

Abstract
The RNA genomes of viruses likely undergo multiple functionally important conformational changes during their replication cycles, changes that are poorly understood at present. We used two complementary in-solution RNA structure probing strategies (SHAPE-MaP and RING-MaP) to examine the structure of the RNA genome of satellite tobacco mosaic virus inside authentic virions and in a capsid-free state. Both RNA states feature similar three-domain architectures in which each major replicative function – translation, capsid coding, and genome synthesis – fall into distinct domains. There are, however, large conformational differences between the in-virion and capsid-free states, primarily in one arm of the central T domain. These data support a model in which the packaged capsid-bound RNA is constrained in a local high-energy conformation by the native capsid shell. The removal of the viral capsid then allows the RNA genome to relax into a more thermodynamically stable conformation. These data support a model in which the RNA architecture of the central T domain changes during capsid assembly and disassembly and may play a role in genome packaging.
Table of Contents
Introduction
Satellite tobacco mosaic virus (STMV) is a T=1 icosahedral plant virus and has a diameter of 17 nm.1,2 The virus particle is composed of 60 identical capsid protein subunits that dimerize and then coalesce to encase a single-stranded genomic RNA that is 1058 nucleotides in length.3,4 The genome encodes the capsid protein mRNA but requires co-infection with a helper virus, typically tobacco mosaic virus, to replicate its RNA genome via a negative strand intermediate.5,6 The capsid protein is then synthesized by the plant translational machinery. Crystallographic analysis of the STMV particle revealed that each protein dimer binds a short helix of six to eight base pairs with an additional base stacked at each 3’ end. An additional non-helical nucleotide was also visualized that is bound by the capsid protein.3,7,8 However, the regions of the RNA genome that link these helices have not been visualized by crystallography.
Many regions within viral RNAs likely sample different secondary structures depending on the RNA functional state. Two important states in the STMV replication cycle include the genomic RNA packaged inside the virion (in virio) and the RNA as released from the capsid inside the cell where translation and replication occur (capsid-free).5 Prior work used SHAPE to develop a secondary structure model for capsid-free RNA, gently extracted from virions, that emphasized long-range base pairing interactions.9 A fully independent SHAPE analysis performed using in vitro transcribed RNA proposed a closely related model, also featuring long-range interactions.10 These SHAPE-directed models were further supported by direct visualization of the genomic RNA using atomic force microscopy9 and cryo-electron microscopy.11 In contrast, multiple models have been proposed for the conformation of the in virio RNA that have emphasized short helices, without long-range base-pairing interactions, based on computational-only secondary structure modeling.2,4,9,12-14 These models vary significantly, reflecting, in part, the difficulty in modeling the secondary structure of a large RNA when extensive protein-RNA interactions are present.9 Here we combine results of several recently developed chemical probing strategies to overcome previous limitations and develop a well-defined model for the secondary structure of the STMV RNA genome as it exists inside authentic virions and in the absence of the capsid.
First, we used SHAPE-MaP (selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling) to chemically probe both the native in virio and deproteinized, capsid-free RNA states. This technology exploits the preferential ability of 2’-hydroxyl-selective reagents to react with conformationally flexible nucleotides to form covalent 2’-O-adducts. The positions of these adducts are encoded as noncomplementary nucleotides during cDNA synthesis.15 The position and frequency of SHAPE adducts are read out by massively parallel sequencing. The single-nucleotide SHAPE reactivity data was used to develop experimentally constrained secondary structure models for both RNA states. Due to better coverage and increased quantitative accuracy afforded by the MaP approach,15,16 these models are more reliable than previously published SHAPE models. In addition, advances in the folding pipeline allowed us to evaluate confidence in the predicted structures by calculating the probability that a given base pair forms across all possible structures in the ensemble of predicted structures. The resulting secondary structure model for the in virio RNA genome is characterized by long-range base-pairing interactions and displays the same overall three domain architecture as the capsid-free model.
Second, we used RING-MaP (RNA interaction groups identified by mutational profiling)17 to detect through-space interactions that occur between nucleotides in both the in virio and capsid-free RNA genome states. This technology exploits the local structural variation or “breathing” of higher order RNA structure. During modification with a chemical probe, some nucleotides become reactive to the probe in a correlated way as the molecule “breathes”. Multiple chemical modifications in a single RNA molecule are again encoded as noncomplementary nucleotides during cDNA synthesis by reverse transcriptase and are detected using massively parallel sequencing. RING-MaP thus identifies nucleotides that are in medium- to long-range structural communication. Protein-RNA interactions do not interfere with the ability of this experiment to detect through-space interactions. RING-MaP can also be used to specifically distinguish interactions that reflect secondary versus tertiary structure elements. RING-MaP data provided direct support for the proposed secondary structure models for both the in virio and capsid-free states. Both the nucleotide-specific and through-space SHAPE and RING approaches captured significant structural differences between the in virio and capsid-free RNA states. These structural differences support the model that the secondary structure of the STMV RNA genome is strongly influenced by the presence of the protein capsid and that the observed conformational change could play a role in packaging and assembly of viral particles.
