- 4. Erqou S, Thompson A, Di Angelantonio E, Saleheen D, Kaptoge S, Marcovina S, Danesh J.. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol 2010;55:2160–2167. [DOI] [PubMed] [Google Scholar]
- 5. Utermann G, Menzel HJ, Kraft HG, Duba HC, Kemmler HG, Seitz C.. Lp(a) glycoprotein phenotypes. Inheritance and relation to Lp(a)-lipoprotein concentrations in plasma. J Clin Invest 1987;80:458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perombelon YF, Soutar AK, Knight BL.. Variation in lipoprotein(a) concentration associated with different apolipoprotein(a) alleles. J Clin Invest 1994;93:1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cohen JC, Chiesa G, Hobbs HH.. Sequence polymorphisms in the apolipoprotein (a) gene. Evidence for dissociation between apolipoprotein(a) size and plasma lipoprotein(a) levels. J Clin Invest 1993;91:1630–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu W, Cheng YC, Chen K, Wang H, Gerhard GS, Still CD, Chu X, Yang R, Parihar A, O’connell JR, Pollin TI, Angles-Cano E, Quon MJ, Mitchell BD, Shuldiner AR, Fu M.. Evidence for several independent genetic variants affecting lipoprotein (a) cholesterol levels. Hum Mol Genet 2015;24:2390–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt K, Noureen A, Kronenberg F, Utermann G.. Structure, function, and genetics of lipoprotein (a). J Lipid Res 2016;57:1339–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Noureen A, Fresser F, Utermann G, Schmidt K.. Sequence variation within the KIV-2 copy number polymorphism of the human LPA gene in African, Asian, and European Populations. PLoS One 2015;10:e0121582.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaisson MJP, Wilson RK, Eichler EE.. Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 2015;16:627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee S, Abecasis GR, Boehnke M, Lin X.. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 2014;95:5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosby O, Alestrom P, Berg K.. Sequence conservation in kringle IV-type 2 repeats of the LPA gene. Atherosclerosis 2000;148:353–364. [DOI] [PubMed] [Google Scholar]
- 14. Weissensteiner H, Forer L, Fuchsberger C, Schöpf B, Kloss-Brandstätter A, Specht G, Kronenberg F, Schönherr S.. mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res 2016;44:W64–W69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emdin CA, Khera AV, Natarajan P, Klarin D, Won H-H, Peloso GM, Stitziel NO, Nomura A, Zekavat SM, Bick AG, Gupta N, Asselta R, Duga S, Merlini PA, Correa A, Kessler T, Wilson JG, Bown MJ, Hall AS, Braund PS, Samani NJ, Schunkert H, Marrugat J, Elosua R, McPherson R, Farrall M, Watkins H, Willer C, Abecasis GR, Felix JF, Vasan RS, Lander E, Rader DJ, Danesh J, Ardissino D, Gabriel S, Saleheen D, Kathiresan S; Charge–Heart Failure Consortium; CARDIoGRAM Exome Consortium. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol 2016;68:2761–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morlan J, Baker J, Sinicropi D.. Mutation detection by real-time PCR: a simple, robust and highly selective method. PLoS One 2009;4:e4584.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steffensen AY, Dandanell M, Jønson L, Ejlertsen B, Gerdes A-M, Nielsen FC, Hansen TV.. Functional characterization of BRCA1 gene variants by mini-gene splicing assay. Eur J Hum Genet 2014;22:1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Catapano AL, Graham I, Backer G, De, Wiklund O, Chapman MJ, Drexel H, Hoes AW, Jennings CS, Landmesser U, Pedersen TR, Reiner Ž, Riccardi G, Taskinen M-R, Tokgozoglu L, Verschuren WMM, Vlachopoulos C, Wood DA, Zamorano JL; Authors/Task Force Members. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J 2016;37:2999–3058. [DOI] [PubMed] [Google Scholar]
- 19. Kraft HG, Windegger M, Menzel HJ, Utermann G.. Significant impact of the +93 C/T polymorphism in the apolipoprotein(a) gene on Lp(a) concentrations in Africans but not in Caucasians: confounding effect of linkage disequilibrium. Hum Mol Genet 1998;7:257–264. [DOI] [PubMed] [Google Scholar]
- 20. Lim ET, Würtz P, Havulinna AS, Palta P, Tukiainen T, Rehnström K, Esko T, Mägi R, Inouye M, Lappalainen T, Chan Y, Salem RM, Lek M, Flannick J, Sim X, Manning A, Ladenvall C, Bumpstead S, Hämäläinen E, Aalto K, Maksimow M, Salmi M, Blankenberg S, Ardissino D, Shah S, Horne B, McPherson R, Hovingh GK, Reilly MP, Watkins H, Goel A, Farrall M, Girelli D, Reiner AP, Stitziel NO, Kathiresan S, Gabriel S, Barrett JC, Lehtimäki T, Laakso M, Groop L, Kaprio J, Perola M, McCarthy MI, Boehnke M, Altshuler DM, Lindgren CM, Hirschhorn JN, Metspalu A, Freimer NB, Zeller T, Jalkanen S, Koskinen S, Raitakari O, Durbin R, MacArthur DG, Salomaa V, Ripatti S, Daly MJ, Palotie A; Sequencing Initiative Suomi (SISu) Project . Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet 2014;10:e1004494.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 2016;17:83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eggington JM, Greene T, Bass BL.. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2011;2:319.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Licht K, Kapoor U, Mayrhofer E, Jantsch MF.. Adenosine to Inosine editing frequency controlled by splicing efficiency. Nucleic Acids Res 2016;44:6398–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nordestgaard BGBG, Langsted A.. Lipoprotein(a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J Lipid Res 2016;57:1953–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kamstrup PR, Nordestgaard BG.. Lipoprotein(a) concentrations, isoform size, and risk of type 2 diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol 2013;1:220–227. [DOI] [PubMed] [Google Scholar]
- 26. Hammond SM, Wood MJA.. Genetic therapies for RNA mis-splicing diseases. Trends Genet 2011;27:196–205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.