

Abstract
The multifaceted involvement of centrosome amplification (CA) in tumorigenesis is coming into focus following years of meticulous experimentation, which have elucidated the powerful abilities of CA to promote cellular invasion, disrupt stem cell division, drive chromosomal instability (CIN), and perturb tissue architecture, activities that can accelerate tumor progression. Integration of the extant in vitro, in vivo, and clinical data suggests that in some tissues CA may be a tumor-initiating event, in others a consequential “hit” in multistep tumorigenesis, and in still others non-tumorigenic. However, in vivo data are limited, do not specifically include breast models, and primarily focus on PLK4 (which has CA-independent mechanisms by which it promotes aggressive cellular phenotypes). In vitro breast cancer models suggest that CA can promote tumorigenesis in breast cancer cells in the setting of p53 loss or mutation, which can both trigger CA and promote cellular tolerance to its tendency to slow proliferation and induce aneuploidy. It is thus our perspective that CA is likely an early hit in multistep breast tumorigenesis that may sometimes be lost to preserve aggressive karyotypes acquired through centrosome clustering-mediated CIN, both numerical and structural. We also envision that the robust link between p53 and CA may underlie, to a considerable degree, racial health disparity in breast cancer outcomes. This question is clinically significant because, if it is true, then analysis of centrosomal profiles and administration of centrosome declustering drugs could prove highly efficacious in risk stratifying breast cancers and treating African American (AA) women with breast cancer.
An Overview of Centrosome Abnormalities
The centrosome, consisting of two centrioles and surrounding pericentriolar material (PCM), is a microtubule- and microfilament-organizing center of animal cells (Farina, et al. 2016). It regulates cell shape, polarity, migration, and the DNA-damage response and serves as a bustling hub of cellular signaling due to its ability to recruit and concentrate cell cycle, checkpoint, and signaling proteins and other molecules (Conduit, et al. 2015; Lerit and Poulton 2015). Its usefulness as a prognostic biomarker in cancer and contribution to tumorigenesis are currently subjects of intense research. Of particular interest is the phenomenon of centrosome amplification (CA), now considered a hallmark of cancer, especially breast cancer (Godinho and Pellman 2014). At least 75% of malignant breast tumors exhibit CA, although the proportion of intratumoral cells with CA varies widely, from 1–100% (Chan 2011). Centrosome abnormalities are classified as voluminous centrosomes and amplified centrosomes (i.e., >2 centrosomes). (pictured in Figure 1). Abnormal amplification of centrosome volume arises from excessive recruitment of PCM, a highly ordered yet dynamic matrix of hundreds of proteins and nucleic acids. PCM size is regulated by centrioles, free cytoplasmic αβ-tubulin, centrobin, kinases like PLK1 and CHK1, and several coiled-coil proteins like pericentrin and CPAP (Antonczak, et al. 2015; Conduit, et al. 2010; Woodruff, et al. 2014). The mechanisms undergirding abnormal amplification of centrosome volume in cancer are still poorly defined, although several stimulators of PCM assembly are overexpressed in cancer (e.g., PLK1 (Degenhardt and Lampkin 2010) and CHK1 (Zhang and Hunter 2014)), cancer cells often harbor supernumerary centrioles (which could then recruit excessive PCM), and the PCM expands following DNA damage (Mullee and Morrison 2015). Centrosomes may also appear to have an increased size but actually consist of multiple centrosomes clustered together, called “megacentrosomes” (Mittal, et al. 2015) or “speckling” (Denu, et al. 2016), with individual centrosomes only readily discerned by imaging of centrioles. Nearly half of breast tumors exhibit centrosomes with abnormal morphology, which confers significantly worse all-cause overall, breast cancer-specific overall, and recurrence-free survival in Kaplan-Meier analyses (Denu et al. 2016).
Figure 1.

Centrosome amplification (A) and abnormally augmented centrosome volume (B) in malignant breast tissue.
Centrosomes in breast tumor cells frequently exhibit augmented volume compared with cells from normal breast tissue (Denu et al. 2016; Lingle, et al. 2002a; Lingle, et al. 1998; Pannu, et al. 2015b). Furthermore, centrosomes from unstable aneuploid breast tumors have ~75% increased volume compared with stable aneuploid breast tumors (Lingle et al. 2002a), and centrosomal volume correlates with CIN in invasive breast tumors (Lingle, et al. 2002b). Centrosomes from breast tumor cells of the highly aggressive triple-negative subtype have ~60% greater volume than centrosomes from grade-matched non-triple-negative tumor cells (Pannu et al. 2015b). These data suggest that larger centrosomes are associated with more aggressive phenotypes in breast cancer. Abnormal amplification of centrosome volume is found in ductal carcinomas in situ, suggesting it may be an early event in breast tumorigenesis (Lingle et al. 2002b). Numerical CA (>1 centrosome before S-phase and >2 centrosomes after S-phase) can arise from various abnormal processes, such as cytokinesis failure, templated overduplication, de novo formation, or cell-cell fusion, such as can be induced by all known human oncogenic viruses (Gao and Zheng 2011; Godinho and Pellman 2014). Because abnormal centrosome numbers correlate with ploidy in breast cancer, cell doubling events are a likely cause of numerical CA in breast cancer, which has been estimated as explaining at least 15% of CA (i.e., 15% of cells with ploidy >3 have CA, although because chromosomes may subsequently be lost, this statistic may underrepresent the true prevalence of CA caused by doubling events) (Denu et al. 2016). The molecules and pathways responsible for numerical CA have been comprehensively reviewed elsewhere (Anderhub, et al. 2012; Brownlee and Rogers 2013; Godinho and Pellman 2014; Korzeniewski, et al. 2013; Marina and Saavedra 2014; Nigg, et al. 2014; Ogden, et al. 2012, 2013). Average centrosome number/cell correlates with tumor grade, Ki67 index, and CIN in breast cancer and is highest in the aggressive TNBC subtype (nearly two-thirds of which exhibit >2 centrosomes/cell on average), suggesting that numerical centrosome amplification is associated with aggressive breast cancer features (Denu et al. 2016). Induction of CA by PLK4 overexpression in MCF10A breast epithelial cells results in decreased CD24 and increased CD44 expression, suggesting that CA drives cellular dedifferentiation rather than merely co-occurring with it (Denu et al. 2016). In a study of n=362 predominantly white breast cancer patients with at least 5 years follow up, it was found that numerical CA confers worse overall, breast cancer-specific, and recurrence-free survival, although not independent of stage and hormone receptor status (Denu et al. 2016). Clustering of centrosomes, which circumvents spindle multipolarity that jeopardizes cell survival, occurs in more than half of breast tumors and is associated with significantly worse overall and recurrence-free survival; however, the impact of clustering on these survival outcomes after adjusting for potential confounders is unclear (Denu et al. 2016). In sum, CA is associated with more aggressive breast cancer features and may adversely impact survival, although further study to substantiate this paradigm in multivariable models is required.
Centrosome Amplification and Breast Tumorigenesis
Molecular Mechanisms Discerned from the Culture Plate
Much in vitro evidence suggests that CA actively drives tumorigenesis rather than merely being a consequence of it by imparting phenotypes such as chromosomal instability (CIN), which can promote aggressive disease features (Funk, et al. 2016). Whether CIN promotes or inhibits tumorigenesis depends on the type of cell (some being inherently more tolerant of DNA damage and aneuploidy than others), its genetic background (e.g., pre-existing p53 mutations), the specific karyotype that is acquired (e.g., gain vs. loss of an oncogene), and the rate of CIN (with moderate levels tolerated better than extreme levels). Overexpression of Aurora Kinase A (AURKA), which causes CA, results in CIN that precedes tumor formation in mouse mammary epithelium, the incidence of which is increased by a p53+/− background (Wang, et al. 2006). Thus, CA may drive breast tumor evolution, especially in the setting of abnormal p53. CA causes CIN by promoting the construction of a multipolar spindle in prophase, since supernumerary centrosomes are not initially clustered together (Ganem, et al. 2009). This abnormal spindle geometry predisposes kinetochores to attach to microtubules emanating from two spindle poles, which is termed merotely. Merotelic attachments can arise from syntelic attachments (where sister kinetochores attach to microtubules from the same spindle pole), which are converted to merotelic attachments to satisfy the spindle assembly checkpoint, or they can be formed from the outset. The proportion of microtubules involved in merotelic attachments dictates the behavior of the merotelically attached chromosome (Thompson and Compton 2011). If few microtubules are oriented to the “wrong” spindle pole (pauci-merotely), chromosome segregation proceeds without apparent impairment. If a roughly equal number of microtubules are attached to the right and wrong spindle poles (equi-merotely), the chromosome lags during anaphase due to strong, opposite poleward forces, but ultimately it tends to segregate to the right cell as a micronucleus. Nevertheless, lagging chromosomes can become trapped in and damaged by the cleavage furrow, resulting in breaks and unbalanced translocations (Janssen, et al. 2011). Trapped chromosomes are either removed from the cleavage site or the cleavage furrow regresses, resulting in polyploidization, which may itself be tumorigenic (Steigemann, et al. 2009), along with further CA. Moreover, even if the lagging chromosome segregates to the right cell as a micronucleus, micronuclear DNA replicates aberrantly and asynchronously with primary nuclear DNA (Crasta, et al. 2012), resulting in rapid accrual of complex, clustered chromosome rearrangements (Holland and Cleveland 2012; Leibowitz, et al. 2015; Zhang, et al. 2013; Zhang, et al. 2015). Chromoanasynthesis can occur because micronuclear replication forks are prone to stalling and collapse, resulting in template switching and microhomology-mediated break-induced replication, respectively, which promote intricate, local chromosome rearrangement. Chromothripsis can occur when the cell enters mitosis despite the fact that micronuclear chromosomes are still slowly replicating, causing the micronuclear chromosomes to prematurely condense and shatter. Rearrangement then occurs when the fragments are stitched back together. In addition, under-replication of micronuclear DNA results in copy-number asymmetry. Thus, equi-merotely can result in “all-at-once,” catastrophic mutagenesis, which permits rapid karyotype evolution and might be an important cause of rapidly-progressing, interval breast cancers such as TNBCs (Goncalves, et al. 2014). If many microtubules are attached to the wrong pole (multi-merotely) (Thompson and Compton 2011), the force pulling the chromosome to the wrong pole is strong, causing it to missegregate without lagging, which results in aneuploidy (Figure 2).
Figure 2.
