

Abstract
Purpose of review
The task of cataloging human genetic variation and its relation to disease is rapidly approaching completion. The new challenge is to discover the function of disease-associated genes and to understand the pathways that lead to human disease. We propose that achieving this new level of understanding will increasingly rely on the use of model organisms. We discuss the advantages of the mouse as a model organism.
Recent findings
The collection of available mouse strains represents as much genetic and phenotype variation as is found in the human population. However, unlike humans, mice can be subjected to experimental breeding protocols and the availability of tissues allows for a far greater and deeper level of phenotyping. New methods for gene editing make it relatively easy to create mouse models of known human mutations. The distinction between genetic and epigenetic inheritance can be studied in great detail. Fecal transplant protocols enable the exploration of the role of the microbiome in physiology and disease.
Summary
We propose that there will be an interdependence between human and model organism research. Technological advances and new genetic screening platforms in the mouse have greatly improved the path to gene discovery and mechanistic studies of gene function.
Keywords: Mouse genetics, obesity, diabetes, systems biology, metabolic disease
INTRODUCTION
Most common human diseases involve loss of homeostatic control across multiple organ systems. Various metabolic organs, such as brain, stomach, liver, adipose, muscle, interact in complex, poorly understood ways to integrate and coordinate activities throughout life and across environments. The ongoing challenge is to identify the genetic variants that control function of individual elements and their complex interactions (1,2).
Rapid improvements in DNA sequencing technology and phenotyping capacity have accelerated the pace at which human genetic variation is characterized. Through the efforts of research groups across the globe, the formidable task of cataloging human allelic and haplotype variation is rapidly reaching completion (3–5). Now, the challenge is to leverage these technologies and resources to systematically define the ways in which genotypes define phenotypes in health and the ways that dysfunction leads to disease.
We propose that an understanding of health and disease depends on experimental examination of genotype-phenotype relations in model organisms. As our understanding of human disease moves past “gene discovery”, we predict that the groundbreaking discoveries required to unravel the complex basic biology of human health and disease will increasingly rely on research in model organisms. To support our argument, we review selected highlights of past discoveries from studies related to obesity and diabetes in mouse models and then look ahead at new developments in model systems that will help us push beyond the limitations of the past.
WHY STUDY MICE?
Mouse models have made significant contributions to our understanding of human biology and disease over the past century (6). These contributions have been based on the similarity between humans and mice in development, physiology and genome organization, as well as access to engineered mutants, phenotyping platforms, and genetic reference populations. The efficacy of such cross-species studies was recently demonstrated for ∼4,500 metabolic, physiological, pharmacological and behavioral traits (7)
Homology
Homologous physiology and morphology between humans and mice enables the study of human diseases in a mammalian surrogate that is amenable to laboratory experimentation. Even when phenotypic differences are evident, the underlying pathways are remarkably similar. For example, there is deep homology in insulin biology that is found throughout the animal kingdom. Insulin has the same molecular structure in humans, mice and fruit flies. The insulin secretory pathway is also remarkably similar, involving the storage of a secretory pool of insulin in granules and their trafficking to the plasma membrane after an appropriate stimulus. Homology is also found in the insulin signaling system, involving the insulin receptor, its tyrosine kinase activity, its downstream substrates, and the regulation of growth and metabolism.
Genomics
Various attributes make the laboratory mouse a good model for studying genetics of human biology in health and disease. The relevance of model organisms to studies of human biology is evident in the highly conserved genetic content, protein functions and metabolic and signaling pathway\ topologies. The important exceptions contribute to discrepancies between genotype-phenotype relations. It is important to note that conclusions about the relevance of mouse models to human biology are often based on studies a small number of strains, and often only one strain. Generalizations across strains is risky given the considerable phenotypic variation across strains, which is documented in the Mouse Phenome Database (jax.org/phenome).
Mutations
Another important attribute is the ability to engineer mutations (8). With CRISPR/Cas9 technologies, a remarkable variety of mutations can be made in a highly efficient and cost-effective manner. These include gain- and loss-of-function, inducible, conditional and tissue-specific mutations as well as reporter constructs to signal when and where are gene is expressed. Targets include both protein coding genes as well as non-coding regulatory RNAs and structural elements. International efforts are underway to establish a comprehensive public resource with cells and mice with engineered mutations as well as associated phenotypes. These and other technologies can also be used to ‘humanize’ mice by introducing specific human DNA sequence variants as a way to establish causation and functional relevance as well as undertake mechanistic studies (9–11).
Tissue access
The mouse also provides opportunities to obtain tissues from test and control groups in ways that can be logistically challenging or ethically inappropriate in humans. In many cases, access to specific tissues at particular developmental and life-history stages is needed. For many metabolic conditions however, organ and tissue samples such as liver biopsies from healthy individuals for comparison to disease states are usually difficult to obtain. In most instances, mice resolve this dilemma, with materials from precisely defined test and control groups.
Phenotyping
Systematic high-throughput phenotyping programs have been established based on common protocols, ontologies, technologies and analyses, as ways to improve generality and reproducibility (9–11).
