

Abstract
Bullous pemphigoid (BP) is an autoimmune and inflammatory skin disease associated with subepidermal blistering and autoantibodies directed against the hemidesmosomal components BP180 and BP230. Animal models of BP were developed by passively transferring anti-BP180 IgG into mice, which recapitulates the key features of human BP. By using these in vivo model systems, key cellular and molecular events leading to the BP disease phenotype are identified, including binding of pathogenic IgG to its target, complement activation of the classical pathway, mast cell degranulation, and infiltration and activation of neutrophils. Proteinases released by infiltrating neutrophils cleave BP180 and other hemidesmosome-associated proteins, causing DEJ separation. Mast cells and mast cell-derived mediators including inflammatory cytokines and proteases are increased in lesional skin and blister fluids of BP. BP animal model evidence also implicates mast cells in the pathogenesis of BP. However, recent studies questioned the pathogenic role of mast cells in autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, and epidermolysis bullosa acquisita. This review highlights the current knowledge on BP pathophysiology with a focus on a potential role for mast cells in BP and mast cell-related critical issues needing to be addressed in the future.
Keywords: autoantibodies, bullous pemphigoid, hemidesmosome, mast cells, skin autoimmunity
Mast Cells (MCs) and MC Receptors
Mast cells are derived from hematopoietic progenitor cells and have been considered as a central player in functional interaction between innate and adaptive immunity. MCs are initially located in the blood vessel and the lymphatic system before homing to tissues, where they acquire their final effector characteristics (1). There are at least two subpopulations of murine MCs based on the composition of chymases and tryptases within their granules. While MCT cells are the prominent MC type within the mucosa of the respiratory and gastrointestinal tracts, MCTC cells are localized within connective tissues including the dermis, submucosa of the conjunctivae, gastrointestinal tract, heart, and perivascular tissues (2). The maturation of MCs in the tissue mainly relies on stem cell factor (SCF) expressed on the homing tissue, which is the ligand of KIT (1).
Mast cells express KIT (CD117) and FcεRI on their surface, which are the receptors of SCF and IgE, respectively. MCs also express other cell surface receptors, including IgG receptors (FcγRIII, FcγRIIa, and FcγRI), C3a and C5a receptors (C5aRs), Toll-like receptors, and receptors for many cytokines/chemokines (3). These receptors mediate activation of MCs. Upon activation, MCs release their mediators to the homing sites, which act in host defense and various pathological conditions (4). Mediators produced by MCs are divided into two categories: preformed and newly synthesized (5). Many mediators are preformed and stored in granules, such as histamine, serine proteases (tryptase and chymase), and TNF-α (6). Upon activation of MCs, these preformed mediators are released into the extracellular environment within minutes (7–9).
After the initial activation, the synthesized bioactive metabolites of arachidonic acid, prostaglandins, leukotrienes (LTs), and cytokines/chemokines will be released into the affected tissue sites rapidly. The second release of granules will amplify the immediate hypersensitivity reaction through the interaction with local cells and infiltrating immune cells (4).
MCs in Non-Skin Autoimmune Diseases Multiple Sclerosis (MS) and Rheumatoid Arthritis
Mast cells have been considered as key effector cells in many immune activities, especially IgE-associated immune responses, including host defense to parasites, allergic diseases, chronic inflammatory disorders (10, 11), and cancer (12, 13). MCs have also been implicated in autoimmune diseases (14–19), such as MS, rheumatoid arthritis (RA), and the autoimmune skin blistering diseases bullous pemphigoid (BP) and epidermolysis bullosa acquisita (EBA).
Multiple sclerosis is an autