

Abstract
Multi-target inhibitors have become increasing popular as a means to leverage the advantages of poly-pharmacology while simplifying drug delivery. Here, we describe dual inhibitors for soluble epoxide hydrolase (sEH) and fatty acid amide hydrolase (FAAH), two targets known to synergize when treating inflammatory and neuropathic pain. The structure activity relationship (SAR) study described herein initially started with t-TUCB (trans-4-[4-(3-trifluoromethoxyphenyl-l-ureido)-cyclohexyloxy]-benzoic acid), a potent sEH inhibitor that was previously shown to weakly inhibit FAAH. Inhibitors with a 6-fold increase of FAAH potency while maintaining high sEH potency were developed by optimization. Interestingly, compared to most FAAH inhibitors that inhibit through time-dependent covalent modification, t-TUCB and related compounds appear to inhibit FAAH through a time-independent, competitive mechanism. These inhibitors are selective for FAAH over other serine hydrolases. In addition, FAAH inhibition by t-TUCB appears to be higher in human FAAH over other species; however, the new dual sEH/FAAH inhibitors have improved cross-species potency. These dual inhibitors may be useful for future studies in understanding the therapeutic application of dual sEH/FAAH inhibition.
Keywords: Soluble Epoxide Hydrolase, Fatty Acid Amide Hydrolase, Urea Inhibitors, Neuropathic Pain
Graphical abstract
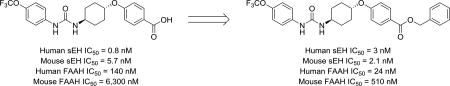
Chronic pain is poorly managed by current treatment options. The available therapies, including non-steroidal anti-inflammatory drugs (NSAIDs) and opioids, are not effective on all types of pain, can be debilitating or have a high potential for abuse.1 Furthermore, few new therapies have come to the market in the past decade. One recent approach towards designing analgesics with high efficacy and reduced side effects has been the combination of inhibitors for two or more targets known to regulate pain, known as poly-pharmacology.2 These multi-target inhibitors have the potential for higher efficacy and reduced drawbacks arising from the use of a single-target drug or a combination of multiple drugs.3 In particular, enzymes involved in the regulation of signaling lipids, including soluble epoxide hydrolase (sEH) and fatty acid amide hydrolase (FAAH), have been proposed as suitable targets for the application of poly-pharmacology for pain treatment.4,5
The sEH is responsible for the regulation of lipid epoxides acting as potent chemical mediators such as epoxyeicosatrienoic acids (EETs).6 These signaling lipids are responsible for mediating a number of biological processes including nociception,7,8 inflammation9 and hypertension.10,11 By converting the biologically active epoxides to their respective largely inactive diols, sEH negatively regulates the activity of the EETs. The in vivo stability of EETs and other chemically stable epoxy-fatty acids is low due to the high catalytic efficiency of sEH.12 Thus, sEH inhibition has been the major approach for studying the biological role of lipid epoxides in numerous disease states including neuropathic and inflammatory pain.13,14 Treating with sEH inhibitors reduces both forms of pain in a manner that may be dependent, in part, on cannabinoid signaling,15 endoplasmic reticulum stress16 and/or other mechanisms. Several of these inhibitors have been developed as IND candidates that have reached Phase I (GSK2256294A) and Phase II (AR9281) clinical trials for COPD and hypertension, respectively (Figure 1).17–19 GSK2256294A has not progressed to further stages of clinical trials and AR9281 was unable to demonstrate efficacy in human patients.
Figure 1.

A. Structures of several sEH inhibitors (t-TUCB, TPPU, GSK2256294A, AR9281) and FAAH inhibitors (URB597, PF-3845, PF-04457845) B. Modifications of t-TUCB skeleton were tested at the trifluoromethoxyphenyl group (“Ring A”), the trans cyclohexyl group (“Ring B”) or the benzoic acid (“Ring C”).