Methods
STMV virions and RNA
Native STMV virions were prepared exactly as described.9 Briefly, STMV virions were purified from leaves of infected tobacco plants, crystalized from 25% ammonium sulfate,7 dialyzed (10,000 MWCO) against SHAPE folding buffer [50 mM HEPES (pH 8.0), 200 mM NaCl, 5 mM MgCl2], and stored at -80 °C. Aliquots were thawed immediately prior to use. Capsid-free RNA was prepared by treating dialyzed STMV particles with proteinase K (1 mg/mL) and 1% (w/vol) sodium dodecyl sulfate for 1 h at 37 °C. Purified genomic RNA was then isolated by three extractions with 2× volume phenol-chloroform-isoamylalcohol (equilibrated with virion dialysis buffer) and five extractions with chloroform. The extracted RNA was exchanged into SHAPE buffer using G25 columns (Microspin, Illustra). In virio RNA was treated with SHAPE reagent prior to removal from the capsid (see below) and was extracted in the same way except that only three chloroform extractions were performed. These protocols avoid use of denaturants or conditions that would aggressively disrupt STMV RNA structure.
SHAPE-MaP
For in virio analyses, dialyzed virions were incubated in SHAPE folding buffer at 37 °C for 20 min, modified by treating with one tenth volume 100 mM 1-methyl-7-nitroisatonic anhydride (1M7) in DMSO, and then incubated 37 °C for 5 min. A no-reagent control (using neat DMSO) was performed in parallel. The modified RNA was then extracted from virions as described above. For capsid-free analyses, RNA was extracted from virions and then modified with 1M7 as described above. A denatured control was performed by incubating capsid-free RNA in 50% (vol/vol) formamide and 4 mM EDTA, heating to 95 °C for 1 min, treating the sample with 100 mM 1M7, and incubating at 95 °C for 1 min. Reverse transcription was then performed under MaP conditions using random primers16 to produce a cDNA library. Second-strand synthesis was used to generate double-stranded DNA from the cDNA, and sequencing libraries were prepared by tagmentation (Nextera XT DNA Sample Preparation Kit; Illumina, FC-131-1024). Libraries16 were sequenced on an Illumina MiSeq using a v2 reagent kit with 300 cycles.
RNA structure modeling
Mutation rates for each sample (1M7, DMSO, and denaturing control) were determined by aligning nucleotide-resolution SHAPE-MaP data to the STMV sequence using ShapeMapper.16 To calculate SHAPE reactivities, data from the no-reagent (DMSO) control were subtracted from the 1M7-modified data and this difference was divided by the denatured control data. These SHAPE reactivities were incorporated as pseudo-free energy change terms into the RNAstructure software package.16,19,20 The SuperFold program was employed to fold the large STMV RNA using a windowed algorithm applying the current optimum parameters: slope = 1.8, intercept = -0.6, max base pairing distance = 50016. The SuperFold program also uses the SHAPE reactivities to calculate base pairing probabilities, which we plot as probability arcs.15,16
RING-MaP
For in virio analyses, thawed virus aliquots were exchanged into RING folding buffer [300 mM sodium cacodylate (pH 7.0), 10 mM MgCl2] using Microspin G25 columns (Illustra). The sample was incubated at 37 °C for 20 minutes, treated with one-tenth volume 1.7 M DMS in ethanol, and incubated at 37 °C for 6 minutes. Reactions were quenched by addition of an equal reaction volume of neat β-mercaptoethanol. A no-reagent control (using neat ethanol) was performed in parallel. Modified RNA was extracted from the capsid (TRIzol; Ambion) and precipitated with ethanol. For capsid-free analyses, RNA was treated with proteinase K and SDS followed by phenol/chloroform extraction and exchanged into RING buffer prior to incubation and modification by DMS or neat ethanol. Reverse transcription under MaP conditions, second-strand synthesis, preparation of DNA libraries, and sequencing was carried out as described above. RING-MaP sequencing data were aligned to the STMV sequence using ShapeMapper.16 The resulting mutation data were used to measure internucleotide correlations using custom software updated to allow for analysis of randomly-primed reverse transcription data.17,18 To detect DMS mutation interdependencies, the Yates’ corrected version of the Pearson’s χ2 test was carried out on all possible pairs of nucleotides. If χ2 ≥ 20 (P < 0.00001), the two nucleotides were taken to have a statistically significant mutation correlation.17 Only statistically significant correlations (χ2 ≥ 20) with (|ρ| – standard deviation) ≥ 0.025 and a predicted coincidence > 20 were considered in further analysis. Note that detection of pairwise through-space RING interactions was limited by the length of sequencing reads to less than approximately 280 nucleotides.