Genetic resources
Finally, by using designed populations of mice, we can minimize environmental heterogeneity, population stratification, unknown ancestral haplotypes, and unbalanced allele frequencies, which can complicate human genome-wide association studies (GWAS) in humans. With newly-developed mouse resources, it is now possible to achieve power and mapping resolution of tens of thousands of humans with only hundreds of mice.
IMPOSSIBLE WITHOUT A MUTANT MOUSE
Two classic discoveries
Leptin; insightful physiology, genetic triumph, epistatic interactions
Long before gene discovery was feasible, physiological studies led to insights not only about satiety control, but also about hormonal regulation of multi-organ systems and gene interaction networks that coordinate metabolic activities. Two spontaneous mutations arose in mice at the Jackson Laboratory, ob in 1950 (12) and db in 1965 (13). The mutant mice are both obese and hyperinsulinemic (14). Douglas Coleman conducted parabiosis experiments where the circulatory systems of db or ob mice were surgically connected to that of normal mice (test) or to each other (control) (15). When the db mouse was paired with a normal mouse or an ob mouse, the ob mouse died of starvation (Figure 1A). But when an ob mouse was paired with a normal mouse, the normal mouse was unaffected. Coleman hypothesized that the ob mouse lacked a circulating satiety factor and the db mouse lacked the receptor for that factor. This hypothesis proved to be prescient with the cloning of leptin and its receptor decades later (16,17).
Figure 1. Parabiosis experiments.
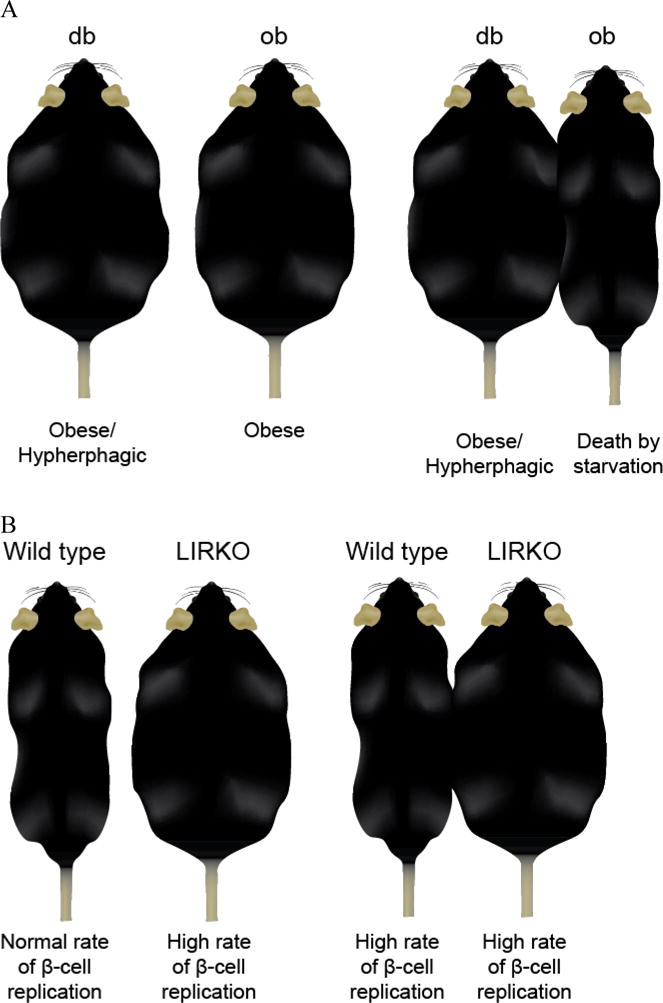
Parabiosis experiments connect the circulatory systems of two mice. A) The db and ob mice are genetically obese due to mutations at different gene loci. When Coleman connected the circulations of the two mutant mice, the db mouse remained obese while the ob mouse starved to death (15). Coleman concluded that the ob mouse lacked a circulating satiety factor and the db mouse lacked its receptor. B) The liver insulin receptor knockout (LIRKO) mouse is severely insulin resistant and has a high rate of β-cell replication. Kulkarni connected the circulatory systems of wildtype and LIRKO mice and observed a high rate of β-cell replication in both mice, suggesting that a circulating factor that is responsive to the insulin resistant state is responsible for the increase in β-cell replication (18,19). Original figure.
The parabiosis paradigm has proved useful in other contexts. For example, Kulkarni and co-workers discovered that mice with a liver-specific deletion of the insulin receptor experienced a dramatic enhancement in β-cell proliferation (19). Parabiosis was used to determine whether a soluble factor, presumably liver-derived, stimulates β-cell proliferation (Figure 1B). Their studies eventually led to the identification of Serpin B1 as a circulating mitogenic factor (20).
Leptin is produced in adipose tissue. Prior to its discovery, adipose tissue had not been regarded as an endocrine organ. The discovery of additional adipose-derived hormones; e.g. adiponectin (21), RBP4 (22) and resistin (23), blurred the boundaries between endocrine and non-endocrine organs. Soon, other hormones were identified, secreted by tissues not usually regarded as endocrinal, such as muscle, bone, macrophages, liver, stomach, and the intestine. This has led to a paradigm shift where most tissues and organs are now considered to be endocrine tissues.