FAAH is a separate enzyme that is studied as a potential therapeutic target for neuropathic and inflammatory pain.20–22 This enzyme hydrolyzes arachidonoyl ethanolamide (AEA), an endocannabinoid that regulates nociception and other physiologies through activation of the cannabinoid receptors.22,23 Like EETs, AEA is quickly metabolized in vivo by FAAH and therefore in vivo studies investigating AEA require FAAH inhibitors. Although activation of cannabinoid receptors has numerous undesired effects including hypothermia, catalepsy and hyperphagia, treatment with FAAH inhibitors or AEA alone is not sufficient for producing these effects.24,25 Several FAAH inhibitors have been developed. Among them, PF-04457845 reached Phase II clinical trials without success due to lack of efficacy despite its excellent target engagement.26 Recently, BIA 10–2474 was also pulled from a Phase I clinical trial after the death of a study subject,27 which was independent of FAAH inhibition.28
Concurrent inhibition of both sEH and FAAH synergistically reduces both inflammatory and neuropathic pain.29 Interestingly, the sEH inhibitor trans-4-[4–(3-trifluoromethoxyphenyl-lureido)-cyclohexyloxy]-benzoic acid (t-TUCB), which was thought to be a selective potent sEH inhibitor (IC50 = 0.4 nM), was recently identified as a weak FAAH inhibitor (human FAAH IC50 = 260 nM). t-TUCB demonstrates excellent efficacy with multiple indications including neuropathic pain, but it was not clear whether its high efficacy is derived from its poly-pharmacology. This excellent efficacy has led to its use as a tool to treat various diseases30–32 in several animal species.33,34 Despite the extensive use of this compound for studying sEH biology, the contribution of FAAH inhibition to results in the experimental or clinical disease models has not been explored. Thus, our primary goal was to produce novel inhibitors with improved potency towards sEH and FAAH. Our secondary goal was to test the plausibility of FAAH inhibition contributing to the observed beneficial effects of t-TUCB and related compounds by defining the potency for dual inhibition in other species.
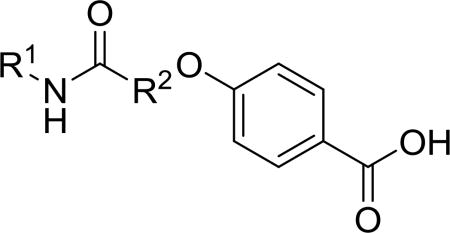
Synthesis of all inhibitors was done according to established procedures (described in detail in the Supplementary Material). Recombinant enzyme preparations were used with fluorescent-based substrates to quantify potency of inhibitors on sEH and FAAH (described in the Supplementary Material). All of the newly synthesized inhibitors are relatively potent towards sEH (IC50 < 50 nM) as expected. Thus, we primarily focused on determining the chemical structures essential for optimizing potency on FAAH. Compared to the known FAAH inhibitors PF-3845 and URB597, t-TUCB is 233-fold and 6-fold less potent, respectively (Table 1).20,35 Rings “A” and “B” and substituents on the 4-position of “C” were modified on t-TUCB to determine the portion of the structure that primarily confers potency on FAAH (Figure 1B). Urea-based FAAH inhibitors described previously have an aromatic substitution on one side of the urea, similar to ring “A” on t-TUCB. Since these compounds had higher potency for the 4-fluoro or unsubstituted rings than the 4-trifluoromethoxy substituent,36 the 4-trifluoromethoxy group on t-TUCB was replaced by a hydrogen (2), fluoride (3) or chloride (4). Potency on FAAH decreased as the size and hydrophobicity of the para position substituent increased, with 4-trifluoromethoxy (1) being the most potent. Substituting the aromatic ring for a cyclohexane (5) or adamantane (6) resulted in a complete loss in activity against FAAH. Switching the cyclohexane linker of ring “B” to a cis conformation (c-TUCB) resulted in a 20-fold loss of potency while replacing it with a butane chain (9) resulted in a completely inactive compound. Modification of the cyclohexane to an aromatic linker (10) had essentially no effect on potency for FAAH relative to t-TUCB. Although many potent urea-based FAAH inhibitors use a piperidine as the carbamoylating nitrogen,21,37,38 the modification to piperidine-incorporated tri-substituted urea reduced potency 13-fold (13). Together, these changes on ring “B” indicate the trans-cyclohexyl ring provides the exact fit in the active site of FAAH essential for t-TUCB’s inhibitory potency.
