

Abstract
Although EGFR is a highly sought-after drug target, inhibitor resistance remains a challenge. As an alternative strategy for kinase inhibition, we sought to explore whether allosteric activation mechanisms could effectively be disrupted. The kinase domain of EGFR forms an atypical asymmetric dimer via head-to-tail interactions and serves as a requisite for kinase activation. The kinase dimer interface is primarily formed by the H-helix derived from one kinase monomer and the small lobe of the second monomer. We hypothesized that a peptide designed to resemble the binding surface of the H-helix may serve as an effective disruptor of EGFR dimerization and activation. A library of constrained peptides was designed to mimic the H-helix of the kinase domain and interface side chains were optimized using molecular modeling. Peptides were constrained using peptide “stapling” to structurally reinforce an alpha-helical conformation. Peptide stapling was demonstrated to notably enhance cell permeation of an H-helix derived peptide termed EHBI2. Using cell-based assays, EHBI2 was further shown to significantly reduce EGFR activity as measured by EGFR phosphorylation and phosphorylation of the downstream signaling substrate Akt. To our knowledge, this is the first H-helix-based compound targeting the asymmetric interface of the kinase domain that can successfully inhibit EGFR activation and signaling. This study presents a novel, alternative targeting site for allosteric inhibition of EGFR.
Keywords: constrained peptide, EGFR, allosteric inhibitor, kinase dimerization, stapled peptide
Graphical abstract
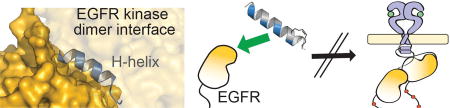
1. Introduction
Human epidermal growth factor receptor (EGFR/HER1/ErbB1) is a member of the receptor tyrosine kinase family and contains an extracellular receptor, a transmembrane helix, a juxtamembrane region and an intracellular kinase domain [1, 2]. Dimerization of EGFR is required for kinase activation, and this can be accomplished by homodimerization or heterodimerization with other members of the ErbB family including ErbB2, ErbB3 and ErbB4 [3]. Ligand-stimulated activation by growth factors such as epidermal growth factor (EGF) induces a significant conformational change, leading to dimerization of the receptor (Figure 1a) [4]. Upon receptor dimerization, the intracellular juxtamembrane region and kinase domains also dimerize [4–7] and one tyrosine kinase domain acts as an allosteric “activator” of the partner tyrosine kinase domain, which functions as the “receiver” [7]. While many kinases dimerize in a symmetrical head-to-head fashion and require phosphorylation of the activation loop for activation, EGFR is not dependent on activation loop phosphorylation but rather requires a distinct asymmetric dimerization interface for kinase activation [4, 7]. In the case of the EGFR kinase dimer, the C-lobe of the activator kinase interacts with the N-lobe of the receiver kinase to form an asymmetric interface and this conformation is required for receptor activation [7, 8]. As part of the interface, the C-lobe of the activator kinase forms multiple contacts derived from the H-helix including residues M945, V948, and M952 to form a stable dimer interface with the N-lobe of the receiver kinase [7]. In an active state, the tyrosine-rich C-terminals of EGFR are phosphorylated and these phospho sites subsequently serve as docking sites for proteins bearing domains such as Src Homology 2 (SH2) or Phosphotyrosine-Binding Domain (PTB) to promote downstream signaling [7, 9, 10].
Figure 1. Targeting disruption of EGFR activation.

a) Ligand-dependent activation of EGFR results in dimerization through multiple interfaces including the extracellular ligand-binding domain, intracellular juxtamembrane segments and intracellular tyrosine kinase domains. The kinase domains form an asymmetric dimer where one tyrosine kinase domain (“activator”) allosterically activates the partner tyrosine kinase domain (“receiver”). The structure was rendered using PyMOL X11 (PDB 2GS6). b) An alignment of the top hits produced from a BLASTP search of the EGFR protein sequence surrounding the H-helix (residues 940–954; accession number P00533). c) A scheme for the design and synthesis of stapled ErbB H-helix Based Inhibitor (EHBI) peptides. d) A peptide mimic of the EGFR H-helix was designed as a strategy to block asymmetric kinase dimerization and inhibit EGFR activation.