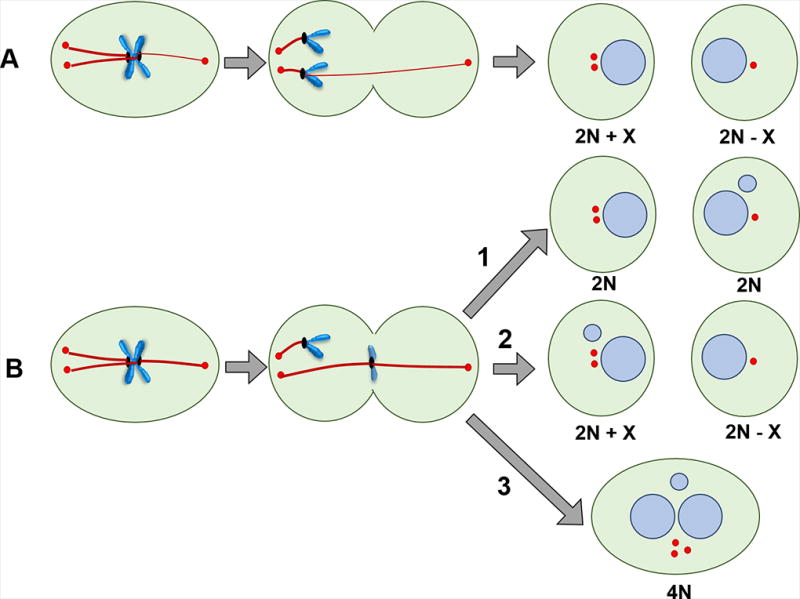
Potential deleterious consequences of CA on chromosome stability based on the class of merotely. (A) Multi-merotely: Many microtubules are attached to the incorrect spindle pole, resulting in chromosome missegregation and aneuploid daughter cells. (B) Equi-merotely: Roughly an equivalent number of microtubules are attached to the two spindle poles, so chromosome lagging occurs due to opposite polar forces. In most cases, the chromosome segregates to the right cell as a micronucleus (1). Occasionally, however, the chromosome missegregates and either forms a micronucleus or rejoins nuclear chromosomes (2). Lagging chromosomes can become trapped in and damaged by the cleavage furrow. They are either removed from the cleavage site or the cleavage furrow regresses, resulting in polyploidization, which may itself be tumorigenic, along with consequent further CA (3).
Although the cell can correct merotelic attachments by converting them to amphitelic ones via Aurora B and MPS1 kinases (Santaguida and Amon 2015), supernumerary centrosomes seem to induce so many merotelic attachments that cellular correction mechanisms are overwhelmed, and persistence of these errors into anaphase results in missegregation of whole chromosomes (Ganem et al. 2009). When centrosomes are clustered into two polar groups, the cell is able to complete bipolar mitosis, but chromosomes missegregate more frequently than in cells with only two centrosomes. Accordingly, severity of CA (i.e., centrosome number and size) correlates with CIN and aneuploidy in breast tumors (Lingle et al. 2002a). Massive genomic alterations occur in punctuated bursts early in breast tumorigenesis (e.g., due to chromothripsis or chromosome missegregation), followed by the gradual accumulation of point mutations over time (e.g., due to defective DNA repair and replication) (Wang, et al. 2014). Thus, it stands to reason that CA is an early event in tumorigenesis and may subsequently be suppressed to inhibit further large-scale stochastic karyotype alterations that may compromise fitness. Indeed, CA is found in pre-invasive breast carcinomas (Lingle, et al. 2002c; Pihan, et al. 2003), and cells spontaneously lose supernumerary centrosomes over time in culture and become chromosomally more stable (Chiba, et al. 2000; Ganem et al. 2009; Oikawa, et al. 2005). However, other studies have found that CA increases from breast hyperplasia to tumor (Bergmann, et al. 2005; Li, et al. 2004), so perhaps, at least in some cases, CA is not eliminated as the tumor evolves but rather its potentially deleterious effects (e.g., severe CIN, slowed proliferation) are suppressed in some manner. It has been demonstrated that centrosomes can be excluded from the mitotic spindle (Kleylein-Sohn, et al. 2012), so they can potentially be borne as passengers without driving CIN. Thus, a specific tumor’s evolutionary trajectory may allow it to evolve to tolerate CA without sacrificing its malignant karyotype, whereas other tumors may not acquire such mechanisms (due to the largely stochastic nature of genome instability) and can only survive by suppressing CA to avoid prolonged, severe CIN.
CA also drives tumorigenesis by CIN-independent mechanisms. Overexpression of PLK4 in MCF10A mammary epithelial cells induces CA and the formation of invasive, matrix-degrading protrusions, unlike a truncated form of PLK4 that retains kinase activity but does not induce CA (Godinho, et al. 2014). Similarly, MCF10A cells exhibiting tetraploidy and CA due to treatment with dihydrocytochalasin B (which causes cytokinesis failure) display invasive protrusions, unlike tetraploid MFC10A cells that have spontaneously lost CA. Invasive features appear to be generated independent of aneuploidy because induction of a similar degree of aneuploidy by depletion of MCAK or MPS1 neither induces CA nor 3D invasion, whereas overexpression of PLK4 or tetraploidization with CA results in the formation of invasive acini. It appears as though increased microtubule nucleation upregulates Rac1 activation, disrupting intercellular adhesions. In addition, CA may disrupt cytoarchitecture, as breast tumors cells with CA lack discernible polarity, and the tissue tends to be highly anaplastic (Lingle and Salisbury 1999). Studies in Drosophila support that CA drives cytoarchitectural disturbances rather than merely being associated with them. Induction of CA by overexpression of SAK (the fly homolog of PLK4) in wing disc epithelial cells strikingly disrupts tissue organization when apoptosis is inhibited (Sabino, et al. 2015), as occurs in cancer cells, which are generally apoptosis reluctant. PLK4 overexpression in murine renal epithelial IMCD3 cells results in nucleation of multiple primary cilia, disruption of ciliary signaling, and perturbation of epithelial organization (e.g., lumen formation, apical and basolateral junction organization, and orientation of cilia) in spheroid cultures (Mahjoub and Stearns 2012). In consonance with this finding, CA correlates with loss of tissue differentiation in breast cancer (Kronenwett, et al. 2005). Supernumerary centrosomes also disrupt asymmetric spindle positioning in stem cells, which causes inappropriate expansion of the stem cell population in Drosophila (Basto, et al. 2008). Finally, CA may promote stem-like features because suppression of the kinase activity of cytoplasmic AURKA decreases the population of CD24low/CD44high in MDA-MB-231 TNBC cells (Zheng, et al. 2016); thus, kinase-mediated activities of cytoplasmic AURKA (which include regulation of centrosome number) may be involved in imparting stem-like phenotype in breast cancer cells. Induction of CA by overexpression of AURKA in MCF-7, T47D, and ZR-75-1 breast cancer cells increases expression of breast cancer stem cell marker ALDH1 (Liu, et al. 2015). Interestingly, the vast majority (96%) of mammosphere-derived AURKA-overexpressing MCF-7 breast cancer cells exhibit spindle bipolarity at least in part due to inactivity of supernumerary centrosomes. That is to say, for some fraction of the cells only two centrosomes were found to have nucleated microtubules that contributed to spindle formation, while the remaining centrosomes appeared to be non-functional passengers in mitosis. In SUM149PT inflammatory breast cancer cells, CA is only found in the stem-like CD24−/low/CD44high subpopulation (Opyrchal, et al. 2014). Altogether, these studies reveal that CA can spearhead an array of processes vital to breast cancer initiation and progression.
In Vivo Work: Seminal Studies and Consequential Controversies
A potential tumor-initiating role for CA in breast tissue was suggested by studies employing a rat model of mammary carcinogenesis (Goepfert, et al. 2002; Goepfert, et al. 2007). It was found that exposure to methylnitrosurea results in CA in not only premalignant and malignant mammary tissue but also in small foci in histologically normal mammary tissue soon after carcinogen exposure. The extents of CA and AURKA expression were found to correlate with the degree of neoplasia and carcinogenic status. A study of mouse mammary tumor virus (MMTV) transgenic mice inducibly expressing K-Ras(G12D) found that CA was evident in premalignant mammary lesions as well as mammary tumors, which was associated with upregulation of the centrosome-regulatory proteins cyclin D1 and Nek2. Thus, CA resulting from oncogenic K-Ras(G12D) signaling may be an early step in breast tumorigenesis, perhaps arising from centrosome cycle deregulation (Zeng, et al. 2010). Another study uncovered that transgene-induced expression of the transcription/translation factor Y-box binding protein-1 (YB-1) in mouse mammary gland epithelial cells results in CA, CIN, enhanced proliferation, and invasive mammary carcinoma formation, suggesting a causative role for CA in breast tumorigenesis (Bergmann et al. 2005). It was later discovered that YB-1 induces cytokinesis failure in human mammary epithelial cells, thereby promoting numerical CA, but also limits centrosome size, thereby restraining abnormal amplification of centrosome volume. It additionally induces aneuploidy, dicentric chromosomes and double minutes, extensive chromosome breakage, defective sister chromatid cohesion, and HER amplification (Davies, et al. 2011), although a subsequent study found that YB-1 induces triple-negative mammary tumor formation (Davies, et al. 2014). Although YB-1 induces numerical CA, it also promotes extensive chromatin remodeling and the development of stem-like properties (Davies et al. 2014), and it binds to the promoter of various genes that drive proliferation (e.g., the genes encoding EGFR, HER2, cyclin A, and cyclin B1) (Davies et al. 2011), so the relative contribution of CA to YB-1-induced breast tumorigenesis is hard to decipher from this study alone. Overexpression of AURKA also induces CA, enhanced proliferation, tetraploidy, genetic instability, mammary hyperplasia, and low-frequency tumor formation after a long latency in mice (Wang et al. 2006). Tumorigenesis is accelerated in the setting of AURKA overexpression by loss of heterozygosity of TP53. Despite the association between CA and mammary tumorigenesis in these studies, it is unclear to what extent CA drives tumorigenesis rather than polyploidy, which is itself a potential initiator and promoter of tumorigenesis (Erenpreisa, et al. 2015; Ogden, et al. 2015; Schoenfelder and Fox 2015). Furthermore, AURKA can induce CIN by increasing microtubule assembly rates, resulting in abnormal spindle geometry and persistent erroneous microtubule-kinetochore attachments, independent of CA (Ertych, et al. 2014). Thus, it is important for future studies to disentangle the relative contributions of CA, polyploidy, and enhancement of microtubule assembly rates to breast tumorigenesis. Given the paucity of research using in vivo breast models, we will next describe literature implementing other models and discuss the relevance of the findings for breast cancer and highlight critical areas of need for breast cancer research, in an effort to build the foundation for such studies.