Table 1.
Potency of inhibitors with modifications on Ring A (R1) and Ring B (R2) against both human sEH and human FAAH.
| ||||
|---|---|---|---|---|
|
| ||||
| R1 | R2 | IC50 (nM) | ||
|
| ||||
| hsEH | hFAAH | |||
| URB597 | 1100 | 23 | ||
| PF-3845 | >10,000 | 0.6 | ||
| t-TUCB |
|
|
0.8 | 140 |
| 2 |
|
|
30 | 9,200 |
| 3 |
|
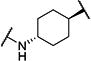
|
18 | 4,600 |
| 4 |
|
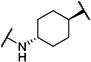
|
7 | 680 |
| 5 |
|
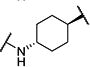
|
6 | >10,000 |
| 6 |
|
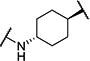
|
3 | >10,000 |
| c-TUCB |
|
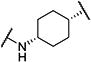
|
2 | 2,800 |
| 9 |
|
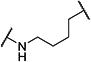
|
15 | >10,000 |
| 10 |
|
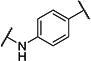
|
7 | 170 |
| 13 |
|
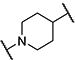
|
8 | 1,800 |
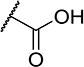
To further explore the relationship between structure and function on the FAAH enzyme, we focused on the substitutions on the 4-position of ring “C” (Table 2). The importance of the terminal carboxylic acid group was explored by testing the potency of the corresponding aldehyde (15) and alcohol (16) in addition to the amide (21) and nitrile (14). Generally, the higher oxidation state of the terminal portion correlates with higher potency towards FAAH. t-TUCB was 10-times more potent than 15 and 50-times more potent than 16. Similarly, the amide (21) was over 100-times more potent than the nitrile (14). Converting the benzoic acid to the phenol (18) had a minor effect on potency. Interestingly, modifying the phenol to the anisole (19) completely removed activity while creating an acetate ester (20) is equipotent to the phenol. Since the substrates for FAAH tend to be relatively hydrophobic lipids, we speculated conversion of the acid and primary amide to the corresponding esters or substituted amides, respectively, would result in improved potency by introducing hydrophobic groups. As expected, the methyl ester (22) had 4-fold improved potency relative to the corresponding acid (t-TUCB). However, incorporating a bulkier substitution than the methyl group such as isopropyl ester (23) showed 11-fold less potency compared to the methyl ester (22). Interestingly, the benzyl ester (24) gives approximately the same potency as the methyl ester, suggesting that π-π stacking between benzylic group and a residue at the active site may be important. Relative to t-TUCB, the methyl (25), ethyl (26) and glycinyl (27) amides all had essentially the same potency; however, the benzyl amide (29) was substantially less potent (16-fold). Between the methyl- (22 and 25) and benzyl-substituted (24 and 29) compounds, the esters generally appear to be more potent than the amides. This difference in the potency may be due to the specific angle of the substituted groups or the increased electron density of the ester carbonyl compared to the amide carbonyl. Generating the methyl ester of the glycinyl amide (28) increased the potency 4-fold compared to the corresponding free acid. Since the amide and esters appeared to be active, the amide bioisostere oxadiazole (17) was tested and had 38-fold less potency than the initial compound. Many compounds in this series could act as prodrugs following esterase or amidase catalyzed hydrolysis.39
Table 2.
Potency of inhibitors with modifications on Ring C (R3) against both human sEH and human FAAH.