Since EGFR is implicated in a variety of diseases including cancer, multiple strategies for inhibiting EGFR activation were previously developed and include blocking growth factor binding, obstructing extracellular receptor-receptor dimerization, or preventing ATP-binding by the kinase domain [11]. For example, gefitinib (Iressa) and erlotinib (Tarceva) are quinazoline derivatives that compete with ATP binding at the kinase domain [12, 13]. In contrast, cetuximab (Erbitux) is a monoclonal antibody that binds to the extracellular region of EGFR, leading to inhibited ligand binding while also impeding the required structural rearrangement of the extracellular domain so as to inhibit receptor dimerization [14]. Many alternative inhibitory strategies have been explored including stapled peptides that target the intracellular juxtamembrane segment of EGFR [6] and constrained beta-loop peptide mimics of the EGFR dimerization arm [15, 16]. In terms of the kinase dimer interface, the human mitogen-inducible gene 6 protein (MIG-6/ERRFI1/RALT/ GENE-33), initially discovered as a potential cell-cycle marker for G1 phase in HL-60 cells [17], has been identified as a negative regulator of EGFR signaling [18]. MIG6 binds to the interface on the C-lobe of EGFR and extends to occupy the substrate-binding site and interact with the activation loop [19, 20]. EGFR can phosphorylate MIG6 at Y394 and Y395, and phosphorylation results in a dual mechanism of inhibition because it continues to occupy the substrate site after phosphorylation, occluding the kinase dimer interface and thereby preventing asymmetric dimerization [20, 21]. With this duality, various MIG6-derived peptides or protein segments have been reported to act as EGFR substrates as well as allosteric inhibitors [19, 20, 22]. As an alternative approach for allosteric inhibition, we wanted to determine whether designing an inhibitor based on the sequence of the H-helix of the kinase domain could target EGFR dimerization, and whether this site alone would suffice to inhibit EGFR activation.
2. Results and Discussion
In order to design a disruptor targeting the protein-protein interface of the kinase dimer, we focused on a key component of the kinase dimer interface: the H-helix of the kinase domain (940–950, TIDVYMIMVKC). This helix, along with the C-terminal flanking residues (951–954, WMID), is evolutionarily conserved in the ErbB family members yet is divergent amongst the kinase superfamily (Figure 1b). A BLASTP search and alignment of this sequence shows significant similarity between the ErbB family members (E-values ≤ 26.0 E-9, Ident.: ≥ 93%). However, the H-helix sequence is divergent amongst the kinase superfamily where the next closest comparisons are Platelet Derived Growth Factor Receptor alpha (PDGFRA) and Janus Kinase 2 (JAK2) with E-values ≥ 7.0 and identities ≤ 80%. Further, since the majority of kinases do not require the H-helix at the dimerization interface, a mimic of this helix may serve as a unique handle for selective targeting of EGFR inhibition. In order to improve the inhibitory properties of a H-helix peptide mimic, molecular modeling and a structural constraint would be applied via peptide “stapling” (Figure 1c) [23]. Ultimately, we hypothesized that disruption of the dimerization interface with an H-helix mimic could prevent allosteric activation of the tyrosine kinase domains and thereby sequester EGFR in an inactive state (Figure 1d).
In order to first establish the optimal position for placement of the hydrocarbon staple, the H-helix (residues 940–954) was synthesized with the staple introduced at various positions on the non-binding, solvent-exposed face of the helix so as to stabilize individual helical turns. Activity of this preliminary library (Figure 2, peptides 1–8) was measured in cells by monitoring EGFR activation after EGF stimulation in the presence of each compound. A cell-based activity assay was used to screen for phosphorylation of EGFR Y1068 and Akt detected by western blotting were used as a readout for EGFR activity. The initial library had notably limited water solubility, and thus phosphorylation results were inconclusive. In order to increase hydrophilicity, a limited number of peptides were modified to contain 3 PEG units at the N-terminus (Figure 2, peptides 9–11). Although water solubility was improved, the peptides did not demonstrate inhibitory effects on EGFR activation (Figure 3a).
Figure 2. H-helix-derived peptide sequences.

The selected EGFR protein sequence (residues 940–954, accession number P00533) is presented as a helical wheel numbered as residues 1–15 (N- to C-terminus). Highlighted in light blue are residues that interact with the EGFR kinase domain (PDB 2GS6). In silico designed peptides (12–18) contain amino acid substitutions that were predicted to have relatively favorable target binding as compared to the wild-type sequence. Additional amino acid substitutions (K) were introduced to the anticipated solvent face of the peptide to improve solubility. The helical wheel was generated from the following site: http://kael.org/helical.htm. Nle = norleucine.
Figure 3. Inhibition of EGFR activation and downstream signaling using H-helix-derived peptides.