Agouti, a novel pathway and pleiotropy
The Agouti mutation is an excellent example of genetics solving a riddle not amenable to a simple hypothesis. The Agouti-yellow (Ay) mutation has pleiotropic effects; it causes obesity and yellow hair. The agouti protein is normally produced in dermal papillae cells and functions in a paracrine fashion to antagonize the action of α-melanocyte stimulating hormone (α-MSH) on melanocytes. This prevents the stimulation of melanin production via the melanocortin-1 receptor, which leads to black hair and results in the production of phaeomelanin, leading to yellow hair. The obesity phenotype is caused by ectopic production of the Agouti protein in the brain, leading to the antagonism of α-MSH binding to the melanocortin-4 receptor. Based on homology to the Agouti protein, the Agouti-related protein (AGRP) was identified and found to be the normal physiological ligand of the melanocortin-4 receptor in the hypothalamus, where it functions to suppress feeding (24). Mutations in the melanocortin-4 receptor (MC4R) are the most common genetic cause of obesity in humans and the receptor is a target for drugs to treat obesity (25).
Genetics of multifactorial metabolic traits
Unlike the ob and db mutations, which have unusually large, consistent phenotypic effects, most genetic variants have relatively modest phenotypic effects in natural populations. By identifying these genes, clues are obtained about molecular and physiological functions. With genes that control complex traits, the powerful technologies and resources of laboratory mice can be used to deconstruct these traits. For example, several genes have been identified that affect different steps in the insulin secretory pathway (26,27), β-cell proliferation (28), and insulin signaling (29).
The vast majority of loci that contribute to phenotype variation are quantitative trait loci (QTLs) where multiple genes acting independently or interactively (epistasis) determine the phenotypic nature of the trait. Until recently, the path from mapping a locus to identifying a causal gene was arduous because the mapping resolution is modest in conventional crosses between pairs of strains. Only with follow-up crosses to capture additional recombinants can the resolution be narrowed to identify candidate genes (30). New developments in genetic resources have revolutionized accelerated the pace QTL detection and gene identification. Similarly, genome sequencing and genetic engineering technologies have accelerated gene identification and validation. As a result, gene discovery, not simply localization, is now routine, exceeding GWAS at a far lower cost (31).
Modifier genes and gene interactions
The pioneering studies by Coleman and colleagues also showed that strain background interacted with the ob mutation to determine whether mice would also become diabetic. In particular, db or ob mice on the C57BL/6J background are non-diabetic, whereas db or ob mice on the C57BL/Ks background become diabetic (32). As the genetic modifiers of phenotypes in db and ob mice suggest, genes rarely act alone, but instead are part of complex webs of interactions, not simply in pathways but also in networks of functional interdependencies that provide resilience to stress, adverse environmental exposures, and suboptimal genetic backgrounds. While evidence for these modifier genes in mice and humans is now considerable (33,34), evidence for more complex gene interactions has been more elusive, especially in humans. However, surveys of hundreds of traits in a CSS panel showed that these interactions are pervasive and strong (35);Spiezio, 2012 #3932;Yazbek, 2011 #3933}. These studies took advantage of the unique genetic architecture of these strains. Now the challenge is finding ways to detect modifier and epistatic effects in mouse crosses and human population. These studies are important because modifiers that suppress disease risk and adverse phenotypic outcomes could be a new class of therapeutic targets given their obvious efficacy and apparent safety.
Epigenetic inheritance
Epigenetic inheritance, which is revolutionizing our understanding of the origins of phenotypic variation, occurs in the absence of inherited genetic variants and without persistent environmental exposures (36–38). In these cases, yet to be identified molecular changes in the germline lead to inherited phenotypic variants that can persist for generations and that can sometimes be reversed with specific crosses. Examples of epigenetic traits related to metabolic conditions involve obesity (39–42) glucose homeostasis (43,44) and lipid metabolism (45), and microbiota (46). Genetic factors also contribute where a genetic variant in a strain that is resistant to diet-induced metabolic conditions epigenetically transferred resistance to a genetically-susceptible strain with resistance persisting for multiple generations and reversible with specific crosses (47). These unexpected results suggest that the ongoing epidemic of metabolic disease may have genetic and environmental origins in early in life and in preceding generations (45). The challenge now is to identify the molecular basis for epigenetic inheritance as well as to establish the chain of causality that leads from somatic exposures to environmental factors and genetic variants through heritable epigenetic changes in the germline to phenotypic variation in later generations. These experiments are complex involving rigorous control of genetics and environment so that specific factors can be manipulated and their consequences evaluated. These remarkable discoveries will no doubt lead to exciting new strategies for therapeutic interventions.
Microbiome
One of the most exciting discoveries involves the intricate ways that gut microbes and host genetics interact to control physiology and metabolism. The more than 600 microbial species alone and together process dietary intake in essential ways to host physiology Microbial dysbiosis is major contributor to host dysfunction through their diverse effects on human metabolism (48). These effects depend heavily on as yet unidentified host genetic factors (49–54). Mouse models will continue to be essential for these studies because combinations of fecal transplants as well as host genetic manipulations can be used to dissect mechanistic relations in controlled ways that are highly relevant to public health but difficult to conduct in human populations.