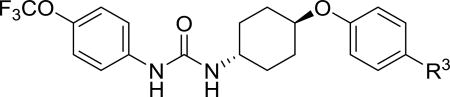
| |||
|---|---|---|---|
|
| |||
| R3 | IC50 (nM) | ||
|
| |||
| hsEH | hFAAH | ||
| t-TUCB |
|
0.8 | 140 |
| 14 |
|
5 | >10,000 |
| 15 |
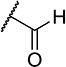
|
4 | 1,100 |
| 16 |
|
3 | 5,800 |
| 17 |
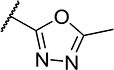
|
4 | 5,300 |
| 18 |
|
2 | 120 |
| 19 |
|
3 | >10,000 |
| 20 |
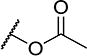
|
4 | 120 |
| 21 |
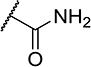
|
2 | 70 |
| 22 |
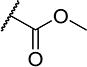
|
7 | 35 |
| 23 |
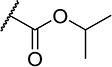
|
5 | 400 |
| 24 |
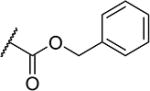
|
3 | 24 |
| 25 |
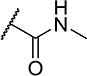
|
2 | 170 |
| 26 |
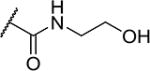
|
2 | 100 |
| 27 |
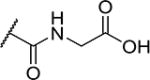
|
2 | 130 |
| 28 |
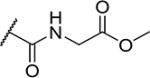
|
3 | 30 |
| 29 |
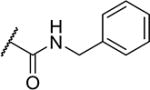
|
5 | 1,100 |
Many FAAH inhibitors work through carbamoylation of the catalytic serine residue.20,36,40 Inhibition through this mechanism is time-dependent because the inhibitory potency depends on the rate of carbamoylation. However, to our surprise, the potency of t-TUCB and two new inhibitors (18 and 24) does not change with time (Figure 2A), suggesting that these inhibitors are unlikely to inhibit through the formation of a covalent intermediate. URB597, an inhibitor known to carbamoylate FAAH, has a 3-fold increase in potency over the same period of time. To further test the mechanism of action of these inhibitors we determined the effect of varying substrate concentrations on the inhibitory potency of t-TUCB (Figure 2B). Results showed an increase in KMapp with only a minor change in vmaxapp indicating that inhibition primarily occurs through a competitive mechanism. Assuming a simple competitive model, the calculated Ki for t-TUCB is 156 ± 85 nM. Using the same analysis, the Ki for 18 and 24 are 9.1 ± 2.5 nM and 43 ± 25 nM, respectively.
Figure 2.

A. t-TUCB, 18, 24 and URB597 were pre-incubated with FAAH enzyme at various time intervals followed by addition of substrate ([S]final = 5 µM). Potency of t-TUCB, 18 or 24 inhibition is independent on pre-incubation time while inhibition by URB597 is time dependent. B. Reaction kinetics of OMP hydrolysis by FAAH was measured at various [OMP] and [t-TUCB] to determine inhibition mechanism. As [t-TUCB] increases the KMapp substantially increases (from KMapp = 30 µM to KMapp >200 µM) and the vmax marginally increases (4.0 to 5.4 RFU/sec), consistent with a primarily competitive mechanism of inhibition.
Since the endogenous substrates, EETs and AEA, for sEH and FAAH, respectively, share the arachidonic acid backbone, it is reasonable to speculate t-TUCB may competitively inhibit FAAH by mimicking and displacing the substrate. Although these lipids are linear carbon chains, a folded orientation in the substrate tunnel may resemble the linked-ring structure of t-TUCB. The crystal structure of methoxy arachidonoyl phosphonate (MAP) bound to FAAH demonstrates the bound substrate is not linear in the active site but bends around the unsaturated bonds.41 To investigate this possibility, we docked t-TUCB, 18 and 24 into the FAAH active site using AutoDock Vina (Figure 3A and Supplementary Figure 1). Docking in the FAAH active site, the polar aromatic groups on t-TUCB, 18 and 24 form hydrogen-bond interactions with the catalytic serine and orient in a manner similar to its lipid substrates when docked in the FAAH active site. In the case of t-TUCB and 18, the A ring occupies the acyl-binding pocket of the active site; while 24 stays in the membrane access channel. The binding orientation from docking t-TUCB and 18 is similar to the co-crystal structure described for covalently bound PF-384520 and is consistent with the SAR described in this manuscript. If the 4-trifluoromethoxyphenyl group on ring A occupies the acyl-binding pocket, then modification of this group to bulky lipophilic groups such as an adamantyl group will be not suitable for this site and modification to smaller groups such as an unsubstituted phenyl will decrease the ability to stably occupy that pocket. Further, modification of ring B will either decrease the stability of that conformation in the case of the butane chain or force the molecule to unfavorable conformation in the case of the cis form of TUCB (c-TUCB) and the tri-substituted urea (13). Finally, modifications to the 4-substituent on ring C determine potency based on the interactions required to generate an acyl-intermediate; thus, we would predict a highly electrophilic organophosphate or trifluoromethylketone analogues that mimic these acyl-intermediates may improve the potency of future inhibitors.42, 43
Figure 3.

