

Abstract
Numerous studies have established a link between individuals with affective disorders and a dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis, most notably characterized by a reduced sensitivity to glucocorticoid negative (−) feedback. Furthermore there is a sex difference in the etiology of mood disorders with incidence in females being two to three times that of males, an association that may be a result of the influence of estradiol (E2) on HPA axis function. In these studies, we have examined the effect of E2 on glucocorticoid-mediated HPA axis (−) feedback during both the diurnal peak and the stress-induced rise in corticosterone (CORT). Young adult female Sprague–Dawley (SD) rats were ovariectomized (OVX) and 1 week later treated subcutaneous (s.c.) with oil or estradiol benzoate (EB) for 4 days. On the 4th day of treatment, animals were injected with a single dose of dexamethasone (DEX), or vehicle. EB treatment significantly increased the evening elevation in CORT and the stress-induced rise in CORT. In contrast, DEX treatment reduced the diurnal and stress induced rise in CORT and adrenocorticotropic hormone (ACTH), and this reduction was not apparent following co-treatment with EB. To determine a potential site of E2’s action, female SD rats were OVX and 1 week later, wax pellets containing E2, the estrogen receptor beta (ERβ) agonist diarylpropionitrile (DPN), or the estrogen receptor alpha (ERα) agonist propylpyrazoletriol (PPT), was implanted bilaterally and dorsal to the paraventricular nucleus of the hypothalamus (PVN). Seven days later, animals were injected s.c. with a single dose of DEX, or vehicle to test for glucocorticoid-dependent (−) feedback. Results show that E2 and PPT increased, while DPN decreased the diurnal peak and stress-induced CORT and ACTH levels as compared to controls. Furthermore, E2 and PPT impaired the ability of DEX to inhibit both the diurnal and the stress-induced rise in CORT and ACTH, whereas DPN had no effect. Neuronal activation was measured by c-fos mRNA expression within the PVN following restraint. E2 and PPT increased c-fos mRNA, and impaired the normal DEX suppression of neuronal activation in the PVN. Taken together, these data indicate that estradiol causes a dysregulation of HPA axis (−) feedback as evidenced by the inability of DEX to suppress diurnal and stress-induced CORT and ACTH secretion. Additionally, the ability of E2 to inhibit glucocorticoid (−) feedback occurs specifically via ERα acting at the level of the PVN.
Keywords: stress, glucocorticoids, estrogen receptor, paraventricular nucleus, anxiety, depression
Major depressive disorder is a leading disability in the United States for young adults (Kessler et al., 2005). Women are twice as likely as men to experience a major depressive episode, and they tend to suffer a more severe form of depression and greater functional impairment (Angold and Worthman, 1993; Weissman et al., 1993; Thase et al., 1994; Kornstein, 1997). Other psychological disorders more prevalent in women include eating disorders and insomnia (Ehlert et al., 2001; Buckley and Schatzberg, 2005). Abnormalities in the hypothalamic–pituitary–adrenal (HPA) axis may underlie such pathologies as they are well documented in many mental disorders (Ehlert et al., 2001; Varghese and Brown, 2001; Barden, 2004). For example, the combined dexamethasone (DEX) suppression test (DST)/corticotropin releasing hormone (CRH) challenge shows a sensitivity of up to 90% in detecting major depressive disorder (Varghese and Brown, 2001). Thus, the apparent dysregulation of the HPA axis in human affective disorders when put in context with a higher prevalence in women could indicate a role for estrogen in HPA axis dysfunction.
Hypothalamic-pituitary-adrenal (HPA) axis activation is a homeostatic mechanism that is triggered by a physical or psychological stressor (see; De Kloet et al., 2005, for review). Stress-related neuronal inputs are integrated at the level of the paraventricular nucleus (PVN) of the hypothalamus to induce the secretion of CRH and arginine vasopressin (AVP) into the hypophyseal portal vasculature. These neuropeptides stimulate the secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary, which subsequently induces corticosteroid production by the adrenal cortex in humans (cortisol) and rodents (corticosterone; CORT). Importantly, HPA axis activation is terminated by a negative feedback action of glucocorticoids. Elevations in circulating CORT inhibit the HPA axis by acting via the glucocorticoid receptor (GR) in a direct and indirect fashion through the anterior pituitary, hypothalamus and hippocampus (Reul and De Kloet, 1985; Ratka et al., 1989). The main effect of elevated CORT is to decrease the synthesis and release of AVP, CRH and ACTH (Jingami et al., 1985).
In the brain, two distinct intracellular receptors have been shown to mediate the cellular responses to CORT (Reul and De Kloet, 1985). The type-I, or mineralocorticoid receptor (MR), is predominantly expressed in limbic structures such as the hippocampus (Reul et al., 1987). Type-II, or glucocorticoid receptor (GR), is expressed throughout the brain with highest amounts in hypothalamic areas such as the PVN and supraoptic nucleus (SON), as well as in the hippocampus. Both receptors are also responsible for mediating the negative feedback effects of CORT on neuroendocrine systems. MR exhibits a 10-fold higher affinity for CORT than GR, thus MR is predominantly occupied by basal CORT levels and is important in regulating basal secretion of CORT (Reul et al., 1987). The GR possesses a lower affinity for CORT and is thought to be the primary CORT receptor that modulates the negative feedback response to elevated CORT levels, although MR is also involved in this (Ratka et al., 1989; Spencer et al., 1998; Pace and Spencer, 2005). Within the PVN, GR have been reported in both CRH and AVP containing neurons although there is much greater evidence for the presence of GR in AVP (Albeck et al., 1994) rather than in CRH neurons (Uht et al., 1988). Nonetheless, CORT acts to inhibit CRH and AVP expression in PVN neurons (Kovacs and Mezey, 1987; Swanson and Simmons, 1989), but in a differential manner (Ma and Aguilera, 1999; Kovacs et al., 2000). Whether glucocorticoid-mediated negative feedback on CRH and AVP neurons is truly direct or indirect is unknown.
Several studies have indicated that in the presence of estradiol there is an enhanced responsiveness of the HPA axis to a stressor which may be due in part to impairment of glucocorticoid negative feedback (Peiffer and Barden, 1987; Turner, 1990, 1992; Peiffer et al., 1991; Burgess and Handa, 1992, 1993; Carey et al., 1995; Ferrini et al., 1995; Patchev et al., 1995). Furthermore, gonad intact female rats have higher, whereas ovariectomized (OVX) females have similar, peak diurnal plasma CORT as compared to gonad intact males (Atkinson and Waddell, 1997; Seale et al., 2004). Potential mechanisms of action include competition between estrogen receptor alpha (ERα) and GR to regulate transcription at an AP-1 site (Uht et al., 1997), and reduction of GR binding and protein levels in the hippocampus, hypothalamus, and anterior pituitary (Burgess and Handa, 1992). In the preoptic area, a region with inhibitory PVN efferents, estradiol has been shown to alter glutamic acid decarboxylase (GAD)67 levels (Curran-Rauhut and Petersen, 2002). In addition to indirect effects on HPA axis, estrogen may directly influence activity within neurosecretory neurons in the PVN. This is suggested by studies showing a greater induction of c-fos mRNA within PVN neurons in response to stress when estradiol is present either systemically or directly administered to the PVN (Yukhananov and Handa, 1996; Rachman et al., 1998; Lund et al., 2004, 2006). Accordingly, studies have indicated that estradiol regulates several PVN neuropeptides (Levin and Sawchenko, 1993), and upregulates restraint-induced CRH mRNA (Lund et al., 2004).
In brain, the actions of estradiol can be mediated by two distinct estrogen receptors, ERα and ERβ. Importantly, this laboratory has recently shown that the HPA enhancing effect of systemic estradiol administration can be mimicked by the local application of 17β-estradiol or a selective ERα agonist to the PVN (Lund et al., 2006). ERα is not expressed within the PVN, yet is expressed in the peri-PVN, a region implicated in HPA axis inhibition (Suzuki and Handa, 2005). Within the PVN, ERβ is found predominantly in pre-autonomic neurons and thus is an unlikely site of a direct estradiol enhancement of HPA activity (Stern and Zhang, 2003). Taken together, these studies suggest that estradiol might impair the glucocorticoid-mediated negative feedback system directly or indirectly to modulate HPA axis activity. However, whether or not estradiol can alter glucocorticoid negative feedback has not been explicitly described.
In the studies described here, we have examined the possibility that estradiol impairs the sensitivity of the HPA axis to glucocorticoid negative feedback, by examining its effect on both the diurnal peak in CORT secretion and following a psychological stressor (restraint). Indeed, we have found that subtype-selective ER agonists when administered into the systemic circulation or locally to the peri-PVN region of the hypothalamus can impair glucocorticoid negative feedback. These data indicate that in the presence of estradiol there is a dysregulation of the HPA axis that is demonstrated by the inability of DEX to suppress diurnal and stress-induced CORT secretion. This impairment in negative feedback occurs specifically via ERα acting at the level of the PVN.
EXPERIMENTAL PROCEDURES
Animals
Adult female Sprague–Dawley (SD) rats (~250 g) were obtained from Charles River Laboratories (Wilmington, MA, USA). The rats were caged in pairs, housed in the Colorado State University vivarium and maintained on a 12-h light/dark cycle (lights on at 07:00 h) with ad libitum access to rat chow and water. One week following arrival, rats were OVX under isoflurane anesthesia. All animal protocols were previously approved by the institutional animal care and use committee (IACUC) at Colorado State University. All procedures conformed to the Public Health Service policy on humane care and use of laboratory animals. Experiments were designed to minimize suffering and number of animals used.
Systemic hormone administration: diurnal experiment
One week following ovariectomy, animals were treated with oil or estradiol benzoate (EB, Sigma, St. Louis, MO, USA, 25 μg/kg; s.c.) daily for 4 days. On the fourth day of treatment, animals were injected s.c. with a single dose of DEX (Sigma) (30 μg/kg, s.c.) at 12:00 h. Controls received oil vehicle. Animals were sacrificed at 18:00 h (lights out at 19:00 h) on the same day; a time that is close to the diurnal peak in plasma CORT in our animal colony. Trunk blood was collected for CORT and ACTH analysis via radioimmunoassay (RIA).