A ground-breaking study more directly supporting a tumorigenic role for CA demonstrated that induction of CA in neural stem cells can initiate tumor formation in flies (Basto et al. 2008). Transgenic Drosophila larval brain cells engineered to overexpress SAK/PLK4 (master regulator of centriole duplication in addition to PCM assembly) exhibit supernumerary centrosomes and can form metastatic tumors when implanted into the abdomens of WT adult hosts, causing premature death (Basto et al. 2008). In this model, CA is associated with only a marginal increase in the percent of cells that are aneuploid, from 0.7 to 1.8%. Although CA could lead to multipolar mitoses and severe aneuploidy, this outcome is avoided because clustering proteins like Ncd/HSET cluster supernumerary centrosomes into two polar groups so that pseudo-bipolar division is possible. Surprisingly, lagging chromosomes are not evident in this model, although CA causes chromosome lagging and CIN in human cell lines and correlates with CIN and aneuploidy in patient tumors (Fukasawa 2005; Ganem et al. 2009; Lingle et al. 2002c). In addition to executing centrosome clustering, Drosophila neuroblasts also partially inactivate supernumerary centrosomes such that they do not appreciably nucleate microtubules and thus do not substantially contribute to spindle morphology (Basto et al. 2008). The mechanism by which centrosomes are inactivated remains poorly defined, but limiting quantities of pericentriolar material components may be the cause. Drosophila neuroblasts with CA exhibit striking defects in spindle alignment, with ~40% of cells failing to align the spindle along the apico-basal polarity axis as defined by the atypical Protein Kinase C crescent. Localization of cortical polarity cues is also disrupted, albeit to a smaller extent. Nonetheless, most of these cells eventually divide asymmetrically, which is necessary to appropriately partition the basally localized cell fate determinants to only one daughter cell, thereby preserving the stem cell pool while expanding the number of more-differentiated ganglion mother cells (Knoblich 2001). However, approximately 10% of larval neuroblasts with CA divide symmetrically, causing an inappropriate increase in the number of self-renewing cells (Basto et al. 2008). Unlike induction of centrosome defects, induction of genomic instability per se does not cause tumor formation in the Drosophila larval brain transplantation assay (Castellanos, et al. 2008). Combined with the very low levels of aneuploidy, it seems unlikely that CA drives tumorigenesis in this model by fostering CIN; rather, illicit stem cell self-renewal may be the culprit. This idea is congruent with the finding that Drosophila neuroblasts with mutations in genes responsible for asymmetric cell division (e.g., numb, raps) form large, invasive tumors in the allograft transplant assay (Caussinus and Gonzalez 2005).
Although it is established that CA can transform Drosophila stem cells in vivo, the same might not be true of mammalian stem cells. For instance, unlike Drosophila neuroblasts, murine neuroblasts with CA due to PLK4 overexpression do not exhibit spindle alignment defects or excessive self-renewal (Marthiens, et al. 2013). Another striking difference from the fly model is that murine neuroblasts with CA exhibit inefficient centrosome clustering, resulting in chromosome lagging and sometimes multipolar mitoses. The outcome of multipolar division is severe aneuploidy and p53-dependent apoptosis, so transgenic mice that overexpress PLK4 in the central nervous system suffer from microcephaly, not cancer. Thus, PLK4-overexpression-induced CA is not oncogenic in murine CNS. Interestingly, mutations in PLK4 that result in markedly decreased functional PLK4 levels and centriole depletion, rather than CA, are associated with microcephaly in humans (Martin, et al. 2014). PLK4-depleted cells with <4 centrioles (i.e., <2 centrosomes) proceed more slowly through mitosis due to the presence of aberrant mitotic spindles, which are thought to underlie impaired tissue growth. It is important to note that these PLK4-depleting mutations disproportionately affect brain size and retinal development, suggesting that the effect of PLK4 perturbation may vary quantitatively or qualitatively by tissue type. This seeming paradox – that both centrosome depletion and CA can induce microcephaly – may be explained by the fact that both of these phenotypes delay mitotic progression, the former in an attempt to resolve monopolar or disorganized spindles, the latter to cluster supernumerary centrosomes into a pseudo-bipolar spindle. However, centrosome clustering also engenders chromosome segregation errors due to the formation of merotelic microtubule-kinetochore attachments during the transient multipolar state that exits in prophase (Ganem et al. 2009). By contrast, cells with PLK4 mutations causing centrosome depletion rarely exhibit chromosome segregation errors during anaphase, multipolar mitoses, or aneuploidy (Martin et al. 2014). Based on these data, it seems that development of the mammalian brain and eye relies on a narrow range of PLK4 expression, with centrosome depletion and CA resulting in impaired growth due, at least in part, to mitotic defects. It is unclear whether PLK4-overexpression-driven CA results in similar phenotypes in mammary stem cells; however, tumor cells from xenografts of MCF-7 cells with constitutively active Raf-1 oncoprotein (MCF-7ΔRaf-1 cells) exhibit mesenchymal phenotype along with upregulated AURKA expression and robust phosphorylation only in CD24−/low (vs. CD24+) cells, which also exhibit CA, so AURKA may promote stem-like features through CA (D'Assoro, et al. 2014). More research is needed to clarify how PLK4-overexpression-induced CA impacts mammary stem cell division and the implications for breast tumorigenesis.
The literature is presently conflicting regarding the role of CA in inducing tumorigenesis outside the central nervous system. Using a Tamoxifen-dependent Cre-inducible PLK4-overexpression model, Vitre et al. demonstrated that PLK4 overexpression alone does not significantly shorten overall survival of transgenic mice, although there was a non-significant trend in that direction (Vitre, et al. 2015). CA was observed in liver and skin epidermis, but not lung, kidney, spleen, or pancreas, suggesting that tissues are selectively permissive for CA. Previously, it was found that PLK4 stabilization induces CA, p53 stabilization, impaired proliferation, and reduced clonogenic survival of hTERT RPE-1 cells (Holland, et al. 2010). The hTERT RPE-1 cells with CA took ~50% longer to progress through mitosis, conceivably due to efforts to organize a pseudo-bipolar spindle, which requires more time than assembling a bipolar spindle with 2 centrosomes (Kwon, et al. 2008). However, despite mitotic delay, the vast majority of the cells with CA nonetheless constructed multipolar spindles secondary to centrosome clustering failure. Different cell types vary considerably in the efficiency of their clustering mechanisms. For example, following CA due to cytokinesis failure, 44% of murine skin fibroblasts cluster centrosomes, whereas only 8% of monkey kidney epithelial cells do (Kwon et al. 2008). Human mammary epithelial cells (HMECs) fall approximately in between, with 24% of cells clustering supernumerary centrosomes. These data are consistent with the finding that PLK4 overexpression can induce CA in mouse epidermis but not kidney. Mammary tissue was not examined in that study, but because HMECs exhibit an intermediate ability to cluster centrosomes, it is conceivable that CA would be better tolerated in breast tissue than in kidney. In an effort to determine what mechanism suppresses CA or otherwise eliminates cells with CA in non-permissive tissues, Vitre et al. examined the effects of partial and total p53 inactivation on CA levels in the kidneys of PLK4-overexpressing and control mice (Vitre et al. 2015). Partial inactivation of p53 did not influence the percentage of cells with CA, measured 2 months after tamoxifen treatment, suggesting that even low levels of p53 can suppress CA. On the other hand, total inactivation of p53 significantly increased CA from ~4% to ~14%, although this was not accompanied by a difference in tumor-free survival. Surprisingly, the percentage of kidney cells with CA in controls (no PLK4 overexpression) was similar in wildtype p53 and p53f/f (floxed) backgrounds (~4% in both), suggesting that in the kidney p53 inactivation does not promote CA. This scenario contrasts strongly with breast epithelial cells, which develop CA following p53 knockout (Croessmann, et al. 2015), so the findings in kidney may not generalize to breast, which seems to possess different mechanisms controlling numerical centrosome homeostasis.
Why the effect of p53 inactivation differs so greatly between the kidney and the breast is not clear, but these striking, fundamental biological differences indicate that a mammary model of tumorigenesis must be tested before it can be concluded in a general way that PLK4-induced CA does not promote p53-dependent tumor formation. This caveat is thrown into stark relief by an analysis of the specific tissues that developed tumors in this study, which was only reported for mice with partial p53 inactivation (as total inactivation results in the development of fatal thymic lymphomas with complete penetrance by 235 d regardless of PLK4 overexpression) (Vitre et al. 2015). Only the PLK4-overexpressing group developed mammary tumors, a remarkable finding because a tamoxifen-inducible system was used, which should suppress the development of estrogen-responsive breast tumors. This finding raises the tantalizing possibility that CA promotes hormone-refractory breast tumor development in the setting of compromised p53, an exciting avenue for future investigations. Thus, although PLK4-mediated induction of CA did not result in significantly worse tumor free survival across tumor types, it might promote mammary tumorigenesis. This seems likely in light of the recent finding that knockdown of PLK4 via shRNA or pharmacologic inhibition (via CFI-400945, a novel PLK4 inhibitor) in xenografted MDA-MB-468 triple-negative breast cancer cells results in smaller tumor volumes (Mason, et al. 2014).
In another study, the same group found that mice overexpressing PLK4 in the basal layer of the epidermis exhibited a 20% increase in basal and suprabasal epidermal cells with CA; an increase in multiciliated basal cells; an increase in spindle alignment defects in basal cells due primarily to uncoupling of the spindle from cortical cues; an increase in spindle multipolarity, aneuploidy, and DNA damage in embryonic epidermal tissue; and defective stratification and growth of embryonic epidermal tissue, which was minimally rescued by p53 knockdown (Kulukian, et al. 2015). Despite these defects, PLK4-overexpressing mice had normal epidermal barrier function and architecture and similar overall and tumor-free survival compared with control mice. By contrast, Serçin et al. found that overexpression of PLK4 in developing embryonic epidermis lead to delayed skin stratification and barrier defects due to apoptosis, and p53 deletion decreased the extent of basal progenitor cell apoptosis and rescued defects in skin stratification and barrier function (Sercin, et al. 2016). In fact, PLK4 overexpression only caused aneuploidy in p53-deficient basal cells, not WT-p53 basal cells. Moreover, PLK4 overexpression with concomitant p53 knockout induced spontaneous skin tumor formation in adult mice with complete penetrance and accelerated tumorigenesis relative to p53 knockout alone. Thus, it seems that p53 compromise is needed for cell survival in the setting of CA, suggesting a twofold role of p53 compromise in CA-driven tumorigenesis – not only as an inducer of CA (which we discuss more later) but also as a potentiator of its effects by permitting the survival of cells sustaining aneuploidy, which may arise from merotelic attachments or low-grade multipolar divisions that otherwise would be incompatible with cellular survival. Altogether, the relationships between CA, p53, and tumorigenesis are multi-layered, may differ by tissue type, and need to be resolved with further meticulous experimentation in variety of model systems.