A. Docking of t-TUCB (yellow) in the active site of FAAH (PDB: 1MT5) using AutoDock Vina and B. Co-crystal structure of t-TUCB (yellow) in the active site of sEH. The key catalytic residues for FAAH (Ser241) and sEH (Asp335) are represented in addition to the proton-donating residue on sEH (Tyr 383 and Tyr466). Hydrogen bonds are represented on the co-crystal structure as yellow dashed lines.
By comparison, the disubstituted urea on t-TUCB is known to form strong hydrogen bonds with the catalytic aspartate residue and the H-bond donating tyrosine residues in the active site of the sEH enzyme.44 In the co-crystallization of t-TUCB in the active site of sEH (Figure 3B) the urea fits between the aspartate and the two tyrosine residues. The flanking pockets on either side of the catalytic site are large and can accommodate a variety of shapes. Thus, consistent with the SAR, a large variety of shapes may be accommodated by sEH on either side of the urea and potency will remain relatively high (IC50 < 50 nM). The atomic coordinates and structure factors (code 6AUM) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
To test the selectivity of these inhibitors toward FAAH, we investigated their ability to inhibit a series of related enzymes, especially serine hydrolases (Table 3). t-TUCB, 18, or 24 did not inhibit the tested carboxylesterases (hCE1 and hCE2), hydrolases involved in xenobiotic detoxification, or paraoxonases (PON1, PON2 and PON3), esterases involved in the regulation of artherosclerosis,45 but 18 did partially inhibit arylacetamide deacetylase (AADAC), a poorly characterized enzyme that is known to metabolize several xenobiotics.46 These results indicate that, unlike most known FAAH inhibitors that have poor selectivity against other serine hydrolases,47, 48 broad off-target inhibition of other serine hydrolases by t-TUCB, 18 and 24 is unlikely.
Table 3.
IC50 values of compounds t-TUCB, 18 and 24 on other human serine hydrolases.
| IC50 (nM) | |||
|---|---|---|---|
|
|
|||
| Enzyme | t-TUCB | 18 | 24 |
|
|
|
||
| FAAH | 140 | 120 | 24 |
| sEH | 0.8 | 2 | 3 |
| mEH | >10,000 | >10,000 | >10,000 |
| hCE1 | >10,000 | >10,000 | >10,000 |
| hCE2 | >10,000 | >10,000 | >10,000 |
| PON1 | >10,000 | >10,000 | >10,000 |
| PON2 | >10,000 | >10,000 | >10,000 |
| PON3 | >10,000 | >10,000 | >10,000 |
| AADAC | >10,000 | 5,400 | >10,000 |
t-TUCB has been used extensively for studying the role of sEH in multiple animal models of pain.33, 34, 49 To test whether potency towards FAAH may account for the efficacy in these animal models, the effectiveness of t-TUCB, 18 and 24 to inhibit FAAH was measured on other species than human using brain microsome preparations. As positive controls, the IC50 values of two well characterized FAAH inhibitors, PF-3845 and URB-597 were measured also (Table 4, Supplementary Data). These two inhibitors are structurally distinct and have excellent selectivity in rat brain microsomes for the FAAH enzyme over other hydrolases.20,38 As expected, both inhibitors blocked >80% of the activity in the microsomes of the different species tested (Supplementary Figure 2). Interestingly, both t-TUCB and 24 are less potent against the FAAH of all other species tested than human FAAH. 18 had comparable IC50s for FAAH from all species tested except from rat, where 18 is 10–fold less potent. The hypothesized mechanism of inhibition based on docking relies on a relatively specific fit in the enzyme active site. Thus, we speculate small modifications to this acyl-binding pocket across species accounts for the substantial species selectivity observed for these inhibitors. By comparison, sEH inhibition relies primarily on strong hydrogen-bonding interactions and less on enzyme fit. t-TUCB, 18 and 24 also had species differences on sEH but all three inhibitors were still relatively potent in all species tested (IC50 < 100 nM). Due to the low potency towards FAAH relative to sEH (Table 4), it is unlikely that FAAH inhibition contributes to the previously observed effects of t-TUCB on pain in a mouse model49.