GENETIC RESOURCES
Over the last 15 years, many mouse genetic resource populations have been reported. These have revolutionized studies of genetically complex traits. They have been used for discovering QTLs, and also for studying epigenetics and systems biology. Developing effective strategies for leveraging these complementary resources will be an important theme in the coming years as we learn how to most effectively utilize these reference populations.
Collaborative Cross (CC)
The Collaborative Cross (CC) is an eight-way mouse recombinant inbred (RI) strain panel that is being developed as a resource for mammalian system genetics (55). The CC strains are derived from eight diverse founder strains, including five classical inbred strains that are important as disease models and three wild-derived strains representing the three Mus musculus subspecies. The eight founder strains capture a level of genetic diversity unsurpassed by any other extant mouse resource. Presently, approximately 70 CC strains have been derived and are available to researchers. The strains have enabled mapping of phenotypes related to diabetes (56), hepatic steatosis (57), drug disposition (58), and other metabolic phenotypes.
Diversity Outbreds (DO)
The Diversity Outbreds (DO) are a heterogeneous stock derived from the same eight founder strains as the CC, resulting in a population with the same array of allelic polymorphisms but present in a wider variety of combinations (59). The DO is maintained as a randomized breeding colony with a population size of 180 pairs. At present, the DO colony has reached 23 generations of outcrossing and can simultaneously achieve high resolution and high power to detect small allelic effects. A drawback of the DO is that each animal is genetically unique and not reproducible. However, genetic loci that are mapped in the DO can be replicated in CC strains or their F1 progeny to provide stable models for validation and in-depth characterization. It is still early days and there are many ongoing studies. A recent study of the genetics of protein abundance demonstrates the power of this resource (60).
Hybrid Mouse Diversity Panel and Recombinant Inbred Strains
When two inbred strains are bred to one another, through meiotic recombination the chromosomes of their offspring contain a mosaic of segments from the founder strains. This mosaic can be fixed by inbreeding, creating panels of unique recombinant inbred (RI) strains. The strains need only be genotyped once and, as inbred strains, offer unlimited biological replication. Panels of inbred strains have been used for systems genetics projects where many phenotypes have been mapped and the data integrated into biological networks. Two recent examples are the BXD panel (from C57BL/6 × DBA) (61–63) and the hybrid mouse diversity panel (HMDP) (64). The HMDP has had remarkable success in identifying genes related to a broad spectrum of human metabolic disease, including atherosclerosis (65), obesity (66), diet responsiveness, insulin resistance and diabetes (67), plasma lipids (66), and hepatic steatosis (68). The ability to carry out unlimited phenotyping on the same panel of RI strains enables the development of causal networks that connect various phenotypes, the ultimate goal of systems biology (64).
Chromosome Substitution Strains (CSSs)
A chromosome substitution strain (CSS, also known as consomic strains) has a chromosome in the host strain replaced with the corresponding strain from a donor strain. The resulting inbred strain is homozygous for the substituted chromosome on an inbred strain background and differs from the original host strain by a single chromosome. A complete panel of substituted chromosomes, which has 22 strains, one for each of the 19 autosomes, the X and Y chromosomes and the mitochondria, neatly partition the genome into non-overlapping units based on the genome’s chromosome structure. Several panels have been made from commonly used inbred strains (69). QTLs can be detected simply by comparing each CSS with the host strain, testing for a significant phenotypic difference that implicate at least one QTL on the substituted chromosome. These strains are inbred, congenic strains are readily derived from them, precisely defined test and control groups can be arranged, and functional studies can be undertaken simultaneously with gene discovery efforts. This enables rigorous study of the molecular, biochemical and physiological mechanisms controlling the phenotypes.
CSSs have been used to study diet-induced metabolic conditions, where genes on the majority of chromosomes have been shown to affect obesity, glucose homeostasis, dyslipidemia, and liver disease and several genes have been identified (31). Also important is discovery of precisely defined comorbidities involving obesity, cholesterol levels, insulin resistance, and fatty liver, where many of the CSSs have unique combinations of these traits (70). Finally, CSSs have been used to identify modifier genes that control phenotypic outcomes, with a recent example being the Moda1 locus, which suppresses diabetes in the HNF1a model of MODY3 (maturity onset diabetes of the young type 3) (71).
Chemical mutagenesis
Chemical mutagenesis has been used to induce mutations in mice. These can cause be both gain-of-function (usually dominant) or loss-of-function (usually recessive) phenotypes. This approach led to the discovery of nicotinamide nucleotide transhydrogenase (Nnt) as a regulator of insulin secretion (72). Follow-up studies showed that the C57BL6/J strain (in contrast to C57BL6/N) has a natural deletion of this gene (73).
Engineered mutations
The ability to delete genes in a tissue-specific manner has produced numerous profound and often surprising new insights. Glut4 is the insulin-regulated glucose transporter expressed in muscle and adipose tissue. Although muscle is the tissue that makes the largest contribution to insulin-stimulated glucose clearance, the deletion of Glut4 in adipose tissue had as much of an effect on insulin resistance as the Glut4 deletion in muscle (74). This is largely due to the endocrine functions of adipose tissue; in response to reduced Glut4 expression, adipose tissue produces hormones that affect whole-body insulin signaling (22).
Early studies showed that administration of insulin in the brain regulates the ability of circulating insulin to suppress hepatic glucose production (75). Deletion of the insulin receptor in the brain abolishes the ability of insulin to suppress hepatic glucose production (76). Now it is well-established that the brain regulates metabolism in numerous organs. Many of the actions of hormones on metabolism occur through the brain. For example, the suppression of glucagon secretion by leptin is primarily through its action in the brain (77). A recent study showed that microbiome production of acetate regulates insulin secretion through an axis that includes the brain (78).
INTEGRATION
Systems studies
These studies seek to define the networks of functional dependencies between genetic variants and multilevel phenotypes from RNA expression patterns to proteins and metabolites and finally to organs and metabolic outcomes. This integrated view complements reductionist approaches (1,2). These reductionist approaches identify the nature and function of elements that compose the foundation of complex biology, whereas systems studies define the context and consequence of the collective and integrated action of these elements focusing on such essential systems properties as robustness, resilience, homeostasis, hysteresis, and criticality (79).
CONCLUSION
The foundation of scientific research is combining discovery with experimentation. Obviously, in humans, discovery is easy. Experimentation, by contrast, is difficult because test and control groups must be controlled in ways so they ideally differ only in single agents. Multifactorial designs are also possible but again rigorous design is needed to manage the genetic and environmental complexities. In model organisms, such control is feasible.
It has been proposed that computational analyses of big-data, together with in vitro studies with human materials, will suffice to provide the understanding that we seek to prevent disease and improve the human condition. Many diseases and most phenotypes are not properties of single cell types in vitro. Insteadm most metabolic conditions, even those resulting from single-gene Mendelian conditions, involve multiple cell types across multiple organs, and thus are problems in systems biology (1,2). We therefore propose that an interdependency between humans and model organisms will emerge as the centerpiece of biomedical research. This relationship will be effective as long as the similarities and differences in development and physiology are fully identified and taken into account when designing studies in model organisms and extrapolating the results back to humans. Pioneering discoveries in the past both challenged and justified investments in technologies and resources now enable these systematic, integrated studies at genome and organismal scale.
A recent commentary predicts that “modeling human complex traits in experimental organisms will become obsolete” (80) The commentary goes on to say that “humans will become a ‘model organism’ through exploiting new technologies such as tissue-specific cell lines and gene editing.” We hope that the foregoing discussion will help to put this notion to rest.
KEY POINTS.
Cataloging of human genetic variation is approaching completion
Discovering gene function and its relation to disease requires model organisms
The mouse has special advantages, largely due to the availability of genetic resources for gene discovery, and technological advances in gene editing and phenotyping
Acknowledgments
None
FINANCIAL SUPPORT AND SPONSORSHIP
Research in the Attie lab is supported by NIH grants DK108259, DK101573, and DK102948.
Footnotes
CONFLICTS OF INTEREST
No conflicts to declare
Contributor Information
Alan D. Attie, Department of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706
Gary A. Churchill, The Jackson Laboratory, Bar Harbor, ME
Joseph H. Nadeau, Pacific Northwest Research Institute, 720 Broadway, Seattle, WA 98119
References
- 1.Argmann CA, Houten SM, Zhu J, Schadt EE. A Next Generation Multiscale View of Inborn Errors of Metabolism. Cell Metab. 2016;23:13–26. doi: 10.1016/j.cmet.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams EG, Auwerx J. The Convergence of Systems and Reductionist Approaches in Complex Trait Analysis. Cell. 2015;162:23–32. doi: 10.1016/j.cell.2015.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International HapMap, C. Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, Ma C, Fontanillas P, Moutsianas L, McCarthy DJ, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536:41–47. doi: 10.1038/nature18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paigen K. One hundred years of mouse genetics: an intellectual history. II. The molecular revolution (1981-2002) Genetics. 2003;163:1227–1235. doi: 10.1093/genetics/163.