A recent report by Levine et al. reveals that CA induced by PLK4 overexpression in the absence of other genetic perturbations (such as TP53 mutation), is capable of initiating spontaneous tumor formation in a mouse model, at least when PLK4 overexpression and CA are relatively mild (Levine, et al. 2017). A key difference between this report and others that did not find CA induces spontaneous tumorigenesis is that the study by Levine et al. induced more modest increases in PLK4 levels by knocking in a single-copy PLK4 transgene into the Col1a1 locus, resulting in more modest increases in CA (only one or two additional centrosomes per cell in a majority of cells within 3 days of induction with doxycycline), similar to what is observed in actual human tumors. All cells with CA exhibited clustering and 10% of anaphases exhibited lagging chromosomes 5 days post induction in murine embryonic fibroblasts from PLK4-transgenic mice. Although mammary gland was not assessed, limiting what conclusions can be drawn from this study for breast tumorigenesis, many tissues exhibited CA with the exception of lung and kidney. PLK4-transgenic mice spontaneously developed lymphomas, squamous cell carcinomas, and sarcomas, which also exhibited karyotypic diversity and aneuploidy, suggesting that CA and its consequence, CIN, were active in their evolution. Thymic tumors exhibited variable but generally decreased levels of p53-target gene expression, suggesting CA itself compromises the p53 pathway. Ultimately, this study suggests that “low-grade” CA (i.e., where cells only exhibit 1 or 2 additional centrosomes) promotes CIN and tumorigenesis in susceptible tissues. Whether mammary gland constitutes one such susceptible tissue remains a question for future studies.
Fan et al. discovered that Kruppel-like factor 14 (KLF14)-mediated suppression of PLK4 expression did not result in spontaneous breast tumor formation; however, it promoted tumorigenesis in an AOM/DSS model of colon cancer (Fan, et al. 2015). An increased number of tumors, increased tumor burden, and greater proportion of high-grade tumors were observed in KLF14-knockout mice. This finding is clinically relevant to breast cancer because KLF14 protein levels are reduced in breast cancer relative to their respective normal tissues, and KLF14 and PLK4 protein levels are inversely correlated. Furthermore, KLF14-knockout mice spontaneous developed tumors with ageing, mostly lymphomas but also lung adenomas. Lymph nodes, spleen, and lung all exhibited a ~20% increase in CA. MEFs from the KLF14-knockout (KO) mice exhibited CIN and aneuploidy and a ~20% increase in the percentage of cells with CA. To determine whether cytokinesis failure was responsible for CA, HeLa cells were arrested in S-phase with hydroxyurea, and MEFs from KLF14-KO mice still exhibited increased CA, suggesting mechanisms other than cytokinesis failure also underlie KLF14-KO-induced CA. KLF4 was found to repress transcription of PLK4 and to inhibit PLK4-directed centriole replication in HeLa cells. KLF14 knockdown induced CA, spindle multipolarity, chromosome missegregation, and aneuploidy, while KLF14 overexpression resulted in centrosome depletion, spindle monopolarity, mitotic arrest, DNA damage, and apoptotic and necrotic cell death that was only partially rescued (~50%) by PLK4 overexpression in HeLa cells. Thus, KLF14 is a tumor suppressor whose loss can drive CA in the breast and which contributes to multistep tumorigenesis in the colon, although whether this is also true in the breast is unknown.
In the future, it will be important to carefully titrate PLK4 levels in cells and comprehensively characterize centrosomal profiles (e.g., centrosome volume, number, maturity, microtubule-nucleating capacity, clustering), as it was recently shown that the manner of CA induced by PLK4 overexpression changes with increasing PLK4 levels (Lopes, et al. 2015). Specifically, mild increases in PLK4 overexpression induce templated centriole overduplication, whereas large increases promote de novo formation in the cytoplasm, although whether the mechanisms by which centrosomes are duplicated impact cell physiology are poorly defined. That is to say, it is not clear whether centrosomes arising de novo are functionally equivalent to those that arise in a templated fashion in human cells. This is also important in light of the findings of Levine et al. that low-grade PLK4 overexpression (i.e., single-copy transgene knock-in) and CA (1–2 extra centrosomes per cell) can spur spontaneous tumorigenesis, whereas studies that may have induced higher-grade PLK4 overexpression and CA found that it did not (Levine, et al.). In addition, PLK4 was recently found to promote tumorigenesis through CA-independent mechanisms, namely the phosphorylation of Arp2, which promotes cancer cell spreading, motility, invasion, and metastasis (Kazazian, et al. 2016; Rosario, et al. 2015), thus raising the question of whether it is CA per se or rather stimulation of actin polymerization that induces spontaneous tumorigenesis in susceptible tissues. This is an important question to resolve with future studies. In summary, the totality of the evidence at present suggests that CA may induce spontaneous tumorigenesis in specific tissues/cell types (e.g., lymphocytes), and in others it may play a role in multi-step carcinogenesis (e.g., following the “hit” of p53 mutation/loss or carcinogen exposure) (Figure 3). However, the transforming capacity of CA has not been directly tested in a breast cancer model that disentangles the effects of the other potential perturbations caused by genetic manipulations (e.g., polyploidy, enhanced actin polymerization), which represents fertile grounds for future research.
Figure 3.
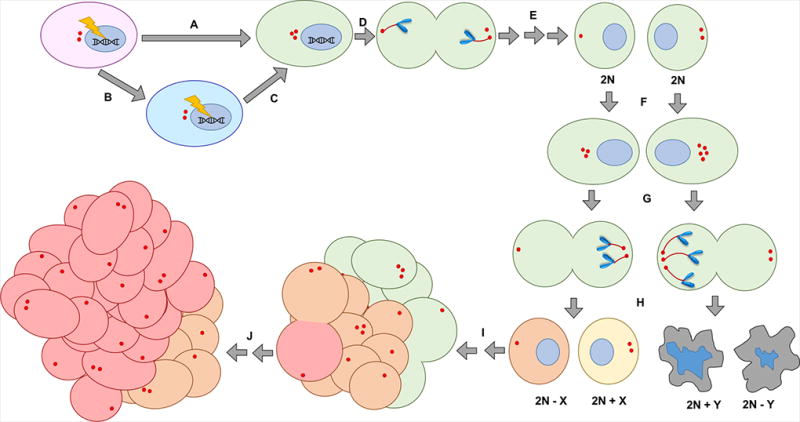
How centrosome amplification (CA) may fuel tumor evolution and generate intratumor heterogeneity in breast cancer. (A) A carcinogenic insult induces CA and also the ability to tolerate its deleterious effects (e.g., TP53 mutation), or (B) a carcinogenic insult induces the ability to tolerate the deleterious effects of CA (e.g., compromised checkpoints, enhanced proliferative potential) followed by (C) a separate insult that induces CA. (D) The cell clusters amplified centrosomes and the cell divides in a pseudobipolar fashion for many divisions. (E) CA does not induce aneuploidy yet (so progeny remain diploid or near-diploid, depending on other phenotypes that may have been acquired during tumor evolution that impact ploidy). (F) Whatever drove CA in the first place persists in the cell, and centrosomes undergo further amplification. (G) In a subsequent mitosis, centrosome clustering eventually results in chromosome missegregation. (H) Some cells may achieve a karyotype that is compatible with survival (orange and yellow cells), whereas others may not (gray apoptotic cells) especially if aneuploidy is too severe. (I) The karyotype of some cells may confer enhanced tumorigenic potential, which results in clonal expansion and population of the tumor. (J) Eventually, subclones may acquire superior karyotypes and also expand (red cells), resulting in a potentially heterogenous tumor population.
Centrosome Amplification and p53 Dysfunction: Dangerous Liaisons
Although p53 mutation is not a prerequisite for CA (D'Assoro, et al. 2002; Pihan et al. 2003), loss of p53 can induce CA depending on the cell type and co-existing cellular stresses (D'Assoro, et al. 2008; D'Assoro, et al. 2004; Fukasawa, et al. 1996). In vitro data clearly support that compromise of p53 promotes CA in breast epithelial cells and breast cancer cells and confers aggressive phenotypes. Knockout of p53 in noncancerous MCF10A breast epithelial cells results in CIN and 2–3-fold increased CA (Croessmann et al. 2015). In hormone-receptor MCF-7 breast cancer cells, induced expression of dominant-negative p53 slightly increases the percentage of cells with CA, but following stimulation with mitogens (E2, EGF, and IG-1) after a period of mitogen withdrawal, a marked increase in CA occurs (D'Assoro et al. 2008). Xenografts of p53-dominant-negative MCF-7 cells give rise to larger, higher-grade tumors that exhibit CA, tamoxifen resistance, triple-negative phenotype, and increased metastatic ability compared with xenografts of control cells with WT p53. Genotoxic stress exacerbates CA in the presence of p53 loss or mutation, as treatment with hydroxyurea or danorubicin significantly increases CA (both the percentage of cells with CA and the size of centrosomes) in both MCF-7 cells with dominant-negative p53 and MDA-MB-231 cells, which are homozygous for a gain-of-function p53 mutant (R280K, which is from exon 8, which has lost the ability to transactivate (the genes for p21Waf1, MDM2, Bax, 14-3-3σ, AIP1, GADD45, Noxa, and p53R2 (Kato, et al. 2003)) and are triple-negative (D'Assoro et al. 2004). TNBCs, which exhibit a high prevalence of TP53 mutations (~64% of TNBC cases vs. ~30% of hormone receptor-positive cases), have ~2 times as many cells with CA as grade-matched non-TNBCs (Pannu et al. 2015b). Thus, CA is a characteristic of this aggressive breast cancer subtype, which is also more common among African American (AA) women than white women (~2 times the odds after adjusting for age, stage, grade, and poverty index) (Lund, et al. 2009). Collectively, these data suggest that in breast cancer, p53 loss or gain-of-function mutation, alone or combined with mitogen stimulation or genotoxic stress, leads to CA and more aggressive phenotypes. Normally p53 maintains centrosome homeostasis by tightly coupling the centrosome duplication and DNA replication cycles. The p53 protein prevents premature and excessive centrosome duplication by inducing expression of p21waf1(Tarapore, et al. 2001), which inhibits cdk2/cyclin E/A (halting the cell cycle at G1/S) and repressing expression of the Anaphase-Promoting Complex/Cyclosome inhibitor Emi1 such that cyclins A and B are degraded (halting the cell cycle at G2/M) (Romanov, et al. 2012). While induced overexpression of p53 restores normal centrosome numbers in p53−/− murine embryonic fibroblasts (MEFs), induced overexpression of p21waf1 results in only partial restoration, suggesting that p53 controls numerical centrosome homeostasis through additional downstream effectors (Tarapore et al. 2001).