Table 4.
IC50 values of compounds t-TUCB, 18 and 24 on sEH and FAAH in multiple species.
| IC50(nM) | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| Enzyme | Species | t-TUCB | 18 | 24 | PF-3845 | URB597 |
|
|
|
|
||||
| FAAH | Human | 140 | 120 | 24 | 0.6 | 23 |
| Mouse | 6,300 | 350 | 510 | 12 | 11 | |
| Rat | >10,000 | 1,700 | >10,000 | 4.4 | 12 | |
| Cat | 1,200 | 92 | 240 | <0.1 | 12 | |
| Dog | 3,900 | 280 | 280 | 0.4 | 22 | |
| Horse | 3,300 | 270 | 87 | 0.3 | 7 | |
|
| ||||||
| sEH | Human | 0.8 | 2 | 3 | - | - |
| Mouse | 5.7 | 0.4 | 2.1 | - | - | |
| Rat | 54 | 1.0 | 1.6 | - | - | |
| Cat | 2.0 | 4.3 | 42 | - | - | |
| Dog | 5.6 | 11 | 28 | - | - | |
| Horse | 19 | 21 | 77 | - | - | |
In conclusion, we have designed dual sEH/FAAH inhibitors with nanomolar potency towards both enzymes. These inhibitors have potency independent of incubation time and a mechanism consistent with competitive inhibition. Furthermore, these inhibitors are selective for FAAH over other serine hydrolases and have relatively low species selectivity. Use of these inhibitors will support future biological studies investigating the importance of dual sEH/FAAH inhibition.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences grant R01 ES002710 and the National Institute of Environmental Health Sciences Superfund Research Program grant P42 ES004699. SDK was supported by a NIGMS-funded Pharmacology Training Program (T32GM099608). MEN and SP acknowledge support from NIH HL107877. Structural data used in this publication were collected at the Gulf Coast Protein Crystallography Beamline at the Center for Advanced Microstructures and Devices. This beamline is supported by the National Science Foundation grant DBI-9871464 with co-funding from the National Institute for General Medical Sciences. We would also like to thank Drs. Patricia Pesavento and Carrie Fino for providing samples of animal tissue used in these studies.
Abbreviations and definitions
- AADAC
arylacetamide deacetylase
- AEA
arachidonoyl ethanolamide
- EETs
epoxyeicosatrienoic acids
- FAAH
fatty acid amide hydrolase
- hCE
human carboxylesterase
- MAP
methoxy arachidonoyl phosphonate
- NSAIDS
non-steroidal anti-inflammatory drugs
- PF-3845
N-(pyridin-3-yl)-4-(3-((5-(trifluoromethyl)pyridin-2-yl)oxy)benzyl)piperidine-1-carboxamide
- PON
paraoxonoase
- sEH
soluble epoxide hydrolase
- t-TUCB
trans-4-[4-(3-trifluoromethoxyphenyl-l-ureido)-cyclohexyloxy]-benzoic acid
- URB597
3'-carbamoyl-[1-1'-biphenyl]-3-yl cyclohexylcarbamate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing financial interests declaration: SDK and BDH have filed a patent through the University of California for compounds described in this text for use as dual sEH/FAAH inhibitors. BDH is founder and SHH is an employee of Eicosis Animal Health, a company developing t-TUCB for use in companion animals.