Systemic hormone administration: stress experiment
One week following ovariectomy animals were treated with oil or EB (25 μg/kg, s.c.) daily for 4 days. On the fourth day of treatment, animals were injected s.c. with a single dose of DEX (30 μg/kg, s.c.) at 04:00 h 6 h prior to restraint stress. Restraint was performed by removing the animal from its home cage and placing it into a Plexiglas restraint tube (Plas-Laboratories, Lansing, MI, USA). Animals were sacrificed directly from the tube 20 min later. Controls received oil vehicle. Control animals for restraint were killed immediately after removal from their home cage. Animals were sacrificed after 20 min of restraint between 10:00 and 11:00 h and trunk blood samples collected for CORT and ACTH analysis via RIA.
Stereotaxic implantation
One week following ovariectomy animals were fitted bilaterally with two 22 gauge stainless-steel cannulae (Small Parts, Miami Lakes, FL, USA) with the aid of a small animal stereotaxic instrument (David Kopf Instruments, Tujunga, CA, USA). The tips of the cannulae were previously packed with one of the following compounds: EB, propylpyrazoletriol (PPT) (ERα agonist; relative binding affinity (RBA)=49; Stauffer et al., 2000; Tocris, Ellisville, MO, USA), or diarylpropionitrile (DPN) (estrogen receptor beta agonist; RBA=18; Meyers et al., 2001; Tocris), dissolved into warmed beeswax to a final concentration of 0.5 μM, and packed to a height of 0.5 mm within the end of the cannulas. Controls received cannulae packed with wax alone. Stereotaxic coordinates to allow placement of the cannula tip to the region just above the PVN were lateral 10° insertion angle to 1.8 mm posterior and 2.0 mm lateral to bregma, and 6.5 mm below the skull surface. A 28 gauge stainless steel wire cut to extend 1.0 mm past the length of the cannulas was inserted into the cannulae and the pellet expelled. Following sacrifice of all animals, confirmation of pellet placement was confirmed in Cresyl Violet–stained brain sections. Animals where both pellets were >0.5 mm away from the PVN were excluded based on previous studies using the same pellet hormone concentration and resulting spread (~0.5 mm for estradiol) (Lund et al., 2005).
Local hormone administration: diurnal experiment
Seven days after stereotaxic surgery, animals were injected s.c. with a single dose of DEX (Sigma) (30 μg/kg, s.c.) at 12:00 h. Controls received oil vehicle. Animals were sacrificed directly from the home cage at 18:00 h (lights out at 19:00 h) the same day, near the diurnal peak in plasma CORT, and trunk blood was collected for CORT analysis using RIA.
Local hormone administration: stress experiment
Seven days after stereotaxic surgery, animals were injected s.c. (04:00 h) with a single dose of DEX (30 μg/kg) 6 h prior to restraint stress (at 10:00 h). Controls received oil vehicle. Animals were sacrificed after 20 min of restraint at 10:20 h and trunk blood samples collected for CORT and ACTH analysis via RIA.
CORT RIA
Trunk blood was centrifuged at 4 °C and plasma removed and stored at −20 °C until assayed. For assay, plasma samples were diluted in 0.01 M PBS (1:25) and corticosteroid binding globulin (CBG) was inactivated by incubation at 65 °C for 1 h. Samples (20 μl) and standards (5–700 ng/ml) were incubated overnight at 4 °C with antiserum (rabbit anti-CORT; MP Biomedicals, Solon, OH, USA) and [3H] CORT (PerkinElmer, Boston, MA, USA) in 0.1% gelatin 0.01 M PBS. Free CORT was separated from antibody-bound CORT with 1.0 ml dextran-coated charcoal. After centrifugation, the supernatant containing antibody-bound CORT was mixed with 4 ml of scintillation fluid and counted with a Packard 2900 TR liquid scintillation counter (Meriden, CT, USA). The intra-assay and inter-assay variance as measured by internal quality controls was 4.5% and 7.8% respectively.
ACTH RIA
For assay, plasma samples were diluted in PBS-albumin (0.01 M PBS, 0.9% NaCl, 0.1% albumin, 100,000 kIUs aprotinin/l). Samples and standards (5–2000 pg per tube) were incubated overnight at 4 °C with antiserum (rabbit anti-ACTH) (Immunostar, Hudson, WI, USA) and 2% normal rabbit serum. The following day, [125I] ACTH (1–39) (Amersham, Piscataway, NJ, USA) in PBS-albumin (100 μl) was added to each tube as the tracer, and incubated overnight at 4 °C. On the third day, goat anti-rabbit gamma globulin (Calbiochem, La Jolla, CA, USA) was added to all tubes except totals, and tubes incubated overnight at 4 °C. On the last day, 3 ml of PBS-albumin was added and tubes were centrifuged (>1000×g). Assay tubes were immediately decanted, blotted dry, and the resulting pellet counted with a Packard cobra II gamma counter. The intra-assay and inter-assay variance as measured by internal quality controls was 3.8% and 6.1% respectively.
In situ hybridization
In situ hybridization for c-fos mRNA was performed as previously described (Lund et al., 2004) utilizing a 48-bp oligonucleotide probe targeted to the rat c-fos gene. The oligonucleotide probe was labeled using 35S-dATP. Following hybridization slides were apposed to autoradiographic film (Kodak BioMax MR, Eastman Kodak, Co., Rochester, NY, USA) and allowed to expose for 10 days. Autoradiograms were analyzed for density using a video camera (Sony XC-77, Tokyo, Japan) attached to a Nikon lens (Melville, NY, USA). Scion Image (Frederick, MD, USA) was utilized to count density per a fixed region encompassing the PVN. For each section, background density was obtained from a region adjacent to the PVN, and subtracted from the PVN measurement. Bilateral measurements were obtained from four separate sections and averaged to obtain the value for each individual animal.
Immunocytochemistry
To examine co-localization of ERα and GAD67, four OVX’d young adult (300 g) female SD rats were intracardially perfused with 4% buffered paraformaldehyde. Brains were postfixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, sectioned at 35 μM on a Leitz cryostat, and stored in cryopreservative at −20 °C until further processed for immunohistochemical detection of ERα and GAD67. After standard washes, the free floating sections were incubated in 6% normal goat serum (NGS) in 0.1 M PBS with 0.1% triton-x (TX) to block nonspecific binding, then incubated for 72 h at 4 °C with the ERα antibody (C1355; detects ligand-bound and unbound ERα; obtained from Dr. M. Shupnik, UVA; 1:10,000 dilution) in 0.1 M PBS with TX in the presence of 2% NGS. Next, the tissue was washed three times for 10 min each in PBS with TX and incubated with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA; 1:500) in PBS with TX in the presence of 2% NGS for 2 h at room temperature. Sections are subsequently washed and processed according to the avidin–biotin–peroxidase procedure (Vector Laboratories; 1:500). After standard washes, the tissue was rinsed in 0.1 M Tris-buffered saline (pH 8.0) for 15 min and then developed with 3,3′ -diaminobenzidine (DAB; 0.5 mg/ml; (Sigma) in 0.1 M Tris-buffered saline) containing 0.2% nickel sulfate and 0.01% hydrogen peroxide for 3–5 min to produce a blue–black reaction product. The reaction is stopped by several washes in 0.1 M PBS. Subsequently, the sections were incubated for 48 h at 4 °C with the mouse GAD67 antibody (Chemicon, Temecula, CA, USA; 1:10,000 dilution) in PBS containing 0.01% TX and 2% NGS. The sections were then washed and processed according to the avidin–biotin–peroxidase procedure (Vector Laboratories; 1:500). After standard washes, the tissue is rinsed in 0.1 M Tris-buffered saline (pH 8.0) for 15 min and then developed with DAB (0.5 mg/ml (Sigma) in 0.1 M Tris-buffered saline) containing 0.01% hydrogen peroxide for 3–5 min to produce a brown reaction product. Control sections, where primary antibody was omitted, were run in parallel. Complete loss of staining for the corresponding antigen was seen in these sections.
Data analysis
Analysis of variance was performed on data from each experiment using the StatView data analysis software (Abacus Concepts, Inc., Berkeley, CA, USA). Fisher’s protected least squares difference (PLSD) analysis was used post hoc where appropriate. Differences were considered significant when P<0.05. Data are expressed as group means±SEM.
RESULTS
Estradiol treatment augments the diurnal rise of CORT and ACTH and prevents DEX suppression of CORT and ACTH
Females have a more robust diurnal CORT rhythm as compared to males, perhaps in part due to altered glucocorticoid negative feedback (Atkinson and Waddell, 1997). Therefore, this experiment was designed to determine the effect of estradiol on glucocorticoid-mediated negative feedback during the diurnal rise in CORT secretion. EB alone significantly (P<0.01) increased plasma CORT and ACTH levels (Fig. 1). DEX treatment also significantly (P<0.01) suppressed serum CORT levels in vehicle-treated animals. In contrast, in EB-treated animals, CORT and ACTH levels were not suppressed by DEX. Thus, in the presence of estradiol, the synthetic glucocorticoid DEX is unable to suppress the diurnal rise in CORT secretion.
Fig. 1.
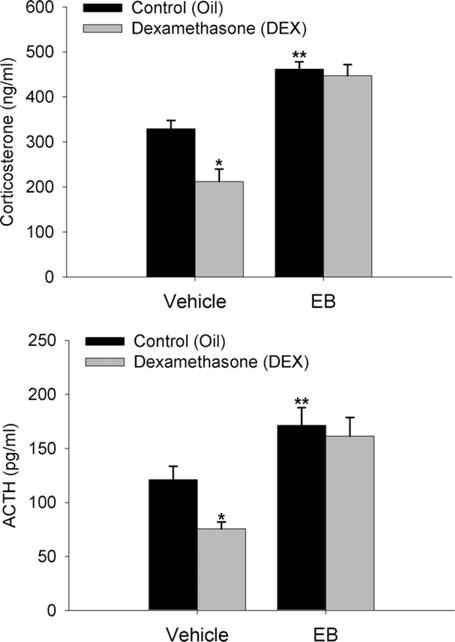
Estradiol impairs DEX suppression of diurnal peak plasma CORT and ACTH. Data are expressed as mean±SEM of seven animals per group. *, ** Indicate significant differences from control animals (P<0.01).