a) PEG3-conjugated H-helix based peptides were tested for their ability to inhibit EGFR phosphorylation as a marker of EGFR inhibition using MDA-MB-231 cells. The ratio of phosphorylated EGFR (pY1068) to the loading control α-tubulin was measured by western blot analysis. Peptides 14 and 17 were found to downregulate phosphorylation. Densitometry is shown in the graph above the western blots using 3 independent experiments performed in triplicate. b) A concentration-dependent cell-based assay was performed using Peptide 17 (EHBI2) and its corresponding scramble control peptide. EHBI2 demonstrated dose-dependent inhibition of EGFR activation while its scramble control had no effect at the same concentrations. Densitometry data represents n=3 experiments performed in triplicate. c) To assess effects on downstream signaling of EGFR, total Akt, phosphorylated Akt (S473) and GAPDH were measured by western blotting. Densitometry data was measured from a triplicate experiment. EHBI2 caused downregulation of Akt phosphorylation, but not its scramble control. One-way ANOVA and a Tukey’s post hoc test (95% confidence interval) were performed on all densitometry data sets. The p-values are as follows: *** indicates p < 0.001, ** indicates p < 0.01, and * indicates p < 0.05 as compared to the positive control (+EGF). Error bars represent standard error of the mean.
To determine whether H-helix analogs could be computationally designed, we used two widely available software packages for peptide and protein design, namely Rosetta [24] and FoldX [25, 26]. We used the mutational scanning procedures implemented in the software to estimate the binding of H-helix with mutations at M945, V948 and M952. We focused on these three positions in the H-helix because these residues are part of the dimer interface and were previously shown to be important for dimerization [7]. Specifically, the programs will fix the backbone structure of the protein complex and exhaustively explore all possible natural amino acids at the specified positions. An empirically determined energy function score for each mutation was reported as a stability measurement of the resultant model (see Methods). Computational mutation scanning indicates that the wild-type sequence, in general, has favorable binding free energy as compared to the modified forms (Supplementary Tables 1–2). However, since the wild-type peptide sequence had no detectable inhibitory properties on EGFR activation, binding free energy values calculated from both programs were rank ordered and only favorable mutations predicted by both Rosetta and FoldX were selected. We then manually inspected the design structures by considering side chain properties (basic, acidic, polar, non-polar and rigidity versus flexibility introduced into the peptide backbone) as well as the interactions, contacts and clashes mediated by the indidivual amino acid substitions. After these procedures, substitutions such as M945Q, V948G and M952R were selected for the design of H-helix analogs and experimental testing.
Based on these predictions, H-helix analogs were synthesized by introducing amino acid substitutions into the sequence, TIDVYMIMVKCWMID (residues 940–954), to generate a library of in silico-optimized variants. A small panel of analogs were tested for inhibition of EGFR activation (Figure 3a, peptides 12–17 and Supplementary Figure S1). Peptides 14 and 17 (ErbB H-helix-Based Inhibitors 1 and 2, EHBI1 and EHBI2) significantly decreased EGFR phosphorylation as compared to the EGF-stimulated cells (lane 2, p<0.001), however, not surprisingly, the stapled peptides required a higher concentration for inhibition as comparison to the small molecule inhibitor gefitinib. Interestingly, these peptides only bore a single amino acid difference between one another at position 6: K or Q, in EHBI1 (KTIDRYKI(Nle)GKC*RID*) and EHBI2 (KTIDRYQI(Nle)GKC*RID*), respectively, in lieu of the native methionine (where stars represent the non-natural olefinic amino acid). These peptides also contained two additional amino acid changes from the parent sequence which are the same between EHBI1 and EHBI2: G at position 9 and R at position 13. Based on these preliminary findings, we focused on EHBI2 for further characterization. To further investigate dose-dependent inhibition of EGFR activation, phosphorylation assays of EGFR and downstream substrates were performed in cells (Figure 3b). A stapled scramble peptide was used as a control (EHBI2 scramble). Over a concentration range of 2.5 to 10 µM, phosphorylation of EGFR pY1068 was monitored by western blot analysis. While EHBI2 demonstrated dose-dependent inhibition of EGFR phosphorylation with maximal inhibition at the highest concentration tested (45% inhibition at 10 µM, p<0.01), the scramble control had no inhibitory effect. To ensure that EHBI2 did not affect total protein levels of EGFR in cells, total EGFR and α-tubulin were used as loading controls. No statistical difference was detected in protein levels for any of the treatment conditions tested (Supplementary Figure S2a). Thus, it appears that EHBI2 inhibits EGFR phosphorylation while not significantly reducing total expression levels of EGFR.