References
- 1.Allerton CMN, Ali Z, Smith DM. CHAPTER 1. The Disease of Pain and Current Market Trends. 2013:1–36. [Google Scholar]
- 2.Reddy AS, Zhang S. Polypharmacology: drug discovery for the future. Expert Rev Clin Pharmacol. 2013;6(1):41–47. doi: 10.1586/ecp.12.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem. 2014;57(19):7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 4.Hwang SH, Wagner KM, Morisseau C, et al. Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J Med Chem. 2011;54(8):3037–3050. doi: 10.1021/jm2001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Migliore M, Habrant D, Sasso O, et al. Potent multitarget FAAH-COX inhibitors: Design and structure-activity relationship studies. Eur J Med Chem. 2016;109:216–237. doi: 10.1016/j.ejmech.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79(24):2311–2319. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner KM, McReynolds CB, Schmidt WK, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther. 2017 doi: 10.1016/j.pharmthera.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102(28):9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Z, Xu F, Huse LM, et al. Soluble Epoxide Hydrolase Regulates Hydrolysis of Vasoactive Epoxyeicosatrienoic Acids. Circ Res. 2000;87(11):992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 11.Ulu A, Lee KSS, Miyabe C, et al. An Omega-3 Epoxide of Docosahexaenoic Acid Lowers Blood Pressure in Angiotensin-II-Dependent Hypertension. J Cardiovasc Pharmacol. 2014;64:87–99. doi: 10.1097/FJC.0000000000000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morisseau C, Inceoglu B, Schmelzer K, et al. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res. 2010;51(12):3481–3490. doi: 10.1194/jlr.M006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inceoglu B, Jinks SL, Ulu A, et al. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105(48):18901–18906. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inceoglu B, Wagner K, Schebb NH, et al. Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP. Proc Natl Acad Sci U S A. 2011;108(12):5093–5097. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner K, Inceoglu B, Hammock BD. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediat. 2011;96(1–4):76–83. doi: 10.1016/j.prostaglandins.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inceoglu B, Bettaieb A, Trindade da Silva C, Lee KSS, Haj FG, Hammock BD. Endoplasmic reticulum stress in the peripheral nervous system is a signiicant driver of neuropathic pain. Proc Natl Acad Sci USA. 2015;112:9082–9087. doi: 10.1073/pnas.1510137112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang L, Cheriyan J, Gutterman DD, et al. Mechanisms of Vascular Dysfunction in COPD and Effects of a Novel Soluble Epoxide Hydrolase Inhibitor in Smokers. Chest. 2017;151(3):555–563. doi: 10.1016/j.chest.2016.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazaar AL, Yang L, Boardley RL, et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016;81(5):971–979. doi: 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen HC, Hammock BD. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem. 2012;55(5):1789–1808. doi: 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn K, Johnson DS, Mileni M, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16(4):411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DS, Stiff C, Lazerwith SE, et al. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med Chem Lett. 2011;2(2):91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piomelli D, Sasso O. Peripheral gating of pain signals by endogenous lipid mediators. Nat Neurosci. 2014;17(2):164–174. doi: 10.1038/nn.3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65(2):849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cravatt BF, Demarest K, Patricelli MP, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98(16):9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fegley D, Gaetani S, Duranti A, et al. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3'-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313(1):352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- 26.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153(9):1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 27.von Schaper E. Bial incident raises FAAH suspicions. Nat Biotechnol. 2016;34(3):223. doi: 10.1038/nbt0316-223a. [DOI] [PubMed] [Google Scholar]
- 28.Kerbrat A, Ferre JC, Fillatre P, et al. Acute Neurologic Disorder from an Inhibitor of Fatty Acid Amide Hydrolase. N Engl J Med. 2016;375(18):1717–1725. doi: 10.1056/NEJMoa1604221. [DOI] [PubMed] [Google Scholar]