Results
Overview of SHAPE and RING RNA structure analyses
The structure of the genome of an RNA virus is likely to be modulated by interactions with numerous protein cofactors. In this work, we investigated the higher order structures of the STMV RNA genome in two states: the in virio state, in which the RNA is packaged inside the intact virus particle and makes extensive interactions with the capsid protein,3,7,8 and a capsid-free state using deproteinized STMV RNA, in which only intramolecular RNA interactions stabilize the structure. Different regions of the STMV RNA likely have varying propensities to form a single stable structure or to sample multiple distinct states. We therefore present the results of our investigations in complementary ways (Figure 1). The most direct and model-free view of the data is in the form of simple SHAPE reactivity plots (Figure 1A). The reactivity data from SHAPE-MaP experiments performed on each RNA state can also be used to develop experimentally constrained, minimum free energy secondary structure models (Figure 1C). These models tend to accurately recapitulate secondary structures in regions with well-determined stable structures20 but do not fully account for RNA motifs that form multiple structures and some tertiary structures.16,21
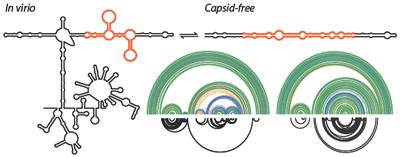
Figure 1.
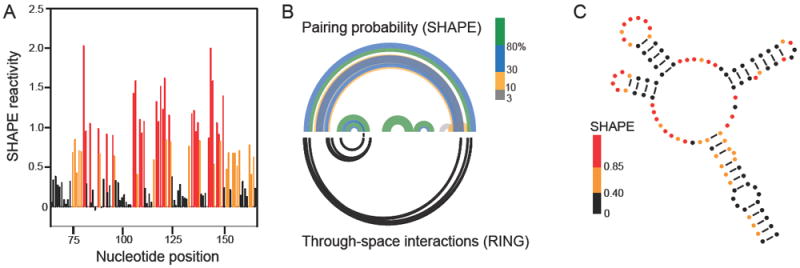
Scheme for using SHAPE-MaP and RING-MaP experiments to visualize both well-folded and conformationally heterogeneous structures in the STMV RNA genome. Data and predictions for a segment in the 5’ domain of the in virio RNA are shown. (A) Model-free SHAPE-MaP reactivity profile with reactivities colored black, orange, or red illustrate low, moderate, and high SHAPE reactivities, respectively. (B) Arc plots showing (top) the base pairing probabilities derived from SHAPE data and (bottom) through-space interactions based on RING correlations. RINGs can measure internucleotide interactions spanning the length of a single sequencing read, ~280 nts in this work. (C) Experimentally constrained, minimum free energy secondary structure model. Nucleotides are colored by experimental SHAPE-MaP reactivity. This secondary structure representation is useful for its simplicity and clarity but often overemphasizes the likelihood of a single structure, as shown by comparing panels B and C.
To evaluate the confidence of these predicted structures and to present the potential structural heterogeneity in a more comprehensive way, we calculated the probability of formation of each base pair across all possible structures in the predicted ensemble15,19 that are consistent with the experimental SHAPE data. These probabilities are visualized as arcs colored according to base pairing probability (Figure 1B, top). This approach highlights regions that are likely to sample a single predominant structure (green arcs) and those regions in which multiple structures likely exist in equilibrium (blue, yellow, and gray arcs).
The pairwise correlation data from the RING experiment are also plotted as arcs (Figure 1B, bottom). The RING data are categorized by the contact distance between two interacting nucleotides. We define the contact distance as the shortest path between two nucleotides skipping nested helices in the SHAPE-predicted secondary structure.17 RING correlations with a contact distance less than 20 tend to report secondary structure interactions; longer contact distances are suggestive of tertiary interactions.17,20 Thus the RING-MaP data could be used to qualitatively characterize interactions as reflecting secondary versus tertiary structure.
Overall architectures of in virio and capsid-free STMV RNAs
SHAPE data indicate that the in virio and capsid-free RNA states are relatively well defined (Figure 2). Both predicted structures can be divided into the three separate domains identified previously.9 The central T domain is the largest of these domains and the best defined; the majority of base pairs in this domain have pairing probabilities greater than 80%. For both the in virio and capsid-free RNA structures, the long-range base pairs that form the anchoring stem of the T shape are predicted with high confidence (Figure 2). This is a key finding as early models proposed only local stem loop structures,4,12,14,22 but current models specifically support these long-range interactions.9-11 We discuss our predicted secondary structures largely in terms of probability arcs, as these best represent the structural variability of some regions and their contrast to the single structures observed in other regions. In order to facilitate conventional visualization of the two conformations, we also constructed traditional minimum free energy secondary structure models from the SHAPE data (Figure 4; Supplemental Figure 1).
Figure 2.
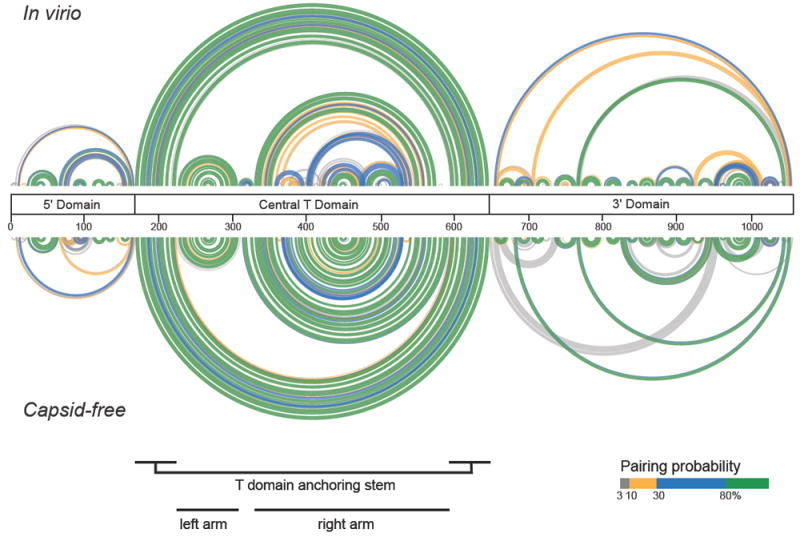
Comparison of base pairing probabilities for the in virio and capsid-free RNA states. Probabilities were calculated from SHAPE data based on the total ensemble of structures using SuperFold.16 Base pairs are colored by their probabilities (legend). The central bar defines the three structural domains that characterize both RNAs; landmarks in the central T domain are indicated below structures.
Figure 4.
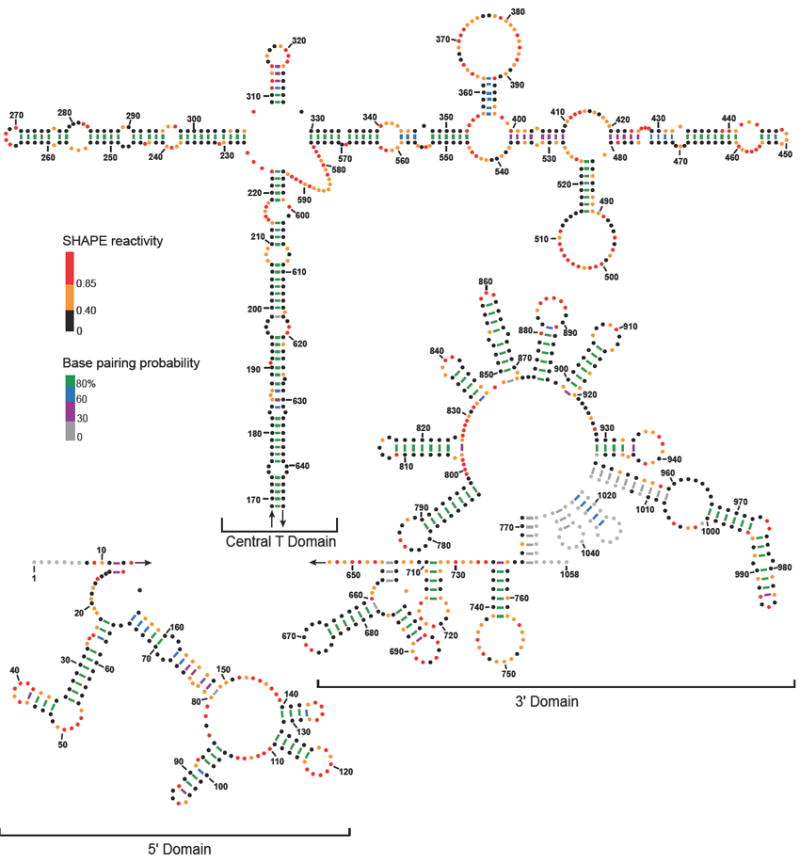
Secondary structure model for the in virio STMV RNA genome. Nucleotides are colored by SHAPE reactivity; black, orange, and red represent low, moderate, and high reactivities, respectively. Lines indicating base pairs are colored by base pair probability (legend). Nucleotides with no SHAPE data are colored grey.
The well-defined series of stacked helices that constitute the “left” arm of the central T domain are identical in both structures. In contrast, the “right” arm of the T domain shows significant structural differences as visualized in the probability arcs for the in virio and capsid-free states (Figure 2, see positions 300-600). The right arm of the capsid-free structure is well defined (high base-pairing probabilities) and consists of a single set of stacked helices linked by short, single-stranded bulges. The in virio “right” arm structure forms helices linked by bulges and also contains two stem loops that branch off the right arm. The predicted base pairs for this segment of the in virio T-domain have varying levels of probability (Figure 2, top), indicative of structural heterogeneity. The less probable predicted base pairs in this region are unique to the in virio structure and do not correspond to helices in the capsid-free structure. These differences suggest that the conformation in the right arm of the in virio and capsid-free states differs substantially.