Estradiol treatment augments restraint-induced CORT and ACTH secretion and prevents DEX suppression of restraint-induced CORT and ACTH
Females also have a more robust hormonal response to a stressor as compared to males, possibly driven by gonadal hormone influences on glucocorticoid negative feedback (Burgess and Handa, 1992; Handa et al., 1994). Therefore, this experiment was designed to determine the effect of estradiol on GR-mediated negative feedback on stress-induced activation of the HPA axis. Estrogen alone significantly (P<0.01) increased stress-induced CORT and ACTH levels (Fig. 2). EB+DEX animals showed significantly (P<0.01) higher plasma CORT and ACTH compared to vehicle+DEX animals. In oil-treated animals, DEX treatment suppressed CORT by an average of 289.6 ng/ml or 62.9% and ACTH by an average of 365.7 pg/ml or 74.9%. In contrast, when pretreated with EB, the DEX-treated animals showed a 199.8 ng/ml or 34.4% on average suppression of CORT and a 281.9 pg/ml or 56.7% suppression of ACTH. Overall, CORT and ACTH values for EB+DEX animals were similar to those of oil+vehicle controls. Thus, in the presence of estradiol, the stress-induced levels of CORT and ACTH are significantly increased and responsiveness to DEX is significantly decreased.
Fig. 2.
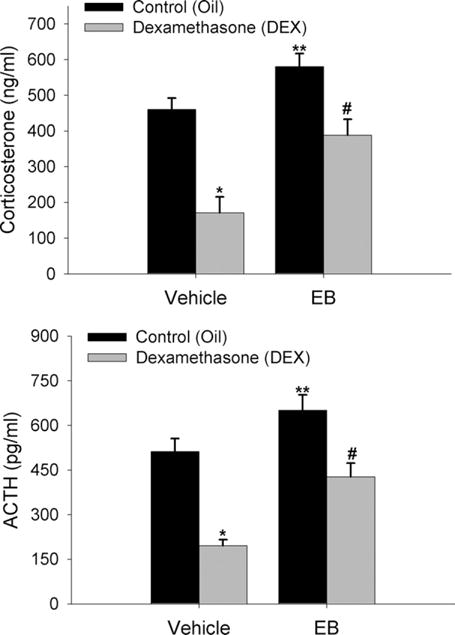
Estradiol impairs DEX suppression of plasma CORT and ACTH following a 20 min restraint stress. Data are expressed as mean±SEM of seven animals per group. *, ** Indicate significant differences from control animals (P<0.01). # indicates significant difference from DEX treated control animals (P<0.01).
Estrogen acts via ERα near the PVN to augment the diurnal rise of CORT and ACTH and to prevent DEX suppression of CORT and ACTH
Given the matching ACTH and CORT data from the previous two experiments, estradiol likely acts at the level of the brain or pituitary to impair glucocorticoid negative feedback. Therefore, this experiment was designed to determine a potential site for estradiol’s effect on glucocorticoid negative feedback during the diurnal rise in CORT, and also to determine whether this effect is through an ERα-or ERβ-dependent mechanism. Animals were stereotaxically implanted with wax pellets containing ER agonists targeted to an area just superior to the PVN (Fig. 3). Estradiol and PPT significantly increased (P<0.01), whereas DPN significantly lowered (P<0.01), CORT and ACTH when compared to controls given a wax-only pellet (Fig. 4). DEX treatment significantly (P<0.01) lowered CORT and ACTH levels in control and DPN-implanted animals, however estradiol and PPT significantly (P<0.01) impaired the suppression by DEX.
Fig. 3.
Representative photomicrograph of Cresyl Violet–stained tissue indicating location of stereotaxically implanted wax pellet.
Fig. 4.
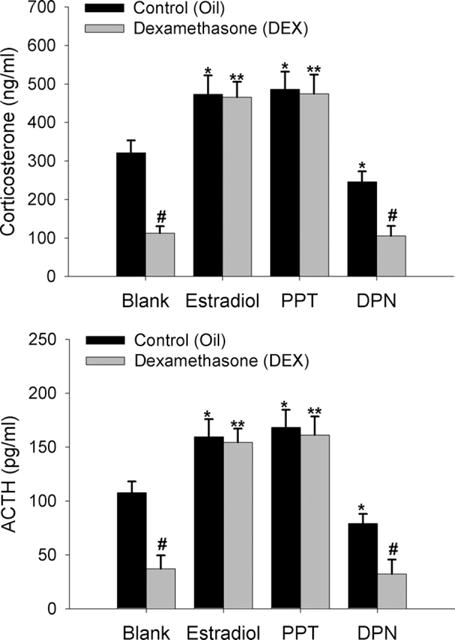
Estradiol impairs DEX suppression of peak diurnal CORT and ACTH through activation of ERα near the PVN of the hypothalamus. Wax pellets containing estradiol, the ERα agonist PPT, the ERβ agonist DPN, or no hormone (control, blank) were implanted to a region just above the PVN. Data are expressed as mean±SEM of 10 animals per group. *, # Indicate significant differences from oil-treated blank control animals (P<0.01). ** Indicates significant difference from DEX-treated control animals (P<0.01).
Estrogen acts via ERα near the PVN to augment restraint-induced CORT and ACTH secretion and to impair DEX suppression of restraint-induced CORT and ACTH
Results from the previous experiment indicated that estradiol may act via ERα at sites near the PVN to impair glucocorticoid dependent HPA negative feedback during the diurnal rise in CORT. We postulated that this may be also found following restraint stress activation of the axis. As in the previous experiment, animals were treated with hormone by stereotaxic implantation of wax pellets containing ER agonists that were targeted to a site just superior to the PVN. Twenty minutes after the start of restraint, estradiol- and PPT-implanted animals had significantly higher (P<0.01) CORT and ACTH than did controls (Fig. 5). DPN-implanted animals had significantly lower (P<0.01) CORT and ACTH following restraint than did controls. Moreover, DEX treatment significantly lowered (P<0.01) CORT and ACTH in both controls and DPN-implanted animals, however estradiol and PPT treatment significantly impaired (P<0.01) the ability of DEX to inhibit CORT and ACTH secretion.
Fig. 5.
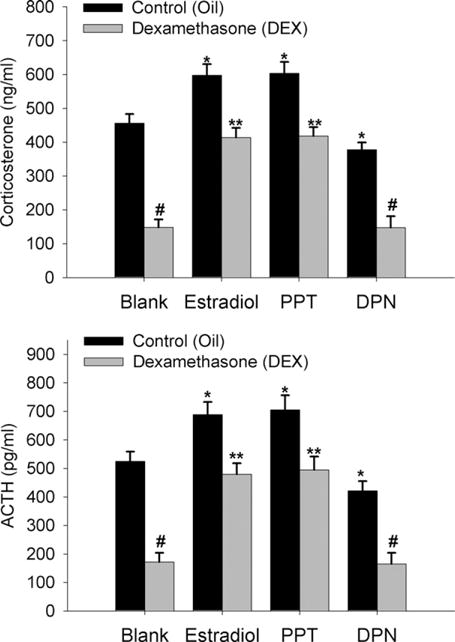
Estradiol impairs DEX suppression of restraint-induced CORT and ACTH through activation of ERα near the PVN of the hypothalamus. Wax pellets containing estradiol, the ERα agonist PPT, the ERβ agonist DPN, or no hormone (control, blank) were implanted to a region just above the PVN. Data are expressed as mean±SEM of six to seven animals per group. *, # Indicate significant differences from oil-treated blank control animals (P<0.01). ** Indicates significant difference from DEX-treated control animals (P<0.01).
Estrogen acts via ERα near the PVN to augment restraint-induced PVN c-fos mRNA expression and to impair DEX suppression of restraint-induced PVN c-fos mRNA expression
During a stressor, neural activity in the PVN is significantly increased, and can be subsequently suppressed with glucocorticoids. It follows then, that estradiol should impair glucocorticoid suppression of neural activity in the PVN. In order to couple our hormonal data with corresponding examination of neuronal activation following the restraint stress, we measured c-fos mRNA expression in the PVN. c-fos mRNA expression was significantly (P<0.05) higher in estradiol- and PPT-implanted animals and lower in DPN-implanted animals as compared to controls (Fig. 6). DEX treatment significantly reduced c-fos mRNA expression following restraint in controls and DPN treated animals, however estradiol and PPT treatment significantly impaired (P<0.01) the ability of DEX to inhibit c-fos mRNA expression.
Fig. 6.
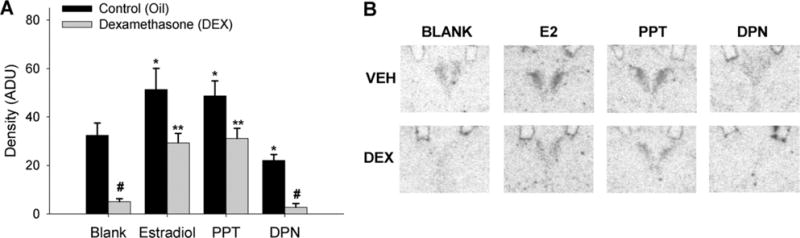
Estradiol impairs DEX suppression of restraint-induced c-fos mRNA expression in the PVN of the hypothalamus through activation of ERα near the PVN. Wax pellets containing estradiol, the ERα agonist PPT, the ERβ agonist DPN, or no hormone (control, blank) were implanted to a region just above the PVN. (A) Relative levels of c-fos mRNA expression. Data are represented as mean arbitrary density units ±SEM. N=6–7 animals per treatment group. *, # Indicate significant differences from oil-treated blank control animals (P<0.05). ** Indicates significant difference from DEX-treated control animals (P<0.01). (B) Representative film autoradiograms.
ERα is expressed in GABAergic neurons surrounding the PVN
To determine the phenotypes of ERα-containing neurons in and around the PVN, we examined ERα immunoreactivity (IR) in OVX young adult female SD rats given replacement estradiol. Nuclear ERα-IR was sparse within the PVN and periventricular PVN, but ERα-IR was seen surrounding the PVN proper especially in the dorsal region (Fig. 7). Moreover, dual-immunocytochemistry revealed that ERα containing neurons in the peri-PVN also contained GAD (67.4%±6.1% of ER positive cells contained GAD67). Thus, ERα is expressed in the PVN surround, and is predominantly found in GABAergic neurons.