Centrosome Amplification and p53 Dysfunction: Racial Disparities
Studies of the centrosome and health disparity in breast cancer have yet to intersect, but it is our perspective that the paths of these two bustling, rapidly progressing fields of research are about to meet. We next present intuitions about potential strategies to reduce racial health disparity in breast cancer through novel risk-stratification schemes and treatment regimens based on CA. Specifically, we hypothesize that CA is more extensive among AA breast tumors than white breast tumors, a likely organellar distinction on which oncologists could capitalize to narrow the troubling gap in clinical outcomes between these two patient populations. To better understand the biologic foundations of racial health disparity, many studies have analyzed molecular features that distinguish AA from white breast tumors, such as genetic variations and gene signatures. However, few reports are available on organelle-level differences. Organelle morphology and function constitute emergent properties that cannot be captured solely through next generation sequencing or microarrays, so disparities that may exist at this tier have largely gone unexamined. However, we present persuasive “circumstantial” evidence suggesting that CA is more prevalent among breast tumors from AA women, which may contribute to their more aggressive disease course.
Unfortunately, racial disparities in breast cancer outcomes appears to be worsening despite the multifarious advancements in breast cancer medicine in recent years. According to SEER data from 2012, the age- and delay-adjusted incidences of invasive breast cancer among white and AA women were 135.0 and 135.2 (per 100,000), respectively, and the age-adjusted mortality rates were 20.7% and 29.4%, respectively (Howlader, et al. 2015). From 1975–2011, AA women never had a greater age- and delay-adjusted incidence of invasive breast cancer than white women, so the year 2012 represents a regrettable inflection point for the health of AA women. Perhaps more disturbingly, between 1975–2012, the age- and delay-adjusted increase in the incidence of breast cancer was greater among AA women than EA women (44.5% vs. 25.8%, respectively) and the decrease in age-adjusted mortality was less among AA women than white women (0.002% and 34.8%, respectively). These data suggest that improvements in breast cancer screening, diagnosis, and treatment disproportionately benefit white women, a trend that has been attributed to the “perfect storm” of aggressive tumor biology among AA women colliding with healthcare inequality (Daly and Olopade 2015). Rates of hormone receptor-positive HER2-negative breast cancer, the least aggressive subtype, are highest among white women, whereas rates of TNBC, advanced-stage disease, and poorly/undifferentiated grade, which are all associated with worse survival, are highest among AA women (Kohler, et al. 2015). Differences in access to healthcare must also be addressed, but so too can clinical strategies to manage inherently more aggressive AA tumor biology, which are presently inadequate. Prognostic tools and treatments tailored to AA women are urgently needed to attenuate racial breast cancer disparity. We believe that assessment of centrosomal profiles and targeting cells with CA represent promising prognostic and therapeutic strategies with exceptional potential for AA women with breast cancer.
Differences in TP53 mutations and levels between AAs and EAs may result in differences in centrosome profiles, which might contribute to racial health disparity in breast cancer. Immunohistochemical analysis of AA and white breast tumors revealed that AA race was associated with ~4 times the odds of low p53 expression as white race even after adjusting for potential confounders (age at diagnosis, tumor grade and stage, biomarker statuses [ER, PR, Her2, and Met], socioeconomic status, severe obesity, and smoking) (Porter, et al. 2004). Consonant with this finding, a study of Brazilian breast cancer patients found that African-Brazilians (n=52) had a significantly higher frequency of TP53 mutation than white Brazilians (n=242) with similar clinicopathologic characteristics (TP53 mutation frequencies of 33% vs. 14%, respectively). African-Brazilians were more likely to have exon 7 mutations, whereas white Brazilians were more likely to have exon 8 mutations, and exon 4 mutations were found exclusively within whites. (Nagai, et al. 2003) However, other studies have not found differences in mutation frequency based on race. For instance, a study of AA women from Michigan (n=47) revealed a similar frequency of mutation compared to EA women from the Midwest (sample size not reported) (Blaszyk, et al. 1994). Similarly, no difference was detected in TP53 exon5-exon8 and intron 5 mutation rates between AA and white breast tumors (n=45 and 47, respectively), but this study did find that these mutations only conferred increased risk of breast cancer mortality in AA patients even after adjusting for potentially confounding clinicopathologic factors (Shiao, et al. 1995). The locations and types of mutations present differed between the races – for instance, only AAs had nonsense mutations, and they did not have any exon 6 mutations – which may explain why TP53 mutation was only prognostic within AAs. The prognostic significance of TP53 mutations depends critically on the type of mutation and its location, so future studies should consider determining the rates of such mutations by race and their prognostic power within groups. A majority of TP53 mutations within exons 5–8 (DNA-binding domain) are missense, but the observed and expected frequencies are similar, whereas there is evidence of preferential selection of nonsense mutations, which are twice as frequent as expected (Petitjean, et al. 2007). Thus, nonsense mutations in the DNA-binding domain may confer worse prognosis than missense mutations here. However, which individual mutations confer worse prognosis in breast cancer are not clear because studies have been underpowered and/or have not accounted for potential confounders.
Exon 4, which contributes to the proline-rich domain and DNA-binding domains of the p53 protein, has been the subject of much study in racial disparity in cancer outcomes. Polymorphism in TP53 along geographic and racial/ethnic lines has been observed, with profound differences in codon 72 (exon 4, CGC→CCC, rs1042522). In Nigeria the most common allele is Pro72 (63% frequency), whereas it is infrequent among for Swedish Saamis (17% frequency) (Birgander, et al. 1996). In certain cancers the Pro72 allele is associated with increased cancer risk and ethnic variation. For instance, in non-small cell lung cancer, the Pro72 allele (which is associated with increased lung cancer risk compared with the Arg72 allele) (Wu, et al. 2002), is significantly more prevalent among AAs (62% frequency) than EAs (30% frequency) (Hu, et al. 2005). Studies of codon 72 polymorphism in breast cancer have yielded conflicting results; however, subgroup analysis of African and white patients, with data for each cohort pooled from case-control studies, did not find codon 72 mutations to be associated with increased risk in either subgroup compared with age-matched controls (Ma, et al. 2011). That being said, further study is needed because the number of African cases was relatively small (3 studies having n=2, n=16 [ref], and n=30 (Mabrouk, et al. 2003) African cases, and a fourth with an unclear number) compared with thousands of white cases. A number of problems have been reported about this meta-analysis, though, calling into question the validity of this study. Inconsistencies between the number of polymorphisms reported in the primary studies and the meta-analysis have been noted, along with cases from the same dataset being included more than once because multiple publications were based on the same dataset (He, et al. 2011). Furthermore, several relevant studies that met the search criteria were excluded, so the conclusions of this study must be met with caution.
A landmark study recently characterized differences between the genomic landscapes of AA and white breast tumors and their relevance to clinical outcomes (Keenan, et al. 2015). TP53 mutations were more common in AA breast tumors than white breast tumors (43% vs. 28%), whereas PIK3CA mutations were less common (20% vs. 34%) (Keenan et al. 2015) AAs had approximately twice the odds of a TP53 mutation and half the odds of a PIK3CA mutation after adjusting for age and stage. Intriguingly, AAs also had approximately twice the risk of recurrence as whites, which mirrored the odds of TP53 mutation. Moreover, statistical significance was lost after adjusting for TP53 mutation, suggesting that racial disparity in breast cancer recurrence arises at least in large part from these mutations. TP53 is the most commonly mutated gene in breast cancer overall and within the HER2 and basal subtypes (37%, 72%, and 80% of cases with predicted non-silent somatic mutations, respectively), although prevalence is equal to PI3KCA mutations within the luminal B subtype (29%), and less than PI3KCA, GATA3, and MAP3K1 mutations in the luminal A subtype (45%, 14%, and 13%, respectively, versus 12% of cases with TP53 mutations) (2012). It is interesting that TP53, but not PIK3CA mutations, influence recurrence, and it is tempting to speculate this is at least partly because p53 can independently impact centrosome homeostasis, unlike PI3K, which can only induce CA in a p53-null background in some contexts, as described next.
Although the role of the PI3K-Akt pathway in CA has not been studied in breast cancer, it has been explored in cervical and colon cancer models. Constitutive activation of the tyrosine kinase receptor Met induces CA through PI3K-Akt in HeLa cells (Nam, et al. 2010). HeLa cells with constitutive Met activation (CA-MET cells) were generated by transfection with a MET construct with a M1268T mutation. Importantly, HeLa cells have low p53 levels despite having WT TP53 because E6 recruits the ubiquitin ligase E6-associated protein (E6AP) to a complex with p53, thereby targeting p53 for proteasomal degradation (Moody and Laimins 2010), so HeLa cells model a functionally p53-null (or p53-low) background. CA-Met HeLa cells exhibited 6–9% CA, whereas control HeLa cells exhibited 2% CA, which was a significant difference (Nam et al. 2010). Pharmacologic inhibition of PI3K in synchronized CA-Met HeLa cells restored CA to control levels, as did siRNA of Akt or expression of dominant-negative Akt, suggesting that Met activation acts through the PI3K-Akt pathway to induce CA in a p53-null background. To clarify the role of p53 in Met-induced CA, centrosome numbers were assessed in synchronous WT and p53−/− HCT116 colon cancer cells following transfection with the CA-Met construct or vector. Constitutive Met activation did not induce CA in p53 WT cells compared with vector controls. Furthermore, whereas p53−/− cells had more than double the percent CA than WT cells (~24% vs. ~11%, respectively), transfection of CA-Met did not significantly increase CA in p53−/− cells, suggesting that p53 loss induces CA through effectors other than Met. Collectively, these results suggest that Met activation cannot induce CA in a p53-WT background, although in a p53-null background it may do so through PI3K-Akt depending on the cellular context (e.g., cervical vs. colon cancer). This may be why TP53 mutations, but seemingly not PIK3CA mutations, confer increased risk of recurrence in breast cancer. Regardless, Met levels have not been shown to differ significantly along racial lines in breast tumors,(Jones, et al. 2004) suggesting that even if a Met/PI3K/Akt axis can induce CA in p53-null breast tumors, this is not a component of racial breast cancer disparity. Because TP53 mutations are more common among AAs, and PIK3CA mutations among EAs, CA may be more prevalent among AAs and may represent an integral component of health disparity in recurrence.
Triple-Negative Breast Cancer: A Mutant p53-Associated Subtype with Racial Bias
Intriguingly, the AA/white survival gap did not manifest until the mid-1980’s, when use of the estrogen receptor antagonist Tamoxifen became widespread (Newman 2014). White women are more likely than AA women tend to have hormone receptor-positive breast cancers, affording them disproportionate opportunity to take advantage of this revolutionary and generally life-saving chemotherapeutic drug. AA women are ~2–3 times more likely than white women to develop TNBC (Dietze, et al. 2015b), which constitutes ~20–46% of AA cases and for which their lifetime risk is ~2% (Dietze et al. 2015b; Kurian, et al. 2010). This breast cancer subtype is characterized by mutations in TP53, which are present in more than half of all TNBCs (Turner, et al. 2013). No targeted therapy is FDA-approved to treat this aggressive breast cancer subtype. Instead, cytotoxic chemotherapy is the standard of care, although it achieves a pathologic complete response in only ~20% of patients (Liedtke, et al. 2008). This aggressive breast cancer subtype is usually diagnosed at a more advanced grade and stage, is more likely to distantly metastasize, and carries a higher five-year risk of mortality relative to non-TNBCs (Dent, et al. 2007). Although the molecular mechanisms underpinning the greater risk of developing TNBC among AA women, sub-Saharan African women have an even greater risk of developing TNBC than AA women, strongly implicating ancestry (Newman 2014).