The models for the 5’ and 3’ domains include multiple well-defined short stem-loop structures and several less probable long-range interactions. Most of the short stem-loop structures are shared between the in virio and capsid-free structures, whereas the long-range (and less probable) interactions tend to differ. There are two well-defined differences between the in virio and capsid-free states in the 5’ and 3’ domains. The in virio 5’ domain contains an additional well-defined medium range helix (spanning roughly positions 70-165) that does not exist in the capsid-free structure. In the 3’ domain, the capsid-free state contains several well-defined longer-range helices that do not appear to exist in the in virio model. Other long-range interactions in the 5’ and 3’ domains of both structures have generally lower probabilities consistent with structural heterogeneity. Interestingly, the different possible long-range conformations rarely disrupt the well-defined local stem loop structures.
Through-space interactions identified by RINGs
The RING-MaP experiment directly maps through-space interactions in RNA. RING-MaP uses dimethyl sulfate (DMS) to modify RNA and therefore is primarily sensitive to correlations involving adenosine and cytosine nucleotides. Nucleotides do not have to base pair directly with one another but need to be involved in a common structural interaction to result in a significant RING correlation.17 RING correlations are measured over the length of a single sequencing read, corresponding to approximately 280 nucleotides in this work. Thus, RING-MaP could not be used to corroborate interactions of nucleotides further apart than 280 nucleotides in primary sequence, such as those that form the stem of the central T domain. Through-space correlations were observed throughout the lengths of both the in virio and capsid-free RNAs (Figure 3, lower arcs). Both RNA states are dominated by correlations that have contact distances of less than 20, indicating that they primarily detect secondary structure in the STMV RNAs. The patterns of the correlations between in virio and capsid-free are different and, intriguingly, these differences lie in exactly the regions proposed to have the largest conformational differences based on the SHAPE-directed base-pairing probabilities.
Figure 3.

Comparison of structure models determined from SHAPE data with RING through-space correlations. Structure models are illustrated as the minimum free energy structure and correspond to the most probable structure from the data shown in Figure 2. (A) In virio and (B) capsid-free RNA states. RING-MaP through-space correlations are illustrated with inverted arcs. The upper row of RINGs (black) displays correlations with contact distances less than 20, which predominantly reflect secondary structure interactions. The bottom row displays correlations with a contact distance greater than 20, potentially reflective of tertiary interactions, Both classes of correlations reflect through-space structural communication.17 Note that minimum free energy structures are shown for simplicity and to facilitate comparison with RING data; however, probability arcs (Figure 2) are the most complete and accurate way to represent structure models.
The correlations with contact distances less than 20 support the proposed SHAPE-directed in virio secondary structure (Figure 3A, compare black arcs with the minimum free energy structure model). In the 5’ domain, the correlations specifically corroborate the presence of several local stem loops and the medium-range helix. In the central T domain, observed correlations support the secondary structure for both arms of the T-shape. The RING correlations mirror the base pairing in the left arm of the T domain structure and provide direct evidence for formation of the two stem loops that branch off of the right arm. Many of the local stem loops predicted in the 3’ domain are also supported to some extent by the RING-MaP correlations.
Very few correlations with contact distances greater than 20 were observed for the in virio state (Figure 3A, bottom; red arcs). Compared to RING correlations observed for RNAs with known tertiary structures,17 these correlations are sufficiently sparse that they do not correspond to significant tertiary structure in the STMV RNA. In addition, most of these correlations have contact distances only marginally above 20. We interpret these correlations as reflective of interactions between helices and neighboring regions of single-stranded nucleotides. Thus, the RING-MaP experiments do not support that significant tertiary structure is present when the RNA genome is packaged in the virion. It is possible that the tertiary structure of the RNA is folded so tightly that the RING-MaP experiment cannot detect the tertiary structure.17 There may also exist tertiary interactions that occur between nucleotides too far apart (> 280 nts) to be detected by RING-MaP.
In the capsid-free RNA state, the RING correlations in the central T domain support the proposed secondary structure. In particular, multiple correlations in the right arm provide strong evidence for a single set of stacked helices separated by single-stranded bulges (Figure 3B). Comparison of the RINGs in the right arm of the in virio and capsid-free states shows large and clear structural differences in this region, further supporting the idea that this segment of the STMV RNA adopts distinct structures in the two states. RING-MaP correlations show modest, but imperfect, support for the base pairs in the 5’ and 3’ domains. The lack of supporting RING correlations in these domains may reflect the potential inaccuracies in the structural model, or sampling of multiple structures in these regions. Very few potential tertiary interaction RINGs were detected in the capsid-free state, and, as for the in virio correlations, contact distances were only slightly more than 20. These correlations generally appear to depict interactions between a helix and a neighboring bulge or loop rather than significant tertiary structures.
Structural model for the STMV RNA genome in virio
We propose an experimentally constrained, minimum free energy secondary structure model of STMV RNA in virio (Figure 4). This structure shares many features with our capsid-free RNA model (Supplemental Figure 1). In both models, the 5’ and 3’ domains consist mainly of short, local stem loops. The main stem of the central, T-shaped domain is dominated by long-range base pairs, with base pairing of nucleotides up to 475 nucleotides apart in primary sequence. The main stem and left arm of the T domain consist of multiple short helices linked by single-stranded bulges. The right arm forms similarly short helices linked by bulges, as seen in our capsid-free model but also includes two stem loops that branch off the right arm at positions 353-400 and 480-528. In the in virio model, 55% of the nucleotides are base paired. Of these base pairs, 70% have a pairing probability greater than or equal to 80% (Figure 4). These data support the view that the majority of the structure is well-defined and significantly samples the predicted minimum free energy conformation. A total of 31 helices of five base pairs or longer exist in the in virio predicted structure.
Using the recently developed ΔSHAPE framework for detecting highly significant differences in SHAPE reactivities,23 we identified nucleotides that showed the greatest differences in reactivity between in virio and capsid-free RNAs (Figure 5A). There is a high frequency of nucleotides with significantly different reactivities in the central T domain, spanning positions 200-600 (Figure 5A). These nucleotides are located in regions that have different predicted structures in the in virio and capsid-free models. Thus, the structural differences in our predicted models are driven by a measurable change in the base pairing of the associated nucleotides. For example, the in virio model contains two stem loops that branch off the main right arm (Figure 5B, top). In contrast, the capsid-free RNA structure model contains a single, long, semi-continuous set of multiple helices linked by short bulges and loops (Figure 5B, bottom). Many of the nucleotides identified as significantly more reactive in virio are located in regions that are predicted to be unpaired in the in virio model but paired in the capsid-free model and vice versa. These data strongly support the idea that there are significant structural differences between the genomic RNA free of capsid proteins and the RNA packaged in the virion.
Figure 5.
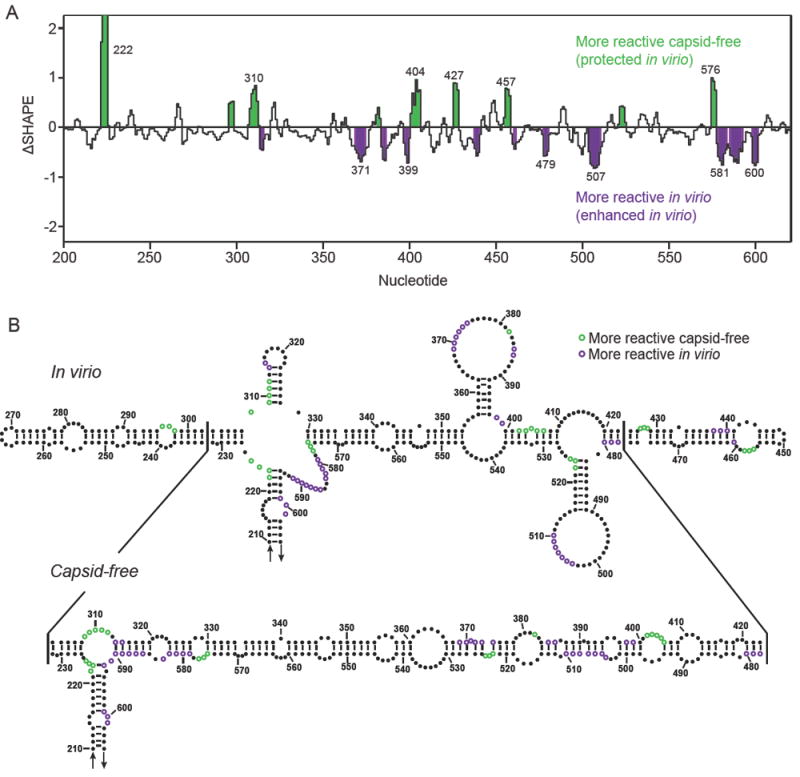
Major conformational differences in the right arm of the T domain that distinguish the in virio and capsid-free RNA states. (A) ΔSHAPE values23 for the central T domain spanning positions 200 to 600 in the genome. Statistically significant changes in SHAPE reactivity between the two RNAs are highlighted in green and purple. (B) Secondary structure models for the central T domain (residues 210-605). Individual nucleotides with significantly different reactivities between the two RNA states are colored green and purple.