Fig. 7.
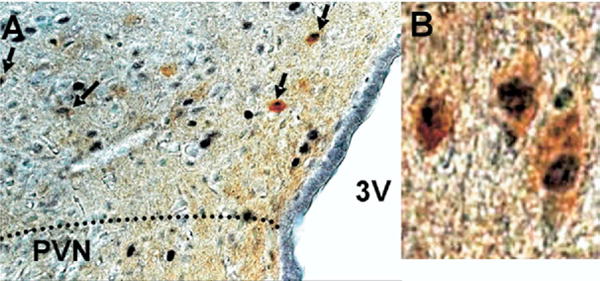
GAD67 is colocalized with ERα in the dorsal peri-PVN region (A); 67.4±6.1% of ERα containing neurons also expressed GAD67 in this region. Cells expressing ERα (dark nuclear staining) and GAD67 (brown cytoplasmic staining) (B).
DISCUSSION
In healthy individuals the physiological product of the stress axis, corticosteroids (cortisol in humans, CORT in rodents), normalize the activity of the axis via negative feedback at multiple sites including the hypothalamus, pituitary gland, and adrenal cortex. Impairment of tonic or phasic HPA axis inhibition results in abnormal endogenous glucocorticoid secretory patterns and levels, which are correlated with several disease states such as hypertension, inflammation, obesity, heart disease and diabetes (Baid and Nieman, 2004; Whitworth et al., 2005; Pasquali et al., 2006; Tomlinson and Stewart, 2007; Tait et al., 2008).
Gonadal steroid hormones play a vital role in modulating HPA axis function. It is well established that basal and stress-induced adrenal steroid secretion is greater in female rodents than in males (Critchlow et al., 1963; Kitay, 1963; Handa et al., 1994), and that the activational effects of gonadal steroids play an integral role in this sex difference (Sencar-Cupovic and Milkovic, 1976). In females, ovariectomy reduces stress-induced CORT and ACTH, and this is reversed by estrogen treatment (Burgess and Handa, 1992; Handa et al., 1994; Suzuki et al., 2001). However, this is not always the case as several groups have reported that estrogen can inhibit responses to stress (Young et al., 2001; Figueiredo et al., 2002; Ochedalski et al., 2007). Furthermore, human studies indicate than men typically respond stronger hormonally to a stressor than women [for review see; Kudielka and Kirschbaum, 2005], however a short-course treatment of estradiol in young men significantly increases cortisol levels following a social stress test (Kirschbaum et al., 1996). Interestingly, women also have higher weekday wakening salivary cortisol levels following an examination stress as compared to men (Weekes et al., 2008). These discrepancies may be due in part to opposing actions of estradiol signaling through ERα and ERβ. However, at present the mechanism whereby estrogen alters HPA axis function is unknown.
In the studies described here, we investigated the effect of estradiol on glucocorticoid-mediated negative feedback. Basal HPA axis activity is circadian in nature, where a diurnal peak in secretion occurs just prior to onset of activity (early morning for humans, and early evening for rodents). Therefore, we examined basal glucocorticoid negative feedback at the diurnal peak in CORT in addition to stress-activated CORT secretion. Furthermore, we determined which estrogen receptor subtype is responsible for this effect and the brain site mediating estradiol’s effects on glucocorticoid-mediated negative feedback. Based on our results, we believe that estradiol exerts its action at the level of the PVN, through an ERα-mediated disruption of GR-mediated negative feedback.
Glucocorticoid-mediated HPA axis negative feedback is dependent upon the dose and the duration of exposure (Abe and Critchlow, 1980). Normal function of the HPA axis is dependent upon permissive and reactive actions of glucocorticoids (for review see; Sapolsky et al., 2000). For example, adrenalectomy (ADX) results in higher PVN neuropeptide synthesis and secretion both at the basal and stressed states. Additionally, feedback can occur either directly at the level of the CRH and AVP neurons of the PVN as well as the corticotrophs of the anterior pituitary, or indirectly though GR and MR containing brain regions that project to the PVN such as the hippocampus and amygdala (Davidson and Feldman, 1967; Bohus and Strashimirov, 1970; Dallman et al., 1987; Sawchenko, 1987). Glucocorticoid effects vary according to duration of exposure. Fast feedback occurs within seconds to minutes, whereas delayed feedback occurs within minutes to hours (Dallman et al., 1987). Fast feedback occurs in a time frame that is too short for de novo protein synthesis, and must be attributable to non-genomic actions of glucocorticoids. These actions have been shown to involve the classical GR and MR as well as endocannabinoids (Widmaier and Dallman, 1984; Hinz and Hirschelmann, 2000; Di et al., 2003, 2005; Patel et al., 2004). Delayed feedback occurs in a time frame that likely involves influences on gene transcription and de novo protein synthesis and is attributable to activation of GR and/or MR, and may be direct or indirect in nature. For the purpose of this study we examined delayed glucocorticoid-mediated HPA axis negative feedback.
In order to more closely relate our findings to the function of the HPA axis in the human condition of depression, we utilized a DST. The DST is a well-established clinical predictor of depression (Carroll and Curtis, 1976), and has been used in rodents as a model for the human test (Lurie et al., 1989). More recently, the combined DEX/CRH test, where DEX suppression in the presence of HPA axis drive (CRH), has been proven to be 90% accurate in diagnosing depressed individuals (Varghese and Brown, 2001). Furthermore, the DEX/CRH test has been used to accurately predict relapse and can be used as a biomarker to positively identify novel treatments for depression (Aubry et al., 2007; Ising et al., 2007). The DST and DEX/CRH test measure common pathology, however the DEX/CRH has better diagnostic reliability (Watson et al., 2006). Therefore, in this study, we examined the DST under the basal state as well as under HPA axis drive (restraint stress) to better match the parameters of the DEX/CRH test utilized in humans.
In our initial set of experiments, we found that the synthetic glucocorticoid DEX, suppressed peak diurnal and restraint-induced CORT in OVX females. The degree of suppression was slightly higher following restraint than during the diurnal peak. This may be in part due to the higher overall activity of the HPA axis following a stressor, or due to decreased glucocorticoid sensitivity during diurnal peak secretion. This decreased sensitivity is thought to be partly responsible for the normal diurnal elevation in CORT (Bradbury et al., 1994). Interestingly, following estradiol replacement to OVX females, the diurnal increase in CORT was significantly increased and DEX suppression of the diurnal peak was completely inhibited. Therefore, estradiol acts to inhibit glucocorticoid negative feedback during the diurnal rise in CORT. Moreover, estradiol treatment increased CORT following a restraint stress and impaired DEX suppression of stress-induced CORT. In both cases, the impairment was not due to estradiol’s actions at the adrenal cortex, as the pattern of plasma CORT closely matched plasma ACTH in all animals. This provides strong evidence that estradiol directly impairs glucocorticoid-mediated negative feedback. These results also suggest that the increased responsiveness to a stressor that is seen in females is likely due to decreased sensitivity to glucocorticoid feedback.
Results from these experiments indicated that the impairment in negative feedback must occur either in the brain or at the level of the pituitary gland. Previous data from this laboratory indicated that estradiol placed centrally near the PVN of the hypothalamus of OVX female rats resulted in a stronger hormonal response to a restraint stress than controls (Lund et al., 2006). Additionally, in a separate earlier study, placement of DEX near the PVN blunted CRH hnRNA transcription following a stressor (Kovacs et al., 1986; Sawchenko, 1987). Therefore, we designed studies to determine the responsible estrogen receptor subtype and the location of estradiol’s effect on glucocorticoid negative feedback. Our data implicate the peri-PVN region as a critical brain site integrating estradiol influence on HPA axis output and glucocorticoid-mediated negative feedback. Our studies utilized the same procedure used previously where wax pellets containing subtype selective agonists PPT (ERα) and DPN (ERβ) in addition to the endogenous ER ligand estradiol were placed bilaterally to a region just above the PVN. This provides for continuous local release confined to a 0.5 mm radius around the wax pellet that allows for the examination of local ER activation independent of peripheral effects (Lund et al., 2006).
We found that activation of ERα in the region of the PVN increased the diurnal gain of the HPA axis and was sufficient to block all suppressive effects of DEX during the diurnal peak in CORT. Following a restraint stress, peri-PVN ERα activation significantly impaired DEX suppression and resulted in CORT and ACTH levels similar to untreated, stressed controls. As expected, c-fos mRNA expression following the restraint stress closely matched the hormonal output of the axis. Animals implanted with estradiol or PPT pellets had stronger activation of PVN neurons following a stressor than control or DPN-implanted animals, and this activation was not diminished by DEX to the same degree as seen in control animals. From these findings we conclude that estradiol impairs glucocorticoid-mediated HPA axis negative feedback during the diurnal peak and following a restraint stress specifically through activation of ERα-containing neurons in the region of the PVN.
The actions of estradiol can be mediated by both ERα and ERβ. Whereas ERβ is found in PVN neurons, ERα is not expressed in CRH or AVP neurons in the PVN (Suzuki and Handa, 2005), thus ruling out a direct action of estradiol through ERα on the function of these neurons. However, ERα is found in stress-responsive areas that provide inhibitory GABA input into the PVN including the bed nucleus of the stria terminalis (BnST) and medial preoptic area (MPOA) (Shughrue et al., 1997) and the peri-PVN region (Herman et al., 2002). In support of a GABAergic mechanism, our studies show co-localization of ERα-IR with GAD67-IR. GAD67 is the rate-limiting enzyme in the production of GABA. Although we found that ERα-IR was largely absent from the PVN, it was present in neurons immediately surrounding the PVN (peri-PVN), especially dorsally. Additionally, a majority of these ERα-IR neurons in the dorsal peri-PVN region also contained GAD67-IR indicating that they are inhibitory interneurons. This region of the peri-PVN contains GAD-IR neurons that project to the parvocellular PVN (Roland and Sawchenko, 1993). Application of a GABA agonist locally to this region can enhance the CORT response to a stressor (Cullinan, 1998), presumably by inhibiting GABAergic transmission to hypophysiotropic CRH neurons. Therefore, it is plausible that estradiol may work via ERα to inhibit GABAergic transmission from these peri-PVN neurons.