The involvement of CA in promoting chromosome instability, invasive behavior, distant metastases, and cytoarchitecture disruption constitutes compelling grounds to suspect its involvement in the etiology of the aggressive behavior of TNBCs, which are characterized by all these features (Dietze, et al. 2015a). TNBCs exhibit remarkably extensive CA: almost two-thirds of cells from patient tumors possess amplified centrosomes, which is about twice the extent found in grade-matched non-TNBCs (Pannu, et al. 2015a). Several corollaries and open questions emerge when considering this line of inquiry. First and foremost, the associations of CA with TNBC and TNBC with AA race suggest that, transitively, CA may be associated with AA race. Hence, differences in centrosome homeostasis may account for some proportion of racial disparity in breast cancer, a novel and potentially impactful idea that merits investigation. A study of breast tumor gene expression data from The Cancer Genome Atlas provides strong circumstantial evidence that CA may differ based on race. Specifically, it was discovered that PLK1 and Aurora C signaling pathways are upregulated in age- and stage-matched breast tumors from AAs compared with EAs (Stewart, et al. 2013). Since PLK1 and Aurora C drive CA (Izumi, et al. 2009; Khan, et al. 2011; Zou, et al. 2014), AA breast tumors may exhibit increased CA relative to white breast tumors. Whether AA breast tumors are enriched in cells with CA is a question of immense clinical importance because centrosome clustering inhibitors are available, some of which are FDA-approved drugs (e.g., griseofulvin, an antifungal). If AA tumors do exhibit disproportionate CA relative to white breast tumors, then therapeutically targeting this organelle-level difference could help to attenuate the ethnicity-based survival gap. Undoubtedly, a range of other molecular and cellular aberrations are involved in racial health disparity; therefore, targeting cells with supernumerary centrosomes alone is unlikely to entirely close the gap. That being said, targeting centrosome clustering mechanisms is nonetheless a highly appealing therapeutic strategy because these mechanisms generally are expendable to normal cells, as they lack CA (Ogden et al. 2012). Another vital and interesting question is whether CA compels breast tumors to evolve into TNBCs, or conversely whether the TNBC phenotype engenders CA. Finally, TNBC is a highly heterogeneous disease that comprises multiple molecular subtypes (Burstein, et al. 2015; Lehmann, et al. 2016). It is unknown whether differences in CA exist between subtypes and, consequently, whether centrosome clustering inhibitors could prove superiorly efficacious in certain TNBC subtypes. AA TNBCs tend to belong to the basal-like 1 subtype, unlike white TNBCs, which tend to belong to the luminal androgen receptor subtype (Lindner, et al. 2013). Thus, it is reasonable to conjecture that the basal-like 1 subtype evinces greater CA than the luminal androgen receptor subtypes, an interesting avenue for future research.
Conclusions
In summary, CA appears to be a correlate of aggressive disease features in breast cancer, but in vivo models are needed to discern whether CA actively drives multi-step mammary tumorigenesis. Given its association with aggressive disease features, CA may be a useful prognostic biomarker in breast cancer, but whether it can independently predict patient outcomes is also still not firmly established and requires further study. Assessing CA could potentially predict responsiveness to centrosome declustering drugs, such as noscapinoids, griseofulvin, and PJ-34 (Ogden et al. 2012), to which TNBC cells are sensitive (Ogden, et al. 2014), an exciting research question. Finally, we are confident that the quest to ameliorate racial disparity in breast cancer outcomes can benefit from a closer examination of racial differences in centrosome biology. Assessment of intratumoral centrosome defects, such as through immunohistochemistry-based assays, may prove valuable in risk-stratifying AA breast cancer patients (especially with TNBC) and guiding their therapy. Conceivably, other organelle-level variations may exist, however, and may represent a fresh and fertile frontier in health disparities research. Tumor biology is the eventual manifestation of a complex playout of both the unique genetic and organellar complements of the constituent tumor cells. Personalization of cancer treatment means to embrace and honor all aspects of this uniqueness, and the tools to do this may very well hold the key to markedly improving breast cancer outcomes.
Acknowledgments
Funding: This work was supported by grants to Ritu Aneja from the National Cancer Institute (U01 CA17967 and R01 CA169127).
Footnotes
Declaration of interests: The authors declare no financial or other competing interests.
Author contributions: AO wrote the manuscript; RA and PCGR critically revised the manuscript; RA supervised the project.
References
- Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderhub SJ, Kramer A, Maier B. Centrosome amplification in tumorigenesis. Cancer Lett. 2012;322:8–17. doi: 10.1016/j.canlet.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Antonczak AK, Mullee LI, Wang Y, Comartin D, Inoue T, Pelletier L, Morrison CG. Opposing effects of pericentrin and microcephalin on the pericentriolar material regulate CHK1 activation in the DNA damage response. Oncogene. 2015 doi: 10.1038/onc.2015.257. [DOI] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. Centrosome Amplification Can Initiate Tumorigenesis in Flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann S, Royer-Pokora B, Fietze E, Jürchott K, Hildebrandt B, Trost D, Leenders F, Claude J-C, Theuring F, Bargou R, et al. YB-1 Provokes Breast Cancer through the Induction of Chromosomal Instability That Emerges from Mitotic Failure and Centrosome Amplification. Cancer Research. 2005;65:4078–4087. doi: 10.1158/0008-5472.CAN-04-4056. [DOI] [PubMed] [Google Scholar]
- Birgander R, Sjalander A, Saha N, Spitsyn V, Beckman L, Beckman G. The codon 31 polymorphism of the p53-inducible gene p21 shows distinct differences between major ethnic groups. Hum Hered. 1996;46:148–154. doi: 10.1159/000154344. [DOI] [PubMed] [Google Scholar]
- Blaszyk H, Vaughn CB, Hartmann A, McGovern RM, Schroeder JJ, Cunningham J, Schaid D, Sommer SS, Kovach JS. Novel pattern of p53 gene mutations in an American black cohort with high mortality from breast cancer. Lancet. 1994;343:1195–1197. doi: 10.1016/s0140-6736(94)92403-1. [DOI] [PubMed] [Google Scholar]
- Brownlee CW, Rogers GC. Show me your license, please: deregulation of centriole duplication mechanisms that promote amplification. Cell Mol Life Sci. 2013;70:1021–1034. doi: 10.1007/s00018-012-1102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21:1688–1698. doi: 10.1158/1078-0432.CCR-14-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos E, Dominguez P, Gonzalez C. Centrosome Dysfunction in <em>Drosophila</em> Neural Stem Cells Causes Tumors that Are Not Due to Genome Instability. Current Biology. 18:1209–1214. doi: 10.1016/j.cub.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Castellanos E, Dominguez P, Gonzalez C. Centrosome dysfunction in Drosophila neural stem cells causes tumors that are not due to genome instability. Curr Biol. 2008;18:1209–1214. doi: 10.1016/j.cub.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Caussinus E, Gonzalez C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat Genet. 2005;37:1125–1129. doi: 10.1038/ng1632. [DOI] [PubMed] [Google Scholar]
- Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122–1144. doi: 10.7150/ijbs.7.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Okuda M, Mussman JG, Fukasawa K. Genomic convergence and suppression of centrosome hyperamplification in primary p53−/− cells in prolonged culture. Exp Cell Res. 2000;258:310–321. doi: 10.1006/excr.2000.4916. [DOI] [PubMed] [Google Scholar]
- Conduit PT, Brunk K, Dobbelaere J, Dix CI, Lucas EP, Raff JW. Centrioles regulate centrosome size by controlling the rate of Cnn incorporation into the PCM. Curr Biol. 2010;20:2178–2186. doi: 10.1016/j.cub.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16:611–624. doi: 10.1038/nrm4062. [DOI] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croessmann S, Wong HY, Zabransky DJ, Chu D, Mendonca J, Sharma A, Mohseni M, Rosen DM, Scharpf RB, Cidado J, et al. NDRG1 links p53 with proliferation-mediated centrosome homeostasis and genome stability. Proceedings of the National Academy of Sciences. 2015;112:11583–11588. doi: 10.1073/pnas.1503683112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Assoro AB, Barrett SL, Folk C, Negron VC, Boeneman K, Busby R, Whitehead C, Stivala F, Lingle WL, Salisbury JL. Amplified centrosomes in breast cancer: a potential indicator of tumor aggressiveness. Breast Cancer Res Treat. 2002;75:25–34. doi: 10.1023/a:1016550619925. [DOI] [PubMed] [Google Scholar]
- D'Assoro AB, Busby R, Acu ID, Quatraro C, Reinholz MM, Farrugia DJ, Schroeder MA, Allen C, Stivala F, Galanis E, et al. Impaired p53 function leads to centrosome amplification, acquired ERalpha phenotypic heterogeneity and distant metastases in breast cancer MCF-7 xenografts. Oncogene. 2008;27:3901–3911. doi: 10.1038/onc.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Assoro AB, Busby R, Suino K, Delva E, Almodovar-Mercado GJ, Johnson H, Folk C, Farrugia DJ, Vasile V, Stivala F, et al. Genotoxic stress leads to centrosome amplification in breast cancer cell lines that have an inactive G1/S cell cycle checkpoint. Oncogene. 2004;23:4068–4075. doi: 10.1038/sj.onc.1207568. [DOI] [PubMed] [Google Scholar]
- D'Assoro AB, liu T, Quatraro C, Amato A, Opyrchal M, Leontovich A, Ikeda Y, Ohmine S, Lingle W, Suman V, et al. The mitotic kinase Aurora-A promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERα(+) breast cancer cells. Oncogene. 2014;33:599–610. doi: 10.1038/onc.2012.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly B, Olopade OI. A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J Clin. 2015;65:221–238. doi: 10.3322/caac.21271. [DOI] [PubMed] [Google Scholar]