- 29.Sasso O, Wagner K, Morisseau C, Inceoglu B, Hammock BD, Piomelli D. Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol Res. 2015;97:7–15. doi: 10.1016/j.phrs.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Yang J, Guo L, et al. Use of a soluble epoxide hydrolase inhibitor in smoke-induced chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2012;46(5):614–622. doi: 10.1165/rcmb.2011-0359OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris TR, Bettaieb A, Kodani S, et al. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicol Appl Pharmacol. 2015;286(2):102–111. doi: 10.1016/j.taap.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun D, Cuevas AJ, Gotlinger K, et al. Soluble epoxide hydrolase-dependent regulation of myogenic response and blood pressure. Am J Physiol Heart Circ Physiol. 2014;306(8):H1146–1153. doi: 10.1152/ajpheart.00920.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guedes AG, Morisseau C, Sole A, et al. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Vet Anaesth Analg. 2013;40(4):440–448. doi: 10.1111/vaa.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ulu A, Appt S, Morisseau C, et al. Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. Br J Pharmacol. 2012;165(5):1401–1412. doi: 10.1111/j.1476-5381.2011.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mor M, Rivara S, Lodola A, et al. Cyclohexylcarbamic Acid 3'- or 4'-Substituted Biphenyl-3-yl Esters as Fatty Acid Amide Hydrolase Inhibitors: Synthesis, Quantitative Structure-Activity Relationships, and Molecular Modeling Studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 36.Keith JM, Apodaca R, Xiao W, et al. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2008;18(17):4838–4843. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- 37.Johnson DS, Ahn K, Kesten S, et al. Benzothiophene piperazine and piperidine urea inhibitors of fatty acid amide hydrolase (FAAH) Bioorg Med Chem Lett. 2009;19(10):2865–2869. doi: 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahn K, Johnson DS, Fitzgerald LR, et al. Novel Mechanistic Class of Fatty Acid Amide Hydrolase Inhibitors with Remarkable Selectivity. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 39.Kim IH, Nishi K, Tsai HJ, et al. Design of bioavailable derivatives of 12-(3-adamantan-1-yl-ureido)dodecanoic acid, a potent inhibitor of the soluble epoxide hydrolase. Bioorg Med Chem. 2007;15(1):312–323. doi: 10.1016/j.bmc.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moreno-Sanz G, Duranti A, Melzig L, et al. Synthesis and structure-activity relationship studies of O-biphenyl-3-yl carbamates as peripherally restricted fatty acid amide hydrolase inhibitors. J Med Chem. 2013;56(14):5917–5930. doi: 10.1021/jm4007017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298(5599):1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 42.Wheelock CE, Nishi K, Ying A, et al. Influence of sulfur oxidation state and steric bulk upon trifluoromethyl ketone (TFK) binding kinetics to carboxylesterases and fatty acid amide hydrolase (FAAH) Bioorg Med Chem. 2008;16(4):2114–2130. doi: 10.1016/j.bmc.2007.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quistad GB, Sparks SE, Casida JE. Fatty acid amide hydrolase inhibition by neurotoxic organophosphorus pesticides. Toxicol Appl Pharmacol. 2001;173(1):48–55. doi: 10.1006/taap.2001.9175. [DOI] [PubMed] [Google Scholar]
- 44.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 45.Chistiakov DA, Melnichenko AA, Orekhov AN, Bobryshev YV. Paraoxonase and atherosclerosis-related cardiovascular diseases. Biochimie. 2017;132:19–27. doi: 10.1016/j.biochi.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Fukami T, Yokoi T. The Emerging Role of Human Esterases. Drug Metab Pharmacokinet. 2012;27(5):466–477. doi: 10.2133/dmpk.dmpk-12-rv-042. [DOI] [PubMed] [Google Scholar]
- 47.Zhang D, Saraf A, Kolasa T, et al. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52(4):1095–1105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 48.Crow JA, Bittles V, Borazjani A, Potter PM, Ross MK. Covalent inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by the carbamates JZL184 and URB597. Biochem pharmacol. 2012;84(9):1215–1222. doi: 10.1016/j.bcp.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner K, Inceoglu B, Dong H, et al. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur J Pharmacol. 2013;700(1–3):93–101. doi: 10.1016/j.ejphar.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.