Our findings are consistent with several other estrogen-sensitive systems that indicate a role for estrogen modulation of GABAergic neurotransmission. These include the actions of estrogens on dendritic spine growth (Murphy et al., 1998) and the positive-feedback response of gonadotropin-releasing hormone neurons (Herbison, 2008). Furthermore, estradiol has been shown to alter GAD67 expression in the AVPV and MPN (Curran-Rauhut and Petersen, 2002). Estradiol-GABA interactions are also critical for proper development of the brain [for review see; McCarthy, 2008]. Given that the direct administration of estradiol or an ERα agonist to the PVN results in impaired glucocorticoid-mediated negative feedback, and the presence of ERα in GAD67 containing neurons surrounding the PVN, these results suggest that ERα might inhibit glucocorticoid negative feedback by reducing an inhibitory GABAergic input into the PVN.
Stress-integration at the level of the PVN involves both inhibitory GABA and excitatory glutamate circuits (see; Herman et al., 2002, 2004; Kovacs et al., 2004, for review). Recent studies have suggested that a substantial part of PVN excitation and inhibition is gated by local circuit neurons in the vicinity of the PVN. Studies have shown that microinjection of glutamate elicits, while local application of a glutamate receptor antagonist inhibits ACTH and CORT secretion (Darlington et al., 1989; Ziegler and Herman, 2000). Inhibitory GABA containing neurons synapse on parvicellular PVN neurons and are present in the immediate surround of the PVN, BnST, MPOA, lateral hypothalamic area (LHA), and anterior hypothalamic area (AHA) (Boudaba et al., 1996; Miklos and Kovacs, 2002). These regions also provide stress responsive afferents surrounding the PVN (Cullinan et al., 1996; Bali et al., 2005). In addition, microinjection of muscimol (GABA-A receptor agonist) into the PVN attenuates, while bicuculline (GABA-A receptor antagonist) augments stress-induced ACTH secretion (Dimicco et al., 1996; Cole and Sawchenko, 2002). Furthermore, studies have shown stress-induced increases in the GABA-synthesizing enzyme GAD67 mRNA in PVN-projecting areas including peri-PVN, MPOA, BnST, arcuate nucleus, and hippocampus (Bowers et al., 1998). The presence of both ionotropic glutamate receptors and GABA-A receptors in CRH and AVP neurons indicates their ability to integrate inhibitory and excitatory inputs (Aubry et al., 1996; Cullinan et al., 1996). This is corroborated by studies showing local GABA-A receptor blockade increases CRH and AVP expression (Bali and Kovacs, 2003). Recent studies by (Di et al., 2005) have shown glucocorticoids secreted in response to a stressor act in a rapid feedback fashion to decrease glutamate and increase GABA release onto PVN neurons. Taken together, these studies suggest that local GABA circuits are key to stress integration at the PVN and play a vital role in glucocorticoid dependent negative feedback although other mechanisms of action for estrogen interference of glucocorticoid-mediated negative feedback must also be considered.
It is possible that our results reflect estradiol’s ability to impair either GR or MR autoregulation and expression (Burgess and Handa, 1992; Patchev and Almeida, 1996), and thus impair glucocorticoid-mediated feedback. Earlier molecular studies have shown that GR and ER can interact in control of a reporter gene regulated through simple hormone response elements (Uht et al., 1997). How this relates to neuronal function is not entirely clear since studies have not definitively colocalized ERα or ERβ with GR or MR in brain regions known to be important in CORT-mediated negative feedback, and ERα has been shown to be absent in GR containing CRH and AVP neurosecretory neurons of the PVN. Until future studies indicate the existence of ERα and GR within the same cell population, it must be postulated that estradiol works indirectly to influence glucocorticoid negative feedback.
Although the effects of estradiol observed here may also be peripheral, causing inhibition of negative feedback at the level of the pituitary or adrenal glands, our results seem to rule out this possibility. The changes in CORT secretion that we observed were mimicked by ACTH secretion, and the local administration of hormone to the peri-PVN region rules out potential actions at peripheral sites. Alternatively, effects could involve alteration of CBG levels or binding capacity. Increased CBG levels might lead to decreased sensitivity to endogenous and exogenous corticosteroids, and thus lower glucocorticoid-mediated negative feedback. Circulating CBG levels are 2.5 times higher in females than in males, however this sex difference appears to be due to the organizational actions of sex hormones (Mataradze et al., 1992). Furthermore, estradiol treatment does not influence CBG levels in females (Mataradze et al., 1992; McCormick et al., 2002), and has been shown to increase or have no effect on CBG levels in males (McCormick et al., 2002; Lund et al., 2004). To circumvent this potential confound, we utilized a synthetic glucocorticoid (DEX), which does not bind CBG (Kolanowski and Pizarro, 1969; Koblinsky et al., 1972). DEX has been shown to inhibit HPA axis activity when administered directly to the PVN (Kovacs and Makara, 1988), and an intraperitoneal injection of femtomolar 3H-DEX produces binding in glucocorticoid-concentrating brain regions within 1 h (Hassan et al., 1999).
An unlikely alternative possibility is that hormone or agonist could have diffused far enough from the pellet to enter the third ventricle, and thus enter cerebrospinal fluid (CSF) circulation. In this case, a di- or tri-synaptic mode of inhibition may be plausible, where estrogen could induce changes in GABA synthesis or release, in areas with indirect connections with the PVN like the hippocampus, lateral septum, or amygdala. We do not think that this is a viable alternative since the diffusion of hormone would result in CSF concentrations too low to appreciably effect ER signaling in distant brain regions.
Yet another possibility is that estradiol could increase GABA receptor levels in brain regions that provide inhibitory input to the PVN, thus increasing potential inhibition of these inhibitory PVN inputs. Indeed, some studies have shown that estrogen can increase GABAA receptor subunit expression in the MPA and BnST (Herbison and Fenelon, 1995). Whether these changes can affect glucocorticoid-mediated HPA negative feedback is uncertain. Such a mechanism is unlikely except at the level of the PVN unless there is a widespread diffusion of hormone away from the pellet, which we have not observed and would likely result in very low levels of hormone in distant brain sites.
Our data indicate that ERα activation in the peri-PVN induces an increase in neuronal activation within the PVN following stress with DEX pretreatment as measured by the induction of c-fos mRNA expression. This argues for a site of action contained to the immediate surround of the PVN. Unfortunately, we currently do not know if there are changes in GAD mRNA and protein levels in ERα-expressing cells within the peri-PVN region following ER agonist treatment in the presence or absence of DEX and/or a stressor. Nor do we know if there are direct projections of these neuronal populations to the PVN. Unfortunately, such studies are technically difficult given the close association of peri-PVN neurons to the PVN proper. It should be mentioned that studies have indicated positive and negative effects of systemically administered estradiol on restraint-induced c-fos expression in the PVN (Lund et al., 2004; Figueiredo et al., 2007). This may be due to extrinsic actions of estradiol on the HPA axis through either ERα or ERβ. However, similar to the results described here, local administration of estradiol to the PVN has been shown to augment c-fos expression (Lund et al., 2006).
While it appears from these studies that the inhibitory effects of estradiol on glucocorticoid negative feedback are mediated through ERα, we did observe that the activation of ERβ in and around the PVN results in a slight but significant suppression in HPA responsivity. DEX suppression was not impaired by ERβ activation. Therefore, it can be proposed that ERβ acts in an opposing fashion to increase glucocorticoid-mediated negative feedback. Further experiments would need to be done to establish a dose-response curve for DEX in the presence or absence of ER agonists. ERβ is expressed in a small number of CRH and AVP containing parvocellular PVN neurons, suggesting a potential direct effect on CRH and AVP transcription (Laflamme et al., 1998; Hrabovszky et al., 2004; Suzuki and Handa, 2005). Furthermore, ERβ has been shown to drive CRH and AVP promoter activity in vitro (Shapiro et al., 2000; Miller et al., 2004; Pak et al., 2007). Alternatively, ERβ may act peripherally as it is expressed within corticotrophs of the anterior pituitary and within the adrenal cortex (Mitchner et al., 1998; de Cremoux et al., 2008). However, our studies report decreased stress responsivity with local peri-PVN administration of an ERβ agonist, which argues against this possibility.
Impairment of glucocorticoid negative feedback is a key characteristic of major depressive disorder in humans (for reviews see; (Swaab et al., 2005; Bao et al., 2008)). Depressed patients have a marked reduction in ability to respond and adapt to a stressor, and exhibit increased neuropeptide levels in the PVN. Furthermore, in these individuals, DEX is unable to suppress the stimulatory actions of CRH (DEX/CRH test), in much the same fashion as those animals treated with estradiol and PPT and subjected to a restraint stress. Interestingly, normal healthy women have higher cortisol responses to a DEX/CRH test than normal healthy men, regardless of age (Heuser et al., 1994). These data may provide a link between the higher incidence of depression and stress sensitivity observed in women.
CONCLUSION
In summary, estradiol directly impairs glucocorticoid-mediated negative feedback through a peri-PVN ERα dependent mechanism. These data suggest that the increased stress sensitivity seen in females might be due to decreased sensitivity to glucocorticoid-mediated HPA axis negative feedback.
Acknowledgments
USPHS: NIH/NINDS R01 NS039951.