Next, we monitored downstream signaling of EGFR by monitoring phosphorylation of Akt at residue S473 (pAkt) [27] using western blotting (Figure 3c) [28]. A significant decrease in pAkt (71% reduction, p < 0.05) was observed when EGF-stimulated cells were treated with 10 µM EHBI2 as compared to the DMSO vehicle control. On the otherhand, its scramble had no inhibitory effects on Akt activation. To confirm that EHBI2 did not affect Akt protein expression levels, total Akt and GAPDH were used as loading controls. No notable difference in protein levels was observed (Supplementary Figure S2b). Consequently, EHBI2 appears to inhibit both activation and downstream signaling of EGFR.
While non-modified peptides are generally not considered to permeate the cell membrane, peptide stapling can greatly improve cellular uptake [29–31]. Further, since the kinase domain of EGFR resides in the cytosol of cells, it is imperative that the inhibitory peptides can access the cytosolic compartment. To monitor cellular uptake of the stapled peptide EHBI2, cells were incubated on a chamber slide for 24 h in complete media containing serum, and then incubated with 5 µM of fluorescein-labeled peptide or DMSO for 7 h. At this point, cells were washed, fixed and imaged for detection of intracellular peptide localization. Relative to the non-stapled control peptide that was largely excluded from the intracellular environment, both EHBI2 and EHBI2 scramble were clearly internalized (Figures 4 and S3). While there is some punctate distribution of EHBI2, presumably in endocytic vesicles, it is also notably distributed throughout the cytoplasm and nucleus. Thus, EHBI2 is cell permeant and can reach the cytosolic space shared by the kinase domain of EGFR.
Figure 4. EHBI2 permeates the cell membrane.

20X confocal microscopy images of MDA-MB-231 cells after a 7 h treatment with 5 µM of fluorescein labeled peptide (green) or DMSO. Nuclei are stained with DAPI (blue). EHBI2 and its scramble control both demonstrate cell permeation while an unstapled analog (15) does not.
3. Conclusions
In summary, we have identified an EGFR kinase domain H-helix analog peptide that can permeate the cell membrane and inhibit EGFR activation and signaling. Others have targeted the asymmetric dimer interface of EGFR; however, much of this work was centered on protein segments or peptides derived from MIG6 which targets multiple sites on the kinase domain [19, 22]. For example, a MIG6-derived peptide was conjugated to gefitinib to create a potent, cell permeable inhibitor that targets multiple sites on the kinase including the ATP binding pocket and substrate binding site [22]. In comparison, the EHBI peptides described in this work demonstrate that an H-helix mimic is sufficient to allosterically inhibit EGFR activation and signaling. Further, although MIG6 targets multiple areas of the kinase domain, MIG6 also targets the kinase dimer interface. Since EHBI2 is proposed to also target this interface, it will be interesting to determine whether or not EHBI2 affects MIG6 binding to EGFR and whether EGFR-mediated phosphorylation of MIG6 is affected. Moreover, since MIG6 is highly expressed in MDA-MB-231 cells [18], it would be interesting to determine whether EHBI2 may work in combination with MIG6 to negatively regulate EGFR activation. A structural model for EGFR kinase oligomerization has also recently been proposed where head-to-tail packing interactions are formed between multiple kinase domains, thereby utilizing the H-helix docking site as part of a complex series of protein-protein interactions that mediate oligomer formation [32]. Based on this model, the EHBI inhibitor peptides may further serve to help downregulate EGFR oligomers.
Since EGFR has proven to be a difficult target where inhibitor resistance is common and small molecule tyrosine kinase inhibitors (TKIs) are often rendered ineffective in clinical settings [11], alternative targeting strategies are needed. By targeting the asymmetric kinase dimer interface rather than the active site of EGFR, this approach may serve as an alternative method for overcoming resistance in EGFR mutants bearing inhibitor insensitive mutations such as T790M [33, 34]. Additionally, since EGFR can heterodimerize with other ErbB family members, it will be important to determine whether EHBI2 can additionally block activation of heterodimeric complexes. While further optimization of the EHBI peptides is needed, this work is an encouraging step towards identifying alternative allosteric targeting sites on this well-studied receptor.
The inhibitory properties of EHBI2 were identified after introducing M945K, V948G and M952R substitutions. Surprisingly, it was previously shown that introducing positively charged single mutations (Arg) at residues in the H-helix that form part of the asymmetric interface, including M945, V948 and M952, inhibited activation as shown by decreased phosphorylation of full-length EGFR [7]. Thus, in the context of full-length EGFR, a positive charge mutation at these sites appears to significantly disrupt the asymmetric dimer interface of EGFR. However, in the context of the designed peptide EHBI2, a single arginine substitution appeared to be favorable for enhancing the inhibitory properties of the peptide as it was experimentally observed to have more potent inhibitory effects as compared to the native H-helix peptide sequence. This finding may be due to a variety of reasons including inherent differences in conformational flexibilities for the full-length protein versus a short peptide when binding the small lobe of the receiver kinase, compensatory differences between individual versus multiple amino acid substitutions on the binding interface or altered interactions that disrupt EGFR activation. Future crystallographic studies of EHBI2 bound to the kinase domain will be needed to clearly elucidate the mechanism for disruption at a molecular level.
The in silico experiments identified multiple substitutions in the binding interface of the H-helix at positions M945, V948, and M952 that may bolster inhibitory activity at the kinase dimer interface despite notable evolutionary conservation in the H-helix. Based on the predicted binding free energy values for individual substitutions, many were predicted to have comparable favorable binding properties and thus further exploration of H-helix analogs may lead to the identification of more potent analogs that target the EGFR kinase dimer interface. Although the initial set of peptides in this work were considerably hydrophobic and were not amenable to direct binding measurements by surface plasmon resonance due to non-specific binding interactions, subsequent development of peptides with improved solubility will facilitate more rapid screening for KD determinations. In addition, combinatorial targeting of other protein-protein interfaces such as the juxtamembrane domain dimer [5] and domain IV dimer [32] may lead to more effective inhibition of EGFR activation. The H-helix mimics described in this work represent a new strategy for EGFR targeting and may serve as useful tools for studying EGFR and ErbB homo- and heterodimerization/oligomerization.
4. Methods
4.1 General Information
The Rink amide methylbenzhydrylamine (MBHA) 100–200 mesh resin and N-α-Fmoc-protected amino acids were purchased from Novabiochem. (S)-N-Fmoc-2-(4’-pentenyl) alanine (S5) was purchased from Okeanos Tech Co., Ltd. Fmoc-11-amino-3,6,9-trioxaundecanoic acid (Fmoc-mini PEG3) and 2-(6-Chloro-1-H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) were purchased from ChemPep Inc. Bis(tricyclohexylphosphine)benzylidine ruthenium(IV) chloride (1st generation Grubbs catalyst) was bought from Sigma Aldrich. 5(6)-carboxyfluorescein was purchased from Gold Acros Organics. All other chemical reagents used for peptide synthesis and purification were purchased from Acros Organics, Fisher, or Sigma Aldrich (unless otherwise noted) and used as supplied. Penicillin/streptomycin (P/S), bovine serum albumin (BSA), and trypan blue were purchased from Amresco. Cell culture media and phosphate buffered saline (PBS) were purchased from Lonza BioWhittaker. Cell Gro 1X trypsin (0.05% trypsin/0.53 mM EDTA in HBSS) was purchased from Corning.
4.2 Peptide Synthesis
Solid phase peptide synthesis was performed on a Rink amide MBHA 100–200 mesh resin with N-α-Fmoc-protected amino acids. Fmoc protecting groups were removed at room temperature for 25 min with 25% (v/v) piperidine in N-methyl-2-pyrrolidinone (NMP). Coupling reactions were setup at room temperature for at least 45 min in NMP with 10 equivalents (eq) amino acid, 9.9 eq HCTU, and 20 eq N,N-diisopropylethylamine (DIPEA). For couplings with (S)-N-Fmoc-2-(4’-pentenyl) alanine (S5) or Fmoc-mini PEG3, 4 eq was used. N-α-Fmoc-L-norleucine was used as a substitute for methionine residues. Before addition of the N-terminal PEG3 linker, ring closing metathesis (RCM) was performed on-resin at room temperature in 1,2-dichloroethane with 0.4 eq of 1st generation Grubbs catalyst twice for 1 h each time. After the RCM reaction, Fmoc-mini PEG3 or β-alanine was added to the N-terminus. All peptides were N-terminally labeled with 5(6)-carboxyfluorescein. N-terminal fluorescein labeling was performed overnight at room temperature with 2 eq of 5(6)-carboxyfluorescein, 1.8 eq HCTU, and 4.6 eq DIPEA in DMF. Peptides were cleaved from resin in 95% (v/v) trifluoroacetic acid (TFA), 2.5% (v/v) triisopropylsilane, 2.5% (v/v) ultra pure water for 4 h at room temperature.
All peptides were characterized by LC-MS (ESI) using an Agilent 1200 with a Zorbax analytical SB-C18 column coupled to an Agilent 6120 Quadrupole mass spectrometer. Peptides were separated over a 10–100% gradient of water:acetonitrile with 0.1% TFA using a linear gradient and a flow rate of 0.5 mL/min. Peptide purification was performed using the same conditions but with a flow rate of 4 mL/min over a semi-preparatory column. Fluorescein-labeled peptides were quantified based on the A495 values measured with a Bio-Tek Synergy 2 plate reader in 10 mM Tris, pH 8 and an extinction coefficient of ε = 68,000 M−1 cm−1 (see reference [15]). Protein Shuffle was used to generate the EHBI2 scramble control (http://www.bioinformatics.org).
Peptide masses for each of the products are as follows: 1 = 2262.0 (expected mass = 2262.6); 2 = 2505.6 (expected mass = 2506.0); 3 = 2243.8 (expected mass = 2243.6); 4 = 2356.6 (expected mass = 2356.8); 5 = 2471.8 (expected mass = 2472.0); 6 = 2243.6 (expected mass = 2243.6); 7 = 2250.6 (expected mass = 2250.7); 8 = 2544.8 (expected mass = 2544.1); 9 = 2624.6 (expected mass = 2624.1); 10 = 2617.4 (expected mass = 2618.1); 11 = 2618.2 (expected mass = 2618.1); 12 = 2624.4 (expected mass = 2625.1); 13 = 2633.1 (expected mass = 2634.1); 14 = 2619.6 (expected mass = 2619.1); 15 = 2624.1 (expected mass = 2625.0); 16 = 2634.2 (expected mass = 2634.1); 17 (EHBI2) = 2619.0 (expected mass = 2619.0); 18 (EHBI2 scr) = 2618.6 (expected mass = 2619.0).
4.3 In Silico Optimization
The starting model for in silico optimization was generated from the crystal structure of EGFR asymmetric dimer (pdb: 2GS6) [7] and the sequence segment corresponding to the H-helix (residues 939–955) in the activator was used as the starting model for the H-helix peptide. Prior to peptide design, energy minimization was performed using the relax function in Rosetta. Foldx version 4 and Rosetta pmut_scan_parallel program was used to perform mutational scan analysis in which the wild-type residue at each of the three positions were mutated to all 19 natural amino acids. To avoid bias towards a single design simulation, 10 different relaxed structures were used. The average binding energies and standard deviations reported by Foldx and Rosetta are shown in Supplementary Tables 1–2.
4.4 Cell Culture
MDA-MB-231 were purchased from the American Type Culture Collection. Cells were cultured on tissue culture plates (CellStar Greiner Bio-One) in RPMI-1640 media with 10% fetal bovine serum (FBS, Thermo Fisher) and penicillin/streptomycin. PBS was used to wash cells multiple times prior to serum starvation.
4.5 Western Blot Analysis
MDA-MB-231 cells were cultured in complete media (RPMI-1640, 10% FBS, penicillin/streptomycin) and plated at 70,000 cells/well on 24-well, tissue culture treated plates (Falcon). Cells were incubated for 24 h at 37 °C, 5.0% CO2 prior to serum starvation. Cells were washed three times with PBS and incubated with serum free RPMI-1640 media. Within the 24 h window for serum starvation, the cells were treated for 9 h with 10 µM of peptide or an equivalent percentage of DMSO (v/v) for the positive and negative controls. As a control, cells were treated with 0.5 µM of gefitinib for 30 min. After the 24 h of serum starvation, cells were left unstimulated or stimulated with 50 ng/mL EGF for 5 min at 37 °C, 5.0 % CO2. Media was immediately aspirated and replaced with loading sample buffer (80 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 0.02% bromophenol blue, 5.3% β-mercaptoethanol) to perform in-well, direct cell lysis. Cell lysates were boiled at 95 °C for 15 min. Cell lysates were separated on 8% polyacrylamide gels using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) in Towbin Buffer (0.1% SDS, 25 mM Tris, 192 mM