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Pandey AK, Mulligan MK, Williams EG, Mozhui K, Li Z, Jovaisaite V, Quarles LD, Xiao Z, Huang J, et al. Joint mouse-human phenome-wide association to test gene function and disease risk. Nat Commun. 2016;7:10464. doi: 10.1038/ncomms10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harms DW, Quadros RM, Seruggia D, Ohtsuka M, Takahashi G, Montoliu L, Gurumurthy CB. Mouse Genome Editing Using the CRISPR/Cas System. Curr Protoc Hum Genet. 2014;83:15 17 11–27. doi: 10.1002/0471142905.hg1507s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514. doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hrabe de Angelis M, Nicholson G, Selloum M, White JK, Morgan H, Ramirez-Solis R, Sorg T, Wells S, Fuchs H, Fray M, et al. Analysis of mammalian gene function through broad-based phenotypic screens across a consortium of mouse clinics. Nat Genet. 2015;47:969–978. doi: 10.1038/ng.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, Salisbury J, Clare S, Ingham NJ, Podrini C, et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell. 2013;154:452–464. doi: 10.1016/j.cell.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered. 1950;41:317–318. doi: 10.1093/oxfordjournals.jhered.a106073. [DOI] [PubMed] [Google Scholar]
- 13.Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153:1127–1128. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- 14.Coleman DL, Hummel KP. Hyperinsulinemia in pre-weaning diabetes (db) mice. Diabetologia. 1974;10(Suppl):607–610. doi: 10.1007/BF01221993. [DOI] [PubMed] [Google Scholar]
- 15.Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9:294–298. doi: 10.1007/BF01221857. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 18.El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, Hou LF, Goodman J, Karampelias C, Qiang GF, et al. SerpinB1 Promotes Pancreatic beta Cell Proliferation. Cell Metabolism. 2016;23:194–205. doi: 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Ouaamari A, Kawamori D, Dirice E, Liew CW, Shadrach JL, Hu J, Katsuta H, Hollister-Lock J, Qian WJ, Wagers AJ, et al. Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states. Cell Rep. 2013;3:401–410. doi: 10.1016/j.celrep.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, Hou L, Goodman J, Karampelias C, Qiang G, et al. SerpinB1 Promotes Pancreatic beta Cell Proliferation. Cell Metab. 2016;23:194–205. doi: 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stern JH, Rutkowski JM, Scherer PE. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016;23:770–784. doi: 10.1016/j.cmet.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz DR, Lazar MA. Human resistin: found in translation from mouse to man. Trends Endocrinol Metab. 2011;22:259–265. doi: 10.1016/j.tem.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, et al. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 25.Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, Inouye M, Freathy RM, Attwood AP, Beckmann JS, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–775. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhatnagar S, Oler AT, Rabaglia ME, Stapleton DS, Schueler KL, Truchan NA, Worzella SL, Stoehr JP, Clee SM, Yandell BS, et al. Positional cloning of a type 2 diabetes quantitative trait locus; tomosyn-2, a negative regulator of insulin secretion. PLoS Genet. 2011;7:e1002323. doi: 10.1371/journal.pgen.1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kebede MA, Oler AT, Gregg T, Balloon AJ, Johnson A, Mitok K, Rabaglia M, Schueler K, Stapleton D, Thorstenson C, et al. SORCS1 is necessary for normal insulin secretory granule biogenesis in metabolically stressed beta cells. J Clin Invest. 2014;124:4240–4256. doi: 10.1172/JCI74072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kluth O, Matzke D, Kamitz A, Jahnert M, Vogel H, Scherneck S, Schulze M, Staiger H, Machicao F, Haring HU, et al. Identification of Four Mouse Diabetes Candidate Genes Altering beta-Cell Proliferation. PLoS Genet. 2015;11:e1005506. doi: 10.1371/journal.pgen.1005506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scherneck S, Nestler M, Vogel H, Bluher M, Block MD, Berriel Diaz M, Herzig S, Schulz N, Teichert M, Tischer S, et al. Positional cloning of zinc finger domain transcription factor Zfp69, a candidate gene for obesity-associated diabetes contributed by mouse locus Nidd/SJL. PLoS Genet. 2009;5:e1000541. doi: 10.1371/journal.pgen.1000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darvasi A. Interval-specific congenic strains (ISCS): an experimental design for mapping a QTL into a 1-centimorgan interval. Mamm Genome. 1997;8:163–167. doi: 10.1007/s003359900382. [DOI] [PubMed] [Google Scholar]
- 31.Buchner DA, Nadeau JH. Contrasting genetic architectures in different mouse reference populations used for studying complex traits. Genome Res. 2015;25:775–791. doi: 10.1101/gr.187450.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coleman DL, Hummel KP. The influence of genetic background on the expression of the obese (Ob) gene in the mouse. Diabetologia. 1973;9:287–293. doi: 10.1007/BF01221856. [DOI] [PubMed] [Google Scholar]