Discussion
Knowledge of the structure of a viral RNA genome at different stages in the virus replication cycle is essential for understanding how function is influenced by higher order RNA structure. In general, the structures of viral RNA genomes packaged inside virions are poorly understood. STMV is a tractable system for the study of genome structure and packaging for icosahedral RNA viruses and represents a kind of “hydrogen atom” for virus assembly. Here we propose a model for the structure of the STMV RNA genome as it exists in authentic virions and show that the capsid environment stabilizes a different structure than predominates in the absence of capsid. The SHAPE-MaP directed structure is well-defined across much of the genome and exhibits multiple long-range base pairing interactions (Figure 4). RING-MaP experiments, which directly detect correlated interactions within approximately 280 nucleotides, provide clear independent support for key structural aspects of the proposed model (Figure 3). The overall architecture of the packaged, in virio RNA shares global features with that of the capsid-free RNA: each forms three clear domains with a large central T domain flanked by 5’ and 3’ domains. The capsid-free RNA structure model is highly similar to that proposed based on older-generation SHAPE experiments. In both cases, the RNA exhibits an unbranched central T structure and the same long-range base-pairing interactions observed in prior work. The 5’ and 3’ domains also share multiple helices with the prior structural model (compare Supplemental Fig. 1 with ref. 9). For both the capsid-free and in virio structures, the three domains correspond closely with the functional components of the virus. The central domain almost exactly spans the coding sequence of the capsid protein, and 5’ and 3’ domains correspond to regulatory regions that direct translation and replication, respectively.5 The structural distinctiveness of the three functional domains supports the idea that higher order RNA structure plays a functional role in viral replication.9
Despite similarities in overall domain structure, SHAPE and RING analyses strongly support the interpretation that the STMV RNA exists in significantly different conformations inside the virion versus free from capsid proteins. Within each structure, 31% of the in virio base pairs and 36% of the capsid-free base pairs are unique to their respective structures. The most well defined conformational difference occurs in the central T domain. In the virion, the right arm is predicted to adopt a structure with two three-helix junctions; when the protein capsid is removed by proteolysis, the RNA forms a more linear structure of roughly continuously stacked helices (Figure 5B). In the central T domain in virio, 266 nucleotides are predicted to be base paired in 15 continuous helices, each containing at least five base pairs. In the capsid-free structure, 312 nucleotides are base paired, and 14 helices are formed. The two states form a similar number of helices with the same average lengths, but the in virio state has more single-stranded nucleotides between the helices. A similar increase in single-stranded connecting elements occurs in the 3’ domain. While the increase in the number of single stranded nucleotides formally results in a local high-energy conformation, these single-stranded regions may confer additional flexibility to the in virio structure. We propose that the in virio RNA exists in a more flexible conformation to allow helices to bind capsid protein dimers and conform to the icosahedral symmetry of the viral particle. When released from the capsid, this flexibility is no longer necessary and additional base pairs form that stabilize the RNA structure.
Our structural data strongly support the interpretation that when the capsid is removed, the RNA relaxes into a more stable, lower energy state than exists inside the virion. These data provide insight into the assembly of the viral particle. If the genome were to fold, unaided, into a structure that is then recognized by the capsid protein, we would expect that the deproteinized RNA and the RNA in the virion would have essentially the same secondary structures. Instead, the predicted in virio and capsid-free structures differ significantly. Thus, the data reported here suggest that the structure of the RNA is altered by the presence of the capsid protein and that the formation of the viral capsid requires the resulting novel RNA structure.
The full mechanism of STMV viral genome packaging remains unclear. If packaging begins before synthesis of the RNA genome is complete, then the capsid protein might trap local secondary structure motifs as they form.4 In this model, key local structures, such as the stem loops branching off the right arm, could be trapped and stabilized by capsid proteins. Additional capsid proteins could then be recruited as the rest of the genome is synthesized. Alternatively, as with other viruses, the entire STMV genome might be synthesized before packaging begins.2 In this case, the capsid protein might refold the RNA to form the conformation compatible with capsid packing. Finally, this work emphasizes that there is a large conformational difference between the in virio and capsid-free RNA, specifically in the right arm of the central T domain. The T=1 capsid structure has a three-fold axis of symmetry,8 and the in virio conformation involves two three-helix junctions that are roughly compatible with this three-fold symmetry. Thus, the structure of the right arm of the T domain might play a key role in virus assembly and may comprise the packaging signal for the virus.
Supplementary Material
Acknowledgments
This work, including the UNC Undergraduate Transcriptome Project, was supported by the National Science Foundation (MCB-1121024 to K.M.W.). Methods development was partially supported by the National Institutes of Health (GM064803). We are indebted to Alex McPherson (UC Irvine) and to many members of the Weeks laboratory for mentorship and discussions.
Footnotes
Supporting Information
One figure showing the secondary structure model for the STMV RNA genome in the capsid-free state. All SHAPE data, RING correlations, and secondary structure models are provided as datasets in the Supporting Information and are freely available on the corresponding author’s web site.