glycine). Proteins were then transferred to polyvinylidene fluoride (PVDF) membranes in Towbin Buffer with 20% methanol. Membranes were blocked overnight in Tris buffered saline-Tween 20 (TBS-T, 4 mM Tris-HCl, 1 mM Tris, 15 mM NaCl, 0.1% Tween 20) with 5% BSA and 0.1% sodium azide. Membranes were probed using primary antibodies in TBS-T with 5% BSA and 0.1% sodium azide. The following primary antibody dilutions were used: 1:1000 pEGFR Y1068 (2HCLC, Thermo Scientific), 1:200 total EGFR (1005, Santa Cruz), 1:200 α-tubulin (12G10, DSHB), 1:500 pAkt Ser473 (D9E, Cell Signaling), 1:500 pan-Akt (11E7, Cell Signaling), and 1:500 GAPDH (GA1R, Thermo Scientific). Secondary antibodies were diluted 1:5000 for anti-rabbit HRP (Pierce) and 1:3000 for anti-mouse HRP (Rockland) in TBS-T with 5% BSA. Membranes were washed three times (10 min/wash) with TBS-T following incubation with primary and secondary antibodies. Membranes that were probed for pEGFR Y1068 or pAkt S473 were stripped and re-probed for total EGFR and total Akt. Membranes were gently rocked in stripping buffer (62.5 mM Tris pH 6.8, 2% SDS, 0.7% β-mercaptoethanol) at 50 °C for 30 min. Afterwards, membranes were washed in TBS-T six times for 5 min/wash followed by an additional three washes for 10 min/wash. Stripped membranes were blocked in 5% BSA TBS-T buffer at room temperature for 1 h before proceeding with primary antibodies. Western blots were imaged using a 10-min integration time with the Odyssey Fc imaging system (LI-COR Biosciences). Image Studio Lite 3.1.4 (LI-COR) was used to collect the densitometry data. GraphPad Prism 5 was used to graph data and perform statistical analyses. Band densities of phosphorylated protein (EGFR Y1068 or Akt S473) were compared as a ratio to total protein or the loading control protein (e.g. pY1068/EGFR or pY1068/α-tubulin). The positive control (+ EGF) ratio was defined as the baseline (set to 1.0). One-way ANOVA and Tukey’s post hoc test with a 95% confidence interval was applied to the densitometry data to determine statistical significance between the samples. Statistically significant differences between the positive control and test conditions were identified with one or more asterisks, depending on the p-value (i.e. *** for p < 0.001, ** for p < 0.01, and * for p < 0.05).
4.6 Cell Permeation Assay
MDA-MB-231 cells were cultured in complete media (RPMI-1640, 10% FBS, penicillin/streptomycin) and plated at 20,000 cells/well in 8-well chamber slides (Thermo Scientific Nunc Lab Tek II). Cells were incubated for 24 h at 37 °C, 5.0 % CO2. Original media was aspirated from each well and replaced with complete media containing either 5 µM of various peptides or DMSO and incubated for 7 h. At this time point, the media was removed and cells were washed once with PBS. Cells were fixed using 2% paraformaldehyde in PBS for 15 min followed by an additional wash with PBS. Cells were permeabilized for 10 min with 0.2 % Triton-X100 in PBS, followed by two additional washes with PBS. Cells were incubated with 4’,6-Diamidino-2-phenylindole (DAPI, 1:10,000, Invitrogen), and 3% BSA in PBS for 35 min, followed by two washes with PBS. PermaFluor Aqueous Mounting Medium (Thermo Scientific) was applied before imaging. Slides were imaged with a Zeiss LSM 710 confocal microscope (20X) and processed using the Zeiss 2012 software (Zen Lite). 5.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful to Dr. Muthugapatti K. Kandasamy at the Biomedical Microscopy Core Facility of UGA for assisting with confocal microscopy imaging. This research was generously funded by the National Institutes of Health (CA154600 and CA188439 to EJK and GM114409 to NK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary figures and methods can be found in the online version of this article.