- 33.Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- 34.Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome research. 2008;18:706–716. doi: 10.1101/gr.074914.107. This article showed highly correlated sets of genes associated with diabetes susceptibility, mainly involved in β-cell replication. A database with gene expression data from 6 tissues of lean, obese, diabetic, and non-diabetic mice associated with this study is widely used in the diabetes field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao H, Burrage LC, Sinasac DS, Hill AE, Ernest SR, O’Brien W, Courtland HW, Jepsen KJ, Kirby A, Kulbokas EJ, et al. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc Natl Acad Sci U S A. 2008;105:19910–19914. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109. doi: 10.1016/j.cell.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rando OJ. Intergenerational Transfer of Epigenetic Information in Sperm. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a022988. The first adequately powered, genome-wide test revealed strong evidence that most traits show unexpectedly strong non-additivity. The pervasive nature for such effects was also found in two panels of CSSs that each involve an inbred strain recently derived from wild mice and also in a panel of rat CSSs. Genetic complexity and non-additivity extended from the chromosome level to many small Mb-scale genetic intervals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaefer S, Nadeau JH. The Genetics of Epigenetic Inheritance: Modes, Molecules, and Mechanisms. Q Rev Biol. 2015;90:381–415. doi: 10.1086/683699. [DOI] [PubMed] [Google Scholar]
- 39.Cropley JE, Eaton SA, Aiken A, Young PE, Giannoulatou E, Ho JW, Buckland ME, Keam SP, Hutvagner G, Humphreys DT, et al. Male-lineage transmission of an acquired metabolic phenotype induced by grand-paternal obesity. Mol Metab. 2016;5:699–708. doi: 10.1016/j.molmet.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Castro Barbosa T, Ingerslev LR, Alm PS, Versteyhe S, Massart J, Rasmussen M, Donkin I, Sjogren R, Mudry JM, Vetterli L, et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol Metab. 2016;5:184–197. doi: 10.1016/j.molmet.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grandjean V, Fourre S, De Abreu DA, Derieppe MA, Remy JJ, Rassoulzadegan M. RNA-mediated paternal heredity of diet-induced obesity and metabolic disorders. Sci Rep. 2015;5:18193. doi: 10.1038/srep18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huypens P, Sass S, Wu M, Dyckhoff D, Tschop M, Theis F, Marschall S, Hrabe de Angelis M, Beckers J. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat Genet. 2016;48:497–499. doi: 10.1038/ng.3527. [DOI] [PubMed] [Google Scholar]
- 43.Jimenez-Chillaron JC, Isganaitis E, Charalambous M, Gesta S, Pentinat-Pelegrin T, Faucette RR, Otis JP, Chow A, Diaz R, Ferguson-Smith A, et al. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes. 2009;58:460–468. doi: 10.2337/db08-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei Y, Yang CR, Wei YP, Zhao ZA, Hou Y, Schatten H, Sun QY. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc Natl Acad Sci U S A. 2014;111:1873–1878. doi: 10.1073/pnas.1321195111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, Bock C, Li C, Gu H, Zamore PD, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, Keller MP, Attie AD, Rey FE, Denu JM. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol Cell. 2016 doi: 10.1016/j.molcel.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yazbek SN, Spiezio SH, Nadeau JH, Buchner DA. Ancestral paternal genotype controls body weight and food intake for multiple generations. Hum Mol Genet. 2010;19:4134–4144. doi: 10.1093/hmg/ddq332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, Forslund K, Hildebrand F, Prifti E, Falony G, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535:376–381. doi: 10.1038/nature18646. These articles describe epigenetic transmission of metabolic phenotypes. [DOI] [PubMed] [Google Scholar]
- 49.Backhed F. Programming of host metabolism by the gut microbiota. Ann Nutr Metab 58 Suppl. 2011;2:44–52. doi: 10.1159/000328042. [DOI] [PubMed] [Google Scholar]
- 50.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujisaka S, Ussar S, Clish C, Devkota S, Dreyfuss JM, Sakaguchi M, Soto M, Konishi M, Softic S, Altindis E, et al. Antibiotic effects on gut microbiota and metabolism are host dependent. J Clin Invest. 2016 doi: 10.1172/JCI86674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKnite AM, Perez-Munoz ME, Lu L, Williams EG, Brewer S, Andreux PA, Bastiaansen JW, Wang X, Kachman SD, Auwerx J, et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS One. 2012;7:e39191. doi: 10.1371/journal.pone.0039191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ussar S, Fujisaka S, Kahn CR. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol Metab. 2016;5:795–803. doi: 10.1016/j.molmet.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ussar S, Griffin NW, Bezy O, Fujisaka S, Vienberg S, Softic S, Deng L, Bry L, Gordon JI, Kahn CR. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab. 2015;22:516–530. doi: 10.1016/j.cmet.2015.07.007. These papers provide an excellent sampling of the dramatic effects of microbiota on host metabolic phenotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nature genetics. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. An introduction to the rationale and design of the Collaborative Cross project. [DOI] [PubMed] [Google Scholar]
- 56.Abu-Toamih Atamni HJ, Ziner Y, Mott R, Wolf L, Iraqi FA. Glucose tolerance female-specific QTL mapped in collaborative cross mice. Mamm Genome. 2016 doi: 10.1007/s00335-016-9667-2. [DOI] [PubMed] [Google Scholar]
- 57.Atamni HJ, Botzman M, Mott R, Gat-Viks I, Iraqi FA. Mapping liver fat female-dependent quantitative trait loci in collaborative cross mice. Mamm Genome. 2016;27:565–573. doi: 10.1007/s00335-016-9658-3. [DOI] [PubMed] [Google Scholar]