Abbreviations
- ACTH
adrenocorticotropic hormone
- AVP
arginine vasopressin
- BnST
bed nucleus of the stria terminalis
- CBG
corticosterone binding globulin
- CORT
corticosterone
- CRH
corticotropin releasing hormone
- CSF
cerebrospinal fluid
- DAB
3,3′-diaminobenzidine
- DEX
dexamethasone
- DPN
diarylpropionitrile
- DST
dexamethasone suppression test
- EB
estradiol benzoate
- ERα
estrogen receptor alpha
- ERβ
estrogen receptor beta
- GAD
glutamic acid decarboxylase
- GR
glucocorticoid receptor
- HPA
hypothalamic-pituitary-adrenal
- IR
immunoreactivity
- MPOA
medial preoptic area
- MR
mineralocorticoid receptor
- OVX
ovariectomized
- PPT
propylpyrazoletriol
- PVN
paraventricular nucleus
- RBA
relative binding affinity
- RIA
radioimmunoassay
- SD
Sprague–Dawley
References
- Abe K, Critchlow V. Delayed feedback inhibition of stress-induced activation of pituitary-adrenal function: effects of varying dose, rate and duration of corticosterone administration and of telencephalon removal. Neuroendocrinology. 1980;31:349–354. doi: 10.1159/000123100. [DOI] [PubMed] [Google Scholar]
- Albeck DS, Hastings NB, McEwen BS. Effects of adrenalectomy and type I or type II glucocorticoid receptor activation on AVP and CRH mRNA in the rat hypothalamus. Brain Res Mol Brain Res. 1994;26:129–134. doi: 10.1016/0169-328x(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Angold A, Worthman CW. Puberty onset of gender differences in rates of depression: a developmental, epidemiologic and neuroendocrine perspective. J Affect Disord. 1993;29:145–158. doi: 10.1016/0165-0327(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Atkinson HC, Waddell BJ. Circadian variation in basal plasma corticosterone and adrenocorticotropin in the rat: sexual dimorphism and changes across the estrous cycle. Endocrinology. 1997;138:3842–3848. doi: 10.1210/endo.138.9.5395. [DOI] [PubMed] [Google Scholar]
- Aubry JM, Bartanusz V, Pagliusi S, Schulz P, Kiss JZ. Expression of ionotropic glutamate receptor subunit mRNAs by paraventricular corticotropin-releasing factor (CRF) neurons. Neurosci Lett. 1996;205:95–98. doi: 10.1016/0304-3940(96)12380-6. [DOI] [PubMed] [Google Scholar]
- Aubry JM, Gervasoni N, Osiek C, Perret G, Rossier MF, Bertschy G, Bondolfi G. The DEX/CRH neuroendocrine test and the prediction of depressive relapse in remitted depressed outpatients. J Psychiatr Res. 2007;41:290–294. doi: 10.1016/j.jpsychires.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Baid S, Nieman LK. Glucocorticoid excess and hypertension. Curr Hypertens Rep. 2004;6:493–499. doi: 10.1007/s11906-004-0046-0. [DOI] [PubMed] [Google Scholar]
- Bali B, Erdelyi F, Szabo G, Kovacs KJ. Visualization of stress-responsive inhibitory circuits in the GAD65-EGFP transgenic mice. Neurosci Lett. 2005;380:60–65. doi: 10.1016/j.neulet.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Bali B, Kovacs KJ. GABAergic control of neuropeptide gene expression in parvocellular neurons of the hypothalamic paraventricular nucleus. Eur J Neurosci. 2003;18:1518–1526. doi: 10.1046/j.1460-9568.2003.02877.x. [DOI] [PubMed] [Google Scholar]
- Bao AM, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev. 2008;57:531–553. doi: 10.1016/j.brainresrev.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Barden N. Implication of the hypothalamic-pituitary-adrenal axis in the physiopathology of depression. J Psychiatry Neurosci. 2004;29:185–193. [PMC free article] [PubMed] [Google Scholar]
- Bohus B, Strashimirov D. Localization and specificity of corticosteroid “feedback receptors” at the hypothalamo-hypophyseal level; comparative effects of various steroids implanted in the median eminence or the anterior pituitary of the rat. Neuroendocrinology. 1970;6:197–209. doi: 10.1159/000121924. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Szabo K, Tasker JG. Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J Neurosci. 1996;16:7151–7160. doi: 10.1523/JNEUROSCI.16-22-07151.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers G, Cullinan WE, Herman JP. Region-specific regulation of glutamic acid decarboxylase (GAD) mRNA expression in central stress circuits. J Neurosci. 1998;18:5938–5947. doi: 10.1523/JNEUROSCI.18-15-05938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury MJ, Akana SF, Dallman MF. Roles of type I and II corticosteroid receptors in regulation of basal activity in the hypothalamo-pituitary-adrenal axis during the diurnal trough and the peak: evidence for a nonadditive effect of combined receptor occupation. Endocrinology. 1994;134:1286–1296. doi: 10.1210/endo.134.3.8119168. [DOI] [PubMed] [Google Scholar]
- Buckley TM, Schatzberg AF. On the interactions of the hypothalamic-pituitary-adrenal (HPA) axis and sleep: normal HPA axis activity and circadian rhythm, exemplary sleep disorders. J Clin Endocrinol Metab. 2005;90:3106–3114. doi: 10.1210/jc.2004-1056. [DOI] [PubMed] [Google Scholar]
- Burgess LH, Handa RJ. Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion, and glucocorticoid receptor-mediated functions in female rats. Endocrinology. 1992;131:1261–1269. doi: 10.1210/endo.131.3.1324155. [DOI] [PubMed] [Google Scholar]
- Burgess LH, Handa RJ. Hormonal regulation of androgen receptor mRNA in the brain and anterior pituitary gland of the male rat. Brain Res Mol Brain Res. 1993;19:31–38. doi: 10.1016/0169-328x(93)90145-f. [DOI] [PubMed] [Google Scholar]
- Carey MP, Deterd CH, de Koning J, Helmerhorst F, de Kloet ER. The influence of ovarian steroids on hypothalamic-pituitary-adrenal regulation in the female rat. J Endocrinol. 1995;144:311–321. doi: 10.1677/joe.0.1440311. [DOI] [PubMed] [Google Scholar]
- Carroll BJ, Curtis GC. Neuroendocrine identification of depressed patients. Aust N Z J Psychiatry. 1976;10:13–20. doi: 10.3109/00048677609159480. [DOI] [PubMed] [Google Scholar]
- Cole RL, Sawchenko PE. Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. J Neurosci. 2002;22:959–969. doi: 10.1523/JNEUROSCI.22-03-00959.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow V, Liebelt RA, Bar-Sela M, Mountcastle W, Lipscomb HS. Sex difference in resting pituitary-adrenal function in the rat. Am J Physiol. 1963;205:807–815. doi: 10.1152/ajplegacy.1963.205.5.807. [DOI] [PubMed] [Google Scholar]
- Cullinan WE. Evidence for a PVN site of action for gamma aminobutyric acid in the regulatory control of the rat stress axis. Physiologist. 1998;41:353. [Google Scholar]
- Cullinan WE, Helmreich DL, Watson SJ. Fos expression in forebrain afferents to the hypothalamic paraventricular nucleus following swim stress. J Comp Neurol. 1996;368:88–99. doi: 10.1002/(SICI)1096-9861(19960422)368:1<88::AID-CNE6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Curran-Rauhut MA, Petersen SL. Regulation of glutamic acid decarboxylase 65 and 67 gene expression by ovarian steroids: identification of two functionally distinct populations of GABA neurones in the preoptic area. J Neuroendocrinol. 2002;14:310–317. doi: 10.1046/j.1365-2826.2002.00780.x. [DOI] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Cascio CS, Darlington DN, Jacobson L, Levin N. Regulation of ACTH secretion: variations on a theme of B. Recent Prog Horm Res. 1987;43:113–173. doi: 10.1016/b978-0-12-571143-2.50010-1. [DOI] [PubMed] [Google Scholar]
- Darlington DN, Miyamoto M, Keil LC, Dallman MF. Paraventricular stimulation with glutamate elicits bradycardia and pituitary responses. Am J Physiol. 1989;256:R112–R119. doi: 10.1152/ajpregu.1989.256.1.R112. [DOI] [PubMed] [Google Scholar]
- Davidson JM, Feldman S. Effects of extrahypothalamic dexamethasone implants on the pituitary-adrenal system. Acta Endocrinol. 1967;55:240–246. doi: 10.1530/acta.0.0550240. [DOI] [PubMed] [Google Scholar]
- de Cremoux P, Rosenberg D, Goussard J, Bremont-Weil C, Tissier F, Tran-Perennou C, Groussin L, Bertagna X, Bertherat J, Raffin-Sanson ML. Expression of progesterone and estradiol receptors in normal adrenal cortex, adrenocortical tumors, and primary pigmented nodular adrenocortical disease. Endocr Relat Cancer. 2008;15:465–474. doi: 10.1677/ERC-07-0081. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology. 2005;146:4292–4301. doi: 10.1210/en.2005-0610. [DOI] [PubMed] [Google Scholar]
- DiMicco JA, Stotz-Potter EH, Monroe AJ, Morin SM. Role of the dorsomedial hypothalamus in the cardiovascular response to stress. Clin Exp Pharmacol Physiol. 1996;23:171–176. doi: 10.1111/j.1440-1681.1996.tb02592.x. [DOI] [PubMed] [Google Scholar]
- Ehlert U, Gaab J, Heinrichs M. Psychoneuroendocrinological contributions to the etiology of depression, posttraumatic stress disorder, and stress-related bodily disorders: the role of the hypothalamus-pituitary-adrenal axis. Biol Psychol. 2001;57:141–152. doi: 10.1016/s0301-0511(01)00092-8. [DOI] [PubMed] [Google Scholar]
- Ferrini M, Lima A, De Nicola AF. Estradiol abolishes autologous down regulation of glucocorticoid receptors in brain. Life Sci. 1995;57:2403–2412. doi: 10.1016/0024-3205(95)02236-3. [DOI] [PubMed] [Google Scholar]
- Figueiredo HF, Dolgas CM, Herman JP. Stress activation of cortex and hippocampus is modulated by sex and stage of estrus. Endocrinology. 2002;143:2534–2540. doi: 10.1210/endo.143.7.8888. [DOI] [PubMed] [Google Scholar]
- Figueiredo HF, Ulrich-Lai YM, Choi DC, Herman JP. Estrogen potentiates adrenocortical responses to stress in female rats. Am J Physiol Endocrinol Metab. 2007;292:E1173–E1182. doi: 10.1152/ajpendo.00102.2006. [DOI] [PubMed] [Google Scholar]