References
- 1.Mirkov TE, Mathews DM, DuPlessis DH, Dodds JA. Nucleotide sequence and translation of STMV-RNA. Virology. 1989;170:139–146. doi: 10.1016/0042-6822(89)90361-9. [DOI] [PubMed] [Google Scholar]
- 2.Schneemann A. The structural and functional role of RNA in icosahedral virus assembly. Annu Rev Micorbiol. 2006;60:51–67. doi: 10.1146/annurev.micro.60.080805.142304. [DOI] [PubMed] [Google Scholar]
- 3.Larson SB, Koszelak S, Day J, Greenwood A, Dodds JA, McPherson A. Double-helical RNA in satellite tobacco mosaic virus. Nature. 1993;361:179–182. doi: 10.1038/361179a0. [DOI] [PubMed] [Google Scholar]
- 4.McPherson A, Larson SB. Satellite tobacco mosaic virus RNA: structure and implications for assembly. Curr Opin Struct Biol. 2001;11:59–65. doi: 10.1016/s0959-440x(00)00166-4. [DOI] [PubMed] [Google Scholar]
- 5.Dodds JA. Satellite tobacco mosaic virus. Annu Rev Phytopathol. 1998;36:295–310. doi: 10.1146/annurev.phyto.36.1.295. [DOI] [PubMed] [Google Scholar]
- 6.Sivanandam V, Mathews D, Rao ALN. Properties of satellite tobacco mosaic virus phenotypes expressed in the presence and absence of helper virus. Virology. 2015;483:163–173. doi: 10.1016/j.virol.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Larson SB, Day J, Greenwood A, McPherson A. Refined structure of satellite tobacco mosaic virus at 1.8 Å resolution. J Mol Biol. 1998;277:37–59. doi: 10.1006/jmbi.1997.1570. [DOI] [PubMed] [Google Scholar]
- 8.Larson SB, Day JS, McPherson A. Satellite tobacco mosaic virus refined to 1.4 Å resolution. Acta Crystallogr D. 2014;70:2316–2330. doi: 10.1107/S1399004714013789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Archer EJ, Simpson MA, Watts NJ, O’Kane R, Wang B, Erie DA, McPherson A, Weeks KM. Long-range architecture in a viral RNA genome. Biochemistry. 2013;52:3182–3190. doi: 10.1021/bi4001535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Athavale SS, Gossett JJ, Bowman JC, Hud NV, Williams LD, Harvey SC. In vitro secondary structure of the genomic RNA of satellite tobacco mosaic virus. PLoS One. 2013;8:e54384. doi: 10.1371/journal.pone.0054384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garmann RF, Gopal A, Athavale SS, Knobler CM, Gelbart WM, Harvey SC. Visualizing the global secondary structure of a viral RNA genome with cryo-electron microscopy. RNA. 2015;21:877–886. doi: 10.1261/rna.047506.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schroder SJ, Stone JW, Bleckley S, Gibbons T, Mathews DM. Ensemble of secondary structures for encapsidated satellite tobacco mosaic virus RNA consistent with chemical probing and crystallography constraints. Biophys J. 2011;101:167–175. doi: 10.1016/j.bpj.2011.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng Y, Larson SB, Heitsch CE, McPherson A, Harvey SC. A model for the structure of satellite tobacco mosaic virus. J Struct Biol. 2012;180:110–116. doi: 10.1016/j.jsb.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroder SJ. Probing viral genomic structure: alternative viewpoints and alternative structures for satellite tobacco mosaic virus RNA. Biochemistry. 2014;53:6728–6737. doi: 10.1021/bi501051k. [DOI] [PubMed] [Google Scholar]
- 15.Siegfried NA, Busan S, Rice GM, Nelson JAE, Weeks KM. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP) Nat Methods. 2014;11:959–965. doi: 10.1038/nmeth.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM. Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile, and accurate RNA structure analysis. Nature Protoc. 2015;10:1643–1669. doi: 10.1038/nprot.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homan PJ, Favorov OV, Lavendar CA, Kursun O, Ge X, Busan S, Dokholyan NV, Weeks KM. Single-molecule correlated chemical probing of RNA. Proc Natl Acad Sci. 2014;111:13858–13863. doi: 10.1073/pnas.1407306111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rice G. Ph D Thesis. University of North Carolina; 2015. High-throughput Experiment Driven Modeling of RNA Interactions and Structures. [Google Scholar]
- 19.Reuter JS, Mathews DH. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci. 2013;110:5498–5503. doi: 10.1073/pnas.1219988110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rice GM, Leonard CW, Weeks KM. RNA secondary structure modeling at consistent high accuracy using differential SHAPE. RNA. 2014;20:846–854. doi: 10.1261/rna.043323.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freddolino PL, Arkhipov AS, Larson SB, McPherson A, Schulten K. Molecular dynamics simulations of the complete satellite tobacco mosaic virus. Structure. 2006;14:437–449. doi: 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Smola MJ, Calabrese JM, Weeks KM. Detection of RNA-protein interactions in living cells with SHAPE. Biochemistry. 2015;54:6867–6875. doi: 10.1021/acs.biochem.5b00977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.