- 58.Nachshon A, Abu-Toamih Atamni HJ, Steuerman Y, Sheikh-Hamed R, Dorman A, Mott R, Dohm JC, Lehrach H, Sultan M, Shamir R, et al. Dissecting the Effect of Genetic Variation on the Hepatic Expression of Drug Disposition Genes across the Collaborative Cross Mouse Strains. Front Genet. 2016;7:172. doi: 10.3389/fgene.2016.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chesler EJ, Gatti DM, Morgan AP, Strobel M, Trepanier L, Oberbeck D, McWeeney S, Hitzemann R, Ferris M, McMullan R, et al. Diversity Outbred Mice at 21: Maintaining Allelic Variation in the Face of Selection. G3 (Bethesda) 2016 doi: 10.1534/g3.116.035527. An introduction to the rationale and design of the Diversity Outbred project. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, Gygi SP. Defining the consequences of genetic variation on a proteome-wide scale. Nature. 2016;534:500–505. doi: 10.1038/nature18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andreux PA, Williams EG, Koutnikova H, Houtkooper RH, Champy MF, Henry H, Schoonjans K, Williams RW, Auwerx J. Systems genetics of metabolism: the use of the BXD murine reference panel for multiscalar integration of traits. Cell. 2012;150:1287–1299. doi: 10.1016/j.cell.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams EG, Wu Y, Jha P, Dubuis S, Blattmann P, Argmann CA, Houten SM, Amariuta T, Wolski W, Zamboni N, et al. Systems proteomics of liver mitochondria function. Science. 2016;352:aad0189. doi: 10.1126/science.aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu Y, Williams EG, Dubuis S, Mottis A, Jovaisaite V, Houten SM, Argmann CA, Faridi P, Wolski W, Kutalik Z, et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell. 2014;158:1415–1430. doi: 10.1016/j.cell.2014.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lusis AJ, Seldin MM, Allayee H, Bennett BJ, Civelek M, Davis RC, Eskin E, Farber CR, Hui S, Mehrabian M, et al. The Hybrid Mouse Diversity Panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. J Lipid Res. 2016;57:925–942. doi: 10.1194/jlr.R066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, Pan C, Packard RR, Eskin E, Yan M, et al. Genetic Architecture of Atherosclerosis in Mice: A Systems Genetics Analysis of Common Inbred Strains. PLoS Genet. 2015;11:e1005711. doi: 10.1371/journal.pgen.1005711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013;17:141–152. doi: 10.1016/j.cmet.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, Castellani LW, Rau CD, Pan C, Phun J, et al. Genetic architecture of insulin resistance in the mouse. Cell Metab. 2015;21:334–346. doi: 10.1016/j.cmet.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hui ST, Parks BW, Org E, Norheim F, Che N, Pan C, Castellani LW, Charugundla S, Dirks DL, Psychogios N, et al. The genetic architecture of NAFLD among inbred strains of mice. Elife. 2015;4:e05607. doi: 10.7554/eLife.05607. These articles (63–68) explain and demonstrate the many advantages of using panels of recombinant inbred mouse strains for mapping essentially unlimited numbers of phenotypes on the same reference population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singer JB, Hill AE, Burrage LC, Olszens KR, Song J, Justice M, O'Brien WE, Conti DV, Witte JS, Lander ES, et al. Genetic dissection of complex traits with chromosome substitution strains of mice. Science. 2004;304:445–448. doi: 10.1126/science.1093139. [DOI] [PubMed] [Google Scholar]
- 70.Sinasac DS, Riordan JD, Spiezio SH, Yandell BS, Croniger CM, Nadeau JH. Genetic control of obesity, glucose homeostasis, dyslipidemia and fatty liver in a mouse model of diet-induced metabolic syndrome. Int J Obes (Lond) 2016;40:346–355. doi: 10.1038/ijo.2015.184. [DOI] [PubMed] [Google Scholar]
- 71.Garcia-Gonzalez MA, Carette C, Bagattin A, Chiral M, Makinistoglu MP, Garbay S, Prevost G, Madaras C, Herault Y, Leibovici M, et al. A suppressor locus for MODY3-diabetes. Sci Rep. 2016;6:33087. doi: 10.1038/srep33087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Freeman H, Shimomura K, Horner E, Cox RD, Ashcroft FM. Nicotinamide nucleotide transhydrogenase: a key role in insulin secretion. Cell Metab. 2006;3:35–45. doi: 10.1016/j.cmet.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 73.Freeman HC, Hugill A, Dear NT, Ashcroft FM, Cox RD. Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes. 2006;55:2153–2156. doi: 10.2337/db06-0358. [DOI] [PubMed] [Google Scholar]
- 74.Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Biol Chem. 2003;278:33609–33612. doi: 10.1074/jbc.R300019200. [DOI] [PubMed] [Google Scholar]
- 75.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–1382. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 76.Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 77.Hidaka S, Yoshimatsu H, Kondou S, Tsuruta Y, Oka K, Noguchi H, Okamoto K, Sakino H, Teshima Y, Okeda T, et al. Chronic central leptin infusion restores hyperglycemia independent of food intake and insulin level in streptozotocin-induced diabetic rats. FASEB J. 2002;16:509–518. doi: 10.1096/fj.01-0164com. [DOI] [PubMed] [Google Scholar]
- 78.Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nadeau JH, Dudley AM. Genetics. Systems genetics. Science. 2011;331:1015–1016. doi: 10.1126/science.1203869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Visscher PM. Human Complex Trait Genetics in the 21st Century. Genetics. 2016;202:377–379. doi: 10.1534/genetics.115.180513. [DOI] [PMC free article] [PubMed] [Google Scholar]