- Handa RJ, Burgess LH, Kerr JE, O’Keefe JA. Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm Behav. 1994;28:464–476. doi: 10.1006/hbeh.1994.1044. [DOI] [PubMed] [Google Scholar]
- Hassan AH, Patchev VK, von Rosenstiel P, Holsboer F, Almeida OF. Plasticity of hippocampal corticosteroid receptors during aging in the rat. FASEB J. 1999;13:115–122. doi: 10.1096/fasebj.13.1.115. [DOI] [PubMed] [Google Scholar]
- Herbison AE. Estrogen positive feedback to gonadotropin-releasing hormone (GnRH) neurons in the rodent: the case for the rostral periventricular area of the third ventricle (RP3V) Brain Res Rev. 2008;57:277–287. doi: 10.1016/j.brainresrev.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE, Fenelon VS. Estrogen regulation of GABAA receptor subunit mRNA expression in preoptic area and bed nucleus of the stria terminalis of female rat brain. J Neurosci. 1995;15:2328–2337. doi: 10.1523/JNEUROSCI.15-03-02328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Mueller NK, Figueiredo H. Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Ann N Y Acad Sci. 2004;1018:35–45. doi: 10.1196/annals.1296.004. [DOI] [PubMed] [Google Scholar]
- Herman JP, Tasker JG, Ziegler DR, Cullinan WE. Local circuit regulation of paraventricular nucleus stress integration: glutamate-GABA connections. Pharmacol Biochem Behav. 2002;71:457–468. doi: 10.1016/s0091-3057(01)00681-5. [DOI] [PubMed] [Google Scholar]
- Heuser IJ, Gotthardt U, Schweiger U, Schmider J, Lammers CH, Dettling M, Holsboer F. Age-associated changes of pituitary-adrenocortical hormone regulation in humans: importance of gender. Neurobiol Aging. 1994;15:227–231. doi: 10.1016/0197-4580(94)90117-1. [DOI] [PubMed] [Google Scholar]
- Hinz B, Hirschelmann R. Rapid non-genomic feedback effects of glucocorticoids on CRF-induced ACTH secretion in rats. Pharmacol Rev. 2000;17:1273–1277. doi: 10.1023/a:1026499604848. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Kallo I, Steinhauser A, Merchenthaler I, Coen CW, Petersen SL, Liposits Z. Estrogen receptor-beta in oxytocin and vasopressin neurons of the rat and human hypothalamus: immunocytochemical and in situ hybridization studies. J Comp Neurol. 2004;473:315–333. doi: 10.1002/cne.20127. [DOI] [PubMed] [Google Scholar]
- Ising M, Horstmann S, Kloiber S, Lucae S, Binder EB, Kern N, Kunzel HE, Pfennig A, Uhr M, Holsboer F. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression—a potential biomarker? Biol Psychiatry. 2007;62:47–54. doi: 10.1016/j.biopsych.2006.07.039. [DOI] [PubMed] [Google Scholar]
- Jingami H, Matsukura S, Numa S, Imura H. Effects of adrenalectomy and dexamethasone administration on the level of preprocorticotropin-releasing factor messenger ribonucleic acid (mRNA) in the hypothalamus and adrenocorticotropin/beta-lipotropin precursor mRNA in the pituitary in rats. Endocrinology. 1985;117:1314–1320. doi: 10.1210/endo-117-4-1314. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschbaum C, Schommer N, Federenko I, Gaab J, Neumann O, Oellers M, Rohleder N, Untiedt A, Hanker J, Pirke KM, Hellhammer DH. Short-term estradiol treatment enhances pituitary-adrenal axis and sympathetic responses to psychosocial stress in healthy young men. J Clin Endocrinol Metab. 1996;81:3639–3643. doi: 10.1210/jcem.81.10.8855815. [DOI] [PubMed] [Google Scholar]
- Kitay JI. Pituitary-adrenal function in the rat after gonadectomy and gonadal hormone replacement. Endocrinology. 1963;73:253–260. doi: 10.1210/endo-73-2-253. [DOI] [PubMed] [Google Scholar]
- Koblinsky M, Beato M, Kalimi M, Feigelson P. Glucocorticoid-binding proteins of rat liver cytosol. II. Physical characterization and properties of the binding proteins. J Biol Chem. 1972;247:7897–7904. [PubMed] [Google Scholar]
- Kolanowski J, Pizarro MA. Critical evaluation of competitive protein-binding radioassay for cortisol. Ann Endocrinol. 1969;30(Suppl):177–182. [PubMed] [Google Scholar]
- Kornstein SG. Gender differences in depression: implications for treatment. J Clin Psychiatry. 1997;58(Suppl 15):12–18. [PubMed] [Google Scholar]
- Kovacs K, Kiss JZ, Makara GB. Glucocorticoid implants around the hypothalamic paraventricular nucleus prevent the increase of corticotropin-releasing factor and arginine vasopressin immunostaining induced by adrenalectomy. Neuroendocrinology. 1986;44:229–234. doi: 10.1159/000124650. [DOI] [PubMed] [Google Scholar]
- Kovacs KJ, Foldes A, Sawchenko PE. Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J Neurosci. 2000;20:3843–3852. doi: 10.1523/JNEUROSCI.20-10-03843.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs KJ, Makara GB. Corticosterone and dexamethasone act at different brain sites to inhibit adrenalectomy-induced adrenocorticotropin hypersecretion. Brain Res. 1988;474:205–210. doi: 10.1016/0006-8993(88)90435-0. [DOI] [PubMed] [Google Scholar]
- Kovacs KJ, Mezey E. Dexamethasone inhibits corticotropin-releasing factor gene expression in the rat paraventricular nucleus. Neuroendocrinology. 1987;46:365–368. doi: 10.1159/000124846. [DOI] [PubMed] [Google Scholar]
- Kovacs KJ, Miklos IH, Bali B. GABAergic mechanisms constraining the activity of the hypothalamo-pituitary-adrenocortical axis. Ann N Y Acad Sci. 2004;1018:466–476. doi: 10.1196/annals.1296.057. [DOI] [PubMed] [Google Scholar]
- Kudielka BM, Kirschbaum C. Sex differences in HPA axis responses to stress: a review. Biol Psychol. 2005;69:113–132. doi: 10.1016/j.biopsycho.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Nappi RE, Drolet G, Labrie C, Rivest S. Expression and neuropeptidergic characterization of estrogen receptors (ERalpha and ERbeta) throughout the rat brain: anatomical evidence of distinct roles of each subtype. J Neurobiol. 1998;36:357–378. doi: 10.1002/(sici)1097-4695(19980905)36:3<357::aid-neu5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Levin MC, Sawchenko PE. Neuropeptide co-expression in the magnocellular neurosecretory system of the female rat: evidence for differential modulation by estrogen. Neuroscience. 1993;54:1001–1018. doi: 10.1016/0306-4522(93)90591-3. [DOI] [PubMed] [Google Scholar]
- Lund TD, Hinds LR, Handa RJ. The androgen 5alpha-dihydrotestosterone and its metabolite 5alpha-androstan-3beta, 17beta-diol inhibit the hypothalamo-pituitary-adrenal response to stress by acting through estrogen receptor beta-expressing neurons in the hypothalamus. J Neurosci. 2006;26:1448–1456. doi: 10.1523/JNEUROSCI.3777-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund TD, Munson DJ, Haldy ME, Handa RJ. Androgen inhibits, while oestrogen enhances, restraint-induced activation of neuropeptide neurones in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol. 2004;16:272–278. doi: 10.1111/j.0953-8194.2004.01167.x. [DOI] [PubMed] [Google Scholar]
- Lund TD, Rovis T, Chung WC, Handa RJ. Novel actions of estrogen receptor-beta on anxiety-related behaviors. Endocrinology. 2005;146:797–807. doi: 10.1210/en.2004-1158. [DOI] [PubMed] [Google Scholar]
- Lurie S, Kuhn C, Bartolome J, Schanberg S. Differential sensitivity to dexamethasone suppression in an animal model of the DST. Biol Psychiatry. 1989;26:26–34. doi: 10.1016/0006-3223(89)90005-x. [DOI] [PubMed] [Google Scholar]
- Ma XM, Aguilera G. Differential regulation of corticotropin-releasing hormone and vasopressin transcription by glucocorticoids. Endocrinology. 1999;140:5642–5650. doi: 10.1210/endo.140.12.7214. [DOI] [PubMed] [Google Scholar]
- Mataradze GD, Kurabekova RM, Rozen VB. The role of sex steroids in the formation of sex-differentiated concentrations of corticosteroid-binding globulin in rats. J Endocrinol. 1992;132:235–240. doi: 10.1677/joe.0.1320235. [DOI] [PubMed] [Google Scholar]
- McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88:91–124. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick CM, Linkroum W, Sallinen BJ, Miller NW. Peripheral and central sex steroids have differential effects on the HPA axis of male and female rats. Stress. 2002;5:235–247. doi: 10.1080/1025389021000061165. [DOI] [PubMed] [Google Scholar]
- Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- Miklos IH, Kovacs KJ. GABAergic innervation of corticotropin-releasing hormone (CRH)-secreting parvocellular neurons and its plasticity as demonstrated by quantitative immunoelectron microscopy. Neuroscience. 2002;113:581–592. doi: 10.1016/s0306-4522(02)00147-1. [DOI] [PubMed] [Google Scholar]
- Miller WJ, Suzuki S, Miller LK, Handa R, Uht RM. Estrogen receptor (ER)beta isoforms rather than ERalpha regulate corticotropin-releasing hormone promoter activity through an alternate pathway. J Neurosci. 2004;24:10628–10635. doi: 10.1523/JNEUROSCI.5540-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchner NA, Garlick C, Ben-Jonathan N. Cellular distribution and gene regulation of estrogen receptors alpha and beta in the rat pituitary gland. Endocrinology. 1998;139:3976–3983. doi: 10.1210/endo.139.9.6181. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Cole NB, Greenberger V, Segal M. Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci. 1998;18:2550–2559. doi: 10.1523/JNEUROSCI.18-07-02550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochedalski T, Subburaju S, Wynn PC, Aguilera G. Interaction between oestrogen and oxytocin on hypothalamic-pituitary-adrenal axis activity. J Neuroendocrinol. 2007;19:189–197. doi: 10.1111/j.1365-2826.2006.01525.x. [DOI] [PubMed] [Google Scholar]