- Davies AH, Barrett I, Pambid MR, Hu K, Stratford AL, Freeman S, Berquin IM, Pelech S, Hieter P, Maxwell C, et al. YB-1 evokes susceptibility to cancer through cytokinesis failure, mitotic dysfunction, and HER2 amplification. Oncogene. 2011;30:3649–3660. doi: 10.1038/onc.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AH, Reipas KM, Pambid MR, Berns R, Stratford AL, Fotovati A, Firmino N, Astanehe A, Hu K, Maxwell C, et al. YB-1 transforms human mammary epithelial cells through chromatin remodeling leading to the development of basal-like breast cancer. Stem Cells. 2014;32:1437–1450. doi: 10.1002/stem.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt Y, Lampkin T. Targeting Polo-like Kinase in Cancer Therapy. Clinical Cancer Research. 2010;16:384–389. doi: 10.1158/1078-0432.CCR-09-1380. [DOI] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- Denu RA, Zasadil LM, Kanugh C, Laffin J, Weaver BA, Burkard ME. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer. 2016;16:47. doi: 10.1186/s12885-016-2083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietze EC, Sistrunk C, Miranda-Carboni G, O'Regan R, Seewaldt VL. Triple-negative breast cancer in African-American women: disparities versus biology. Nat Rev Cancer. 2015a doi: 10.1038/nrc3896. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietze EC, Sistrunk C, Miranda-Carboni G, O'Regan R, Seewaldt VL. Triple-negative breast cancer in African-American women: disparities versus biology. Nat Rev Cancer. 2015b doi: 10.1038/nrc3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erenpreisa J, Salmina K, Huna A, Jackson TR, Vazquez-Martin A, Cragg MS. The "virgin birth", polyploidy, and the origin of cancer. Oncoscience. 2015;2:3–14. doi: 10.18632/oncoscience.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertych N, Stolz A, Stenzinger A, Weichert W, Kaulfuß S, Burfeind P, Aigner A, Wordeman L, Bastians H. Increased microtubule assembly rates mediate chromosomal instability in colorectal cancer cells. Nat Cell Biol. 2014;16:779–791. doi: 10.1038/ncb2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Sun L, Shan P, Zhang X, Huan J, Zhang X, Li D, Wang T, Wei T, Zhang X, et al. Loss of KLF14 triggers centrosome amplification and tumorigenesis. Nat Commun. 2015;6:8450. doi: 10.1038/ncomms9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina F, Gaillard J, Guerin C, Coute Y, Sillibourne J, Blanchoin L, Thery M. The centrosome is an actin-organizing centre. Nat Cell Biol. 2016;18:65–75. doi: 10.1038/ncb3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230:6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Fukasawa K, Choi T, Kuriyama R, Rulong S, Woude GFV. Abnormal Centrosome Amplification in the Absence of p53. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- Funk LC, Zasadil LM, Weaver BA. Living in CIN: Mitotic Infidelity and Its Consequences for Tumor Promotion and Suppression. Developmental Cell. 2016;39:638–652. doi: 10.1016/j.devcel.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Zheng J. Oncogenic virus-mediated cell fusion: new insights into initiation and progression of oncogenic viruses--related cancers. Cancer Lett. 2011;303:1–8. doi: 10.1016/j.canlet.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Godinho SA, Pellman D. Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D. Oncogene-like induction of cellular invasion from centrosome amplification. Nature. 2014;510:167–171. doi: 10.1038/nature13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goepfert TM, Adigun YE, Zhong L, Gay J, Medina D, Brinkley WR. Centrosome Amplification and Overexpression of Aurora A Are Early Events in Rat Mammary Carcinogenesis. Cancer Res. 2002;62:4115–4122. [PubMed] [Google Scholar]
- Goepfert TM, Moreno-Smith M, Edwards DG, Pathak S, Medina D, Brinkley WR. Loss of chromosomal integrity drives rat mammary tumorigenesis. Int J Cancer. 2007;120:985–994. doi: 10.1002/ijc.22420. [DOI] [PubMed] [Google Scholar]
- Goncalves R, Warner WA, Luo J, Ellis MJ. New concepts in breast cancer genomics and genetics. Breast Cancer Res. 2014;16:460. doi: 10.1186/s13058-014-0460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X-F, Su J, Wang W. Need for clarification of data in the recent meta-analysis about p53 codon 72 polymorphism and breast cancer risk. Breast Cancer Res Treat. 2011;129:291–292. doi: 10.1007/s10549-011-1522-2. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. Mechanisms and consequences of localized, complex chromosomal rearrangements in cancer and developmental diseases. Nature medicine. 2012;18:1630–1638. doi: 10.1038/nm.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol. 2010;188:191–198. doi: 10.1083/jcb.200911102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlader NNA, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975–2012. National Cancer Institute; Bethesda, MD: Apr, 2015. http://seer.cancer.gov/csr/1975_2012/, based on November 2014 SEER data submission, posted to the SEER web site. [Google Scholar]
- Hu Y, McDermott MP, Ahrendt SA. The p53 Codon 72 Proline Allele Is Associated with p53 Gene Mutations in Non–Small Cell Lung Cancer. Clinical Cancer Research. 2005;11:2502–2509. doi: 10.1158/1078-0432.CCR-04-1913. [DOI] [PubMed] [Google Scholar]
- Izumi H, Matsumoto Y, Ikeuchi T, Saya H, Kajii T, Matsuura S. BubR1 localizes to centrosomes and suppresses centrosome amplification via regulating Plk1 activity in interphase cells. Oncogene. 2009;28:2806–2820. doi: 10.1038/onc.2009.141. [DOI] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH. Chromosome Segregation Errors as a Cause of DNA Damage and Structural Chromosome Aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- Jones BA, Kasl SV, Howe CL, Lachman M, Dubrow R, Curnen MM, Soler-Vila H, Beeghly A, Duan F, Owens P. African-American/White differences in breast carcinoma. Cancer. 2004;101:1293–1301. doi: 10.1002/cncr.20500. [DOI] [PubMed] [Google Scholar]
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian K, Go CD, Wu H, Brashavitskaya O, Xu RSZ, Dennis JW, Gingras A-C, Swallow C. Plk4 promotes cancer invasion and metastasis through Arp2/3 complex regulation of the actin cytoskeleton. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-16-2060. [DOI] [PubMed] [Google Scholar]
- Keenan T, Moy B, Mroz EA, Ross K, Niemierko A, Rocco JW, Isakoff S, Ellisen LW, Bardia A. Comparison of the Genomic Landscape Between Primary Breast Cancer in African American Versus White Women and the Association of Racial Differences With Tumor Recurrence. Journal of Clinical Oncology. 2015 doi: 10.1200/JCO.2015.62.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Ezan F, Crémet J-Y, Fautrel A, Gilot D, Lambert M, Benaud C, Troadec M-B, Prigent C. Overexpression of Active Aurora-C Kinase Results in Cell Transformation and Tumour Formation. PLoS One. 2011;6:e26512. doi: 10.1371/journal.pone.0026512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein-Sohn J, Pollinger B, Ohmer M, Hofmann F, Nigg EA, Hemmings BA, Wartmann M. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J Cell Sci. 2012;125:5391–5402. doi: 10.1242/jcs.107474. [DOI] [PubMed] [Google Scholar]
- Knoblich JA. Asymmetric cell division during animal development. Nat Rev Mol Cell Biol. 2001;2:11–20. doi: 10.1038/35048085. [DOI] [PubMed] [Google Scholar]
- Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, Boscoe FP, Cronin KA, Lake A, Noone AM, et al. Annual Report to the Nation on the Status of Cancer, 1975–2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst. 2015;107:djv048. doi: 10.1093/jnci/djv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski N, Hohenfellner M, Duensing S. The centrosome as potential target for cancer therapy and prevention. Expert Opin Ther Targets. 2013;17:43–52. doi: 10.1517/14728222.2013.731396. [DOI] [PubMed] [Google Scholar]
- Kronenwett U, Huwendiek S, Castro J, Ried T, Auer G. Characterisation of breast fine-needle aspiration biopsies by centrosome aberrations and genomic instability. Br J Cancer. 2005;92:389–395. doi: 10.1038/sj.bjc.6602246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulukian A, Holland AJ, Vitre B, Naik S, Cleveland DW, Fuchs E. Epidermal development, growth control, and homeostasis in the face of centrosome amplification. Proc Natl Acad Sci U S A. 2015;112:E6311–6320. doi: 10.1073/pnas.1518376112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian AW, Fish K, Shema SJ, Clarke CA. Lifetime risks of specific breast cancer subtypes among women in four racial/ethnic groups. Breast Cancer Res. 2010;12:R99. doi: 10.1186/bcr2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, Pietenpol JA. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE. 2016;11:e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz ML, Zhang CZ, Pellman D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu Rev Genet. 2015;49:183–211. doi: 10.1146/annurev-genet-120213-092228. [DOI] [PubMed] [Google Scholar]
- Lerit DA, Poulton JS. Centrosomes are multifunctional regulators of genome stability. Chromosome Res. 2015 doi: 10.1007/s10577-015-9506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D, et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Developmental Cell. doi: 10.1016/j.devcel.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D, et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev Cell. 2017;40:313–322.e315. doi: 10.1016/j.devcel.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, Li SA. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci U S A. 2004;101:18123–18128. doi: 10.1073/pnas.0408273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- Lindner R, Sullivan C, Offor O, Lezon-Geyda K, Halligan K, Fischbach N, Shah M, Bossuyt V, Schulz V, Tuck DP, et al. Molecular phenotypes in triple negative breast cancer from African American patients suggest targets for therapy. PLoS One. 2013;8:e71915. doi: 10.1371/journal.pone.0071915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A. 2002a;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A. 2002b;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL. Centrosome amplification drives chromosomal instability in breast tumor development. Proceedings of the National Academy of Sciences. 2002c;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: Implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A. 1998;95:2950–2955. doi: 10.1073/pnas.95.6.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Salisbury JL. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol. 1999;155:1941–1951. doi: 10.1016/S0002-9440(10)65513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Sun B, Zhao X, Li Y, Zhao X, Liu Y, Yao Z, Gu Q, Dong X, Shao B, et al. USP44<sup>+</sup>Cancer Stem Cell Subclones Contribute to Breast Cancer Aggressiveness by Promoting Vasculogenic Mimicry. Molecular Cancer Therapeutics. 2015;14:2121–2131. doi: 10.1158/1535-7163.MCT-15-0114-T. [DOI] [PubMed] [Google Scholar]
- Lopes CA, Jana SC, Cunha-Ferreira I, Zitouni S, Bento I, Duarte P, Gilberto S, Freixo F, Guerrero A, Francia M, et al. PLK4 trans-Autoactivation Controls Centriole Biogenesis in Space. Dev Cell. 2015;35:222–235. doi: 10.1016/j.devcel.2015.09.020. [DOI] [PubMed] [Google Scholar]