References
- 1.Ferguson KM. Structure-based view of epidermal growth factor receptor regulation. Annu Rev Biophys. 2008;37:353–373. doi: 10.1146/annurev.biophys.37.032807.125829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kovacs E, Zorn JA, Huang Y, Barros T, Kuriyan J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem. 2015;84:739–764. doi: 10.1146/annurev-biochem-060614-034402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 4.Lemmon MA, Schlessinger J, Ferguson KM. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harbor Perspectives in Biology. 2014;6:a020768. doi: 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang XW, Kuriyan J. Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell. 2009;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinclair JK, Schepartz A. Influence of macrocyclization on allosteric, juxtamembrane-derived, stapled peptide inhibitors of the epidermal growth factor receptor (EGFR) Org Lett. 2014;16:4916–4919. doi: 10.1021/ol502426b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang XW, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Ward MD, Leahy DJ. Kinase Activator-Receiver Preference in ErbB Heterodimers Is Determined by Intracellular Regions and Is Not Coupled to Extracellular Asymmetry. Journal of Biological Chemistry. 2015;290:1570–1579. doi: 10.1074/jbc.M114.612085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batzer AG, Rotin D, Urena JM, Skolnik EY, Schlessinger J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol. 1994;14:5192–5201. doi: 10.1128/mcb.14.8.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner MJ, Stacey MM, Liu BA, Pawson T. Molecular mechanisms of SH2- and PTB-domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb Perspect Biol. 2013;5:a008987. doi: 10.1101/cshperspect.a008987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 13.Roskoski R., Jr ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol Res. 2014;87:42–59. doi: 10.1016/j.phrs.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Hanold LE, Oruganty K, Ton NT, Beedle AM, Kannan N, Kennedy EJ. Inhibiting EGFR Dimerization Using Triazolyl-Bridged Dimerization Arm Mimics. Plos One. 2015;10:e0118796. doi: 10.1371/journal.pone.0118796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanold LE, Watkins CP, Ton NT, Liaw P, Beedle AM, Kennedy EJ. Design of a selenylsulfide-bridged EGFR dimerization arm mimic. Bioorg Med Chem. 2015;23:2761–2766. doi: 10.1016/j.bmc.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wick M, Burger C, Funk M, Muller R. Identification of a Novel Mitogen-Inducible Gene (Mig-6) - Regulation during G(1) Progression and Differentiation. Experimental Cell Research. 1995;219:527–535. doi: 10.1006/excr.1995.1261. [DOI] [PubMed] [Google Scholar]
- 18.Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, Sommergruber W, Kraut N, Ullrich A, Fassler R, Klein R. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation (vol 12, pg 568, 2006) Nature Medicine. 2006;12:862–862. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 19.Zhang XW, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–U713. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park E, Kim N, Ficarro SB, Zhang Y, Lee BI, Cho A, Kim K, Park AKJ, Park WY, Murray B, Meyerson M, Beroukhim R, Marto JA, Cho J, Eck MJ. Structure and mechanism of activity-based inhibition of the EGF receptor by Mig6. Nature Structural & Molecular Biology. 2015;22:703–U784. doi: 10.1038/nsmb.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maity TK, Venugopalan A, Linnoila I, Cultraro CM, Giannakou A, Nemati R, Zhang X, Webster JD, Ritt D, Ghosal S, Hoschuetzky H, Simpson RM, Biswas R, Politi K, Morrison DK, Varmus HE, Guha U. Loss of MIG6 Accelerates Initiation and Progression of Mutant Epidermal Growth Factor Receptor-Driven Lung Adenocarcinoma. Cancer Discov. 2015;5:534–549. doi: 10.1158/2159-8290.CD-14-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill ZB, Perera BGK, Andrews SS, Maly DJ. Targeting Diverse Signaling Interaction Sites Allows the Rapid Generation of Bivalent Kinase Inhibitors. Acs Chemical Biology. 2012;7:487–495. doi: 10.1021/cb200387g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 24.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 27.Humtsoe JO, Kramer RH. Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene. 2010;29:1214–1226. doi: 10.1038/onc.2009.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemmings BA, Restuccia DF. Pi3k-Pkb/Akt Pathway. Cold Spring Harbor Perspectives in Biology. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu Q, Moellering RE, Hilinski GJ, Kim Y-W, Grossmann TN, Yeh JTH, Verdine GL. Towards understanding cell penetration by stapled peptides. MedChemComm. 2015;6:111–119. [Google Scholar]
- 30.Wang Y, Ho TG, Bertinetti D, Neddermann M, Franz E, Mo GC, Schendowich LP, Sukhu A, Spelts RC, Zhang J, Herberg FW, Kennedy EJ. Isoform-selective disruption of AKAP-localized PKA using hydrocarbon stapled peptides. ACS Chem Biol. 2014;9:635–642. doi: 10.1021/cb400900r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Ho TG, Franz E, Hermann JS, Smith FD, Hehnly H, Esseltine JL, Hanold LE, Murph MM, Bertinetti D, Scott JD, Herberg FW, Kennedy EJ. PKA-type I selective constrained peptide disruptors of AKAP complexes. ACS Chem Biol. 2015;10:1502–1510. doi: 10.1021/acschembio.5b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Bharill S, Karandur D, Peterson SM, Marita M, Shi X, Kaliszewski MJ, Smith AW, Isacoff EY, Kuriyan J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. Elife. 2016;5:e14107. doi: 10.7554/eLife.14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 34.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.