- Pace TW, Spencer RL. Disruption of mineralocorticoid receptor function increases corticosterone responding to a mild, but not moderate, psychological stressor. Am J Physiol Endocrinol Metab. 2005;288:E1082–E1088. doi: 10.1152/ajpendo.00521.2004. [DOI] [PubMed] [Google Scholar]
- Pak T, Chung W, Handa R. Estrogen receptor-beta mediates DHT-induced stimulation of the arginine vasopressin promoter in neuronal cells. Endocrinology. 2007;148:33781–33782. doi: 10.1210/en.2007-0086. [DOI] [PubMed] [Google Scholar]
- Pasquali R, Vicennati V, Cacciari M, Pagotto U. The hypothalamic-pituitary-adrenal axis activity in obesity and the metabolic syndrome. Ann N Y Acad Sci. 2006;1083:111–128. doi: 10.1196/annals.1367.009. [DOI] [PubMed] [Google Scholar]
- Patchev VK, Almeida OF. Gonadal steroids exert facilitating and “buffering” effects on glucocorticoid-mediated transcriptional regulation of corticotropin-releasing hormone and corticosteroid receptor genes in rat brain. J Neurosci. 1996;16:7077–7084. doi: 10.1523/JNEUROSCI.16-21-07077.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patchev VK, Hayashi S, Orikasa C, Almeida OF. Implications of estrogen-dependent brain organization for gender differences in hypothalamo-pituitary-adrenal regulation. FASEB J. 1995;9:419–423. doi: 10.1096/fasebj.9.5.7896013. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- Peiffer A, Barden N. Estrogen-induced decrease of glucocorticoid receptor messenger ribonucleic acid concentration in rat anterior pituitary gland. Mol Endocrinol. 1987;1:435–440. doi: 10.1210/mend-1-6-435. [DOI] [PubMed] [Google Scholar]
- Peiffer A, Veilleux S, Barden N. Antidepressant and other centrally acting drugs regulate glucocorticoid receptor messenger RNA levels in rat brain. Psychoneuroendocrinology. 1991;16:505–515. doi: 10.1016/0306-4530(91)90034-q. [DOI] [PubMed] [Google Scholar]
- Rachman IM, Unnerstall JR, Pfaff DW, Cohen RS. Estrogen alters behavior and forebrain c-fos expression in ovariectomized rats subjected to the forced swim test. Proc Natl Acad Sci U S A. 1998;95:13941–13946. doi: 10.1073/pnas.95.23.13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratka A, Sutanto W, Bloemers M, de Kloet ER. On the role of brain mineralocorticoid (type I) and glucocorticoid (type II) receptors in neuroendocrine regulation. Neuroendocrinology. 1989;50:117–123. doi: 10.1159/000125210. [DOI] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Reul JM, van den Bosch FR, de Kloet ER. Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: functional implications. J Endocrinol. 1987;115:459–467. doi: 10.1677/joe.0.1150459. [DOI] [PubMed] [Google Scholar]
- Roland BL, Sawchenko PE. Local origins of some GABAergic projections to the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J Comp Neurol. 1993;332:123–143. doi: 10.1002/cne.903320109. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE. Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in the paraventricular nucleus. Brain Res. 1987;403:213–223. doi: 10.1016/0006-8993(87)90058-8. [DOI] [PubMed] [Google Scholar]
- Seale JV, Wood SA, Atkinson HC, Harbuz MS, Lightman SL. Gonadal steroid replacement reverses gonadectomy-induced changes in the corticosterone pulse profile and stress-induced hypothalamic-pituitary-adrenal axis activity of male and female rats. J Neuroendocrinol. 2004;16:989–998. doi: 10.1111/j.1365-2826.2004.01258.x. [DOI] [PubMed] [Google Scholar]
- Sencar-Cupovic I, Milkovic S. The development of sex differences in the adrenal morphology and responsiveness in stress of rats from birth to the end of life. Mech Ageing Dev. 1976;5:1–9. doi: 10.1016/0047-6374(76)90002-6. [DOI] [PubMed] [Google Scholar]
- Shapiro RA, Xu C, Dorsa DM. Differential transcriptional regulation of rat vasopressin gene expression by estrogen receptor alpha and beta. Endocrinology. 2000;141:4056–4064. doi: 10.1210/endo.141.11.7796. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Spencer RL, Kim PJ, Kalman BA, Cole MA. Evidence for mineralocorticoid receptor facilitation of glucocorticoid receptor-dependent regulation of hypothalamic-pituitary-adrenal axis activity. Endocrinology. 1998;139:2718–2726. doi: 10.1210/endo.139.6.6029. [DOI] [PubMed] [Google Scholar]
- Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J Med Chem. 2000;43:4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- Stern JE, Zhang W. Preautonomic neurons in the paraventricular nucleus of the hypothalamus contain estrogen receptor beta. Brain Res. 2003;975:99–109. doi: 10.1016/s0006-8993(03)02594-0. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Handa RJ. Estrogen receptor-beta, but not estrogen receptor-alpha, is expressed in prolactin neurons of the female rat paraventricular and supraoptic nuclei: comparison with other neuropeptides. J Comp Neurol. 2005;484:28–42. doi: 10.1002/cne.20457. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Lund TD, Price RH, Handa RJ. Sex differences in the hypothalamo–pituitary–adrenal axis: novel roles for androgen and estrogen receptors. Recent Res Dev Endocr. 2001:69–86. [Google Scholar]
- Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. 2005;4:141–194. doi: 10.1016/j.arr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Simmons DM. Differential steroid hormone and neural influences on peptide mRNA levels in CRH cells of the paraventricular nucleus: a hybridization histochemical study in the rat. J Comp Neurol. 1989;285:413–435. doi: 10.1002/cne.902850402. [DOI] [PubMed] [Google Scholar]
- Tait AS, Butts CL, Sternberg EM. The role of glucocorticoids and progestins in inflammatory, autoimmune, and infectious disease. J Leukoc Biol. 2008;84:924–931. doi: 10.1189/jlb.0208104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thase ME, Reynolds CF, III, Frank E, Simons AD, McGeary J, Fasiczka AL, Garamoni GG, Jennings JR, Kupfer DJ. Do depressed men and women respond similarly to cognitive behavior therapy? Am J Psychiatry. 1994;151:500–505. doi: 10.1176/ajp.151.4.500. [DOI] [PubMed] [Google Scholar]
- Tomlinson JW, Stewart PM. Modulation of glucocorticoid action and the treatment of type-2 diabetes. Best Pract Res Clin Endocrinol Metab. 2007;21:607–619. doi: 10.1016/j.beem.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Turner BB. Sex difference in glucocorticoid binding in rat pituitary is estrogen dependent. Life Sci. 1990;46:1399–1406. doi: 10.1016/0024-3205(90)90340-w. [DOI] [PubMed] [Google Scholar]
- Turner BB. Sex differences in the binding of type I and type II corticosteroid receptors in rat hippocampus. Brain Res. 1992;581:229–236. doi: 10.1016/0006-8993(92)90712-i. [DOI] [PubMed] [Google Scholar]
- Uht RM, Anderson CM, Webb P, Kushner PJ. Transcriptional activities of estrogen and glucocorticoid receptors are functionally integrated at the AP-1 response element. Endocrinology. 1997;138:2900–2908. doi: 10.1210/endo.138.7.5244. [DOI] [PubMed] [Google Scholar]
- Uht RM, McKelvy JF, Harrison RW, Bohn MC. Demonstration of glucocorticoid receptor-like immunoreactivity in glucocorticoid-sensitive vasopressin and corticotropin-releasing factor neurons in the hypothalamic paraventricular nucleus. J Neurosci Res. 1988;19:405–411. 468–469. doi: 10.1002/jnr.490190404. [DOI] [PubMed] [Google Scholar]
- Varghese FP, Brown ES. The hypothalamic-pituitary-adrenal axis in major depressive disorder: a brief primer for primary care physicians. Prim Care Companion J Clin Psychiatry. 2001;3:151–155. doi: 10.4088/pcc.v03n0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson S, Gallagher P, Smith MS, Ferrier IN, Young AH. The dex/CRH test—is it better than the DST? Psychoneuroendocrinology. 2006;31:889–894. doi: 10.1016/j.psyneuen.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Weekes NY, Lewis RS, Goto SG, Garrison-Jakel J, Patel F, Lupien S. The effect of an environmental stressor on gender differences on the awakening cortisol response. Psychoneuroendocrinology. 2008;33:766–772. doi: 10.1016/j.psyneuen.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Weissman MM, Bland R, Joyce PR, Newman S, Wells JE, Wittchen HU. Sex differences in rates of depression: cross-national perspectives. J Affect Disord. 1993;29:77–84. doi: 10.1016/0165-0327(93)90025-f. [DOI] [PubMed] [Google Scholar]
- Whitworth JA, Williamson PM, Mangos G, Kelly JJ. Cardiovascular consequences of cortisol excess. Vasc Health Risk Manag. 2005;1:291–299. doi: 10.2147/vhrm.2005.1.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmaier EP, Dallman MF. The effects of corticotropin-releasing factor on adrenocorticotropin secretion from perifused pituitaries in vitro: rapid inhibition by glucocorticoids. Endocrinology. 1984;115:2368–2374. doi: 10.1210/endo-115-6-2368. [DOI] [PubMed] [Google Scholar]
- Young EA, Altemus M, Parkison V, Shastry S. Effects of estrogen antagonists and agonists on the ACTH response to restraint stress in female rats. Neuropsychopharmacology. 2001;25:881–891. doi: 10.1016/S0893-133X(01)00301-3. [DOI] [PubMed] [Google Scholar]
- Yukhananov RY, Handa RJ. Alterations in kappa opioid receptor mRNA levels in the paraventricular nucleus of the hypothalamus by stress and sex steroids. Neuroreport. 1996;7:1690–1694. doi: 10.1097/00001756-199607080-00033. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Herman JP. Local integration of glutamate signaling in the hypothalamic paraventricular region: regulation of glucocorticoid stress responses. Endocrinology. 2000;141:4801–4804. doi: 10.1210/endo.141.12.7949. [DOI] [PubMed] [Google Scholar]