- Lund MJ, Trivers KF, Porter PL, Coates RJ, Leyland-Jones B, Brawley OW, Flagg EW, O'Regan RM, Gabram SG, Eley JW. Race and triple negative threats to breast cancer survival: a population-based study in Atlanta, GA. Breast Cancer Res Treat. 2009;113:357–370. doi: 10.1007/s10549-008-9926-3. [DOI] [PubMed] [Google Scholar]
- Ma Y, Yang J, Liu Z, Zhang P, Yang Z, Wang Y, Qin H. No significant association between the TP53 codon 72 polymorphism and breast cancer risk: a meta-analysis of 21 studies involving 24,063 subjects. Breast Cancer Res Treat. 2011;125:201–205. doi: 10.1007/s10549-010-0920-1. [DOI] [PubMed] [Google Scholar]
- Mabrouk I, Baccouche S, El-Abed R, Mokdad-Gargouri R, Mosbah A, Said S, Daoud J, Frikha M, Jlidi R, Gargouri A. No evidence of correlation between p53 codon 72 polymorphism and risk of bladder or breast carcinoma in Tunisian patients. Ann N Y Acad Sci. 2003;1010:764–770. doi: 10.1196/annals.1299.137. [DOI] [PubMed] [Google Scholar]
- Mahjoub MR, Stearns T. Supernumerary centrosomes nucleate extra cilia and compromise primary cilium signaling. Curr Biol. 2012;22:1628–1634. doi: 10.1016/j.cub.2012.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina M, Saavedra HI. Nek2 and Plk4: prognostic markers, drivers of breast tumorigenesis and drug resistance. Front Biosci (Landmark Ed) 2014;19:352–365. doi: 10.2741/4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, Basto R. Centrosome amplification causes microcephaly. Nat Cell Biol. 2013;15:731–740. doi: 10.1038/ncb2746. [DOI] [PubMed] [Google Scholar]
- Martin CA, Ahmad I, Klingseisen A, Hussain MS, Bicknell LS, Leitch A, Nurnberg G, Toliat MR, Murray JE, Hunt D, et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet. 2014;46:1283–1292. doi: 10.1038/ng.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason Jacqueline M, Lin Dan C-C, Wei X, Che Y, Yao Y, Kiarash R, Cescon David W, Fletcher Graham C, Awrey Donald E, Bray Mark R, et al. Functional Characterization of CFI-400945, a Polo-like Kinase 4 Inhibitor, as a Potential Anticancer Agent. Cancer Cell. 2014;26:163–176. doi: 10.1016/j.ccr.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Mittal K, Pannu V, Rida PCG, Klimov S, Dharma SS, Ketema Hw, Reid MD, Aneja R. Abstract B05: Centrosomal profiles of pancreatic adenocarcinoma from African American and European American patients: A comparative analysis. Cancer Epidemiology Biomarkers & Prevention. 2015;24:B05. [Google Scholar]
- Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- Mullee LI, Morrison CG. Centrosomes in the DNA damage response-the hub outside the centre. Chromosome Res. 2015 doi: 10.1007/s10577-015-9503-7. [DOI] [PubMed] [Google Scholar]
- Nagai MA, Schaer Barbosa H, Zago MA, Araujo Silva W, Jr, Nishimoto IN, Salaorni S, Guerreiro Costa LN, Silva Araujo M, Caldas Oliveira AG, Mourao Neto M, et al. TP53 mutations in primary breast carcinomas from white and African-Brazilian patients. Int J Oncol. 2003;23:189–196. [PubMed] [Google Scholar]
- Nam H-J, Chae S, Jang S-H, Cho H, Lee J-H. The PI3K–Akt mediates oncogenic Met-induced centrosome amplification and chromosome instability. Carcinogenesis. 2010;31:1531–1540. doi: 10.1093/carcin/bgq133. [DOI] [PubMed] [Google Scholar]
- Newman LA. Breast cancer disparities: high-risk breast cancer and African ancestry. Surg Oncol Clin N Am. 2014;23:579–592. doi: 10.1016/j.soc.2014.03.014. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Cajanek L, Arquint C. The centrosome duplication cycle in health and disease. FEBS Lett. 2014;588:2366–2372. doi: 10.1016/j.febslet.2014.06.030. [DOI] [PubMed] [Google Scholar]
- Ogden A, Cheng A, Rida PC, Pannu V, Osan R, Clewley R, Aneja R. Quantitative multi-parametric evaluation of centrosome declustering drugs: centrosome amplification, mitotic phenotype, cell cycle and death. Cell Death Dis. 2014;5:e1204. doi: 10.1038/cddis.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden A, Rida PC, Aneja R. Let's huddle to prevent a muddle: centrosome declustering as an attractive anticancer strategy. Cell Death Differ. 2012;19:1255–1267. doi: 10.1038/cdd.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden A, Rida PC, Aneja R. Heading off with the herd: how cancer cells might maneuver supernumerary centrosomes for directional migration. Cancer Metastasis Rev. 2013;32:269–287. doi: 10.1007/s10555-012-9413-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden A, Rida PC, Knudsen BS, Kucuk O, Aneja R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett. 2015;367:89–92. doi: 10.1016/j.canlet.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Okuda M, Ma Z, Goorha R, Tsujimoto H, Inokuma H, Fukasawa K. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol. 2005;25:4046–4061. doi: 10.1128/MCB.25.10.4046-4061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opyrchal M, Salisbury JL, Iankov I, Goetz MP, McCubrey J, Gambino MW, Malatino L, Puccia G, Ingle JN, Galanis E, et al. Inhibition of Cdk2 kinase activity selectively targets the CD44(+)/CD24(-)/Low stem-like subpopulation and restores chemosensitivity of SUM149PT triple-negative breast cancer cells. Int J Oncol. 2014;45:1193–1199. doi: 10.3892/ijo.2014.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannu V, Mittal K, Cantuaria G, Reid MD, Li X, Donthamsetty S, McBride M, Klimov S, Osan R, Gupta MV, et al. Rampant centrosome amplification underlies more aggressive disease course of triple negative breast cancers. Oncotarget. 2015a;6:10487–10497. doi: 10.18632/oncotarget.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannu V, Mittal K, Cantuaria G, Reid MD, Li X, Donthamsetty S, McBride M, Klimov S, Osan R, Gupta MV, et al. Rampant centrosome amplification underlies more aggressive disease course of triple negative breast cancers. Oncotarget. 2015b;6:10487–10497. doi: 10.18632/oncotarget.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean A, Achatz MIW, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- Pihan GA, Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003;63:1398–1404. [PubMed] [Google Scholar]
- Porter PL, Lund MJ, Lin MG, Yuan X, Liff JM, Flagg EW, Coates RJ, Eley JW. Racial differences in the expression of cell cycle–regulatory proteins in breast carcinoma. Cancer. 2004;100:2533–2542. doi: 10.1002/cncr.20279. [DOI] [PubMed] [Google Scholar]
- Romanov VS, Pospelov VA, Pospelova TV. Cyclin-dependent kinase inhibitor p21(Waf1): contemporary view on its role in senescence and oncogenesis. Biochemistry (Mosc) 2012;77:575–584. doi: 10.1134/S000629791206003X. [DOI] [PubMed] [Google Scholar]
- Rosario CO, Kazazian K, Zih FS, Brashavitskaya O, Haffani Y, Xu RS, George A, Dennis JW, Swallow CJ. A novel role for Plk4 in regulating cell spreading and motility. Oncogene. 2015;34:3441–3451. doi: 10.1038/onc.2014.275. [DOI] [PubMed] [Google Scholar]
- Sabino D, Gogendeau D, Gambarotto D, Nano M, Pennetier C, Dingli F, Arras G, Loew D, Basto R. Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr Biol. 2015;25:879–889. doi: 10.1016/j.cub.2015.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015;16:473–485. doi: 10.1038/nrm4025. [DOI] [PubMed] [Google Scholar]
- Schoenfelder KP, Fox DT. The expanding implications of polyploidy. J Cell Biol. 2015;209:485–491. doi: 10.1083/jcb.201502016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sercin O, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, Le Mercier M, Lambrechts D, Basto R, Blanpain C. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat Cell Biol. 2016;18:100–110. doi: 10.1038/ncb3270. [DOI] [PubMed] [Google Scholar]
- Shiao Y-H, Chen VW, Scheer WD, Wu XC, Correa P. Racial Disparity in the Association of p53 Gene Alterations with Breast Cancer Survival. Cancer Research. 1995;55:1485–1490. [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Stewart PA, Luks J, Roycik MD, Sang Q-XA, Zhang J. Differentially Expressed Transcripts and Dysregulated Signaling Pathways and Networks in African American Breast Cancer. PLoS One. 2013;8:e82460. doi: 10.1371/journal.pone.0082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarapore P, Horn HF, Tokuyama Y, Fukasawa K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene. 2001;20:3173–3184. doi: 10.1038/sj.onc.1204424. [DOI] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors. Proc Natl Acad Sci U S A. 2011;108:17974–17978. doi: 10.1073/pnas.1109720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, D’Incalci M, Piccolo S, Veronesi A, Zambelli A, Del Sal G, et al. Targeting triple negative breast cancer: Is p53 the answer? Cancer Treatment Reviews. 2013;39:541–550. doi: 10.1016/j.ctrv.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Vitre B, Holland AJ, Kulukian A, Shoshani O, Hirai M, Wang Y, Maldonado M, Cho T, Boubaker J, Swing DA, et al. Chronic centrosome amplification without tumorigenesis. Proceedings of the National Academy of Sciences. 2015;112:E6321–E6330. doi: 10.1073/pnas.1519388112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, Deng CX. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25:7148–7158. doi: 10.1038/sj.onc.1209707. [DOI] [PubMed] [Google Scholar]
- Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff JB, Wueseke O, Hyman AA. Pericentriolar material structure and dynamics. Philosophical Transactions of the Royal Society B: Biological Sciences. 2014;369:20130459. doi: 10.1098/rstb.2013.0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhao H, Amos CI, Shete S, Makan N, Hong WK, Kadlubar FF, Spitz MR. p53 Genotypes and Haplotypes Associated With Lung Cancer Susceptibility and Ethnicity. Journal of the National Cancer Institute. 2002;94:681–690. doi: 10.1093/jnci/94.9.681. [DOI] [PubMed] [Google Scholar]
- Zeng X, Shaikh FY, Harrison MK, Adon AM, Trimboli AJ, Carroll KA, Sharma N, Timmers C, Chodosh LA, Leone G, et al. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene. 2010;29:5103–5112. doi: 10.1038/onc.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Leibowitz ML, Pellman D. Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013;27:2513–2530. doi: 10.1101/gad.229559.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. International Journal of Cancer. 2014;134:1013–1023. doi: 10.1002/ijc.28226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Yue C, Li G, He B, Cheng W, Wang X, Yan M, Long Z, Qiu W, Yuan Z, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nature Communications. 2016;7:10180. doi: 10.1038/ncomms10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Zhang D, Qin G, Chen X, Wang H, Zhang D. BRCA1 and FancJ cooperatively promote interstrand crosslinker induced centrosome amplification through the activation of polo-like kinase 1. Cell Cycle. 2014;13:3685–3697. doi: 10.4161/15384101.2014.964973. [DOI] [PMC free article] [PubMed] [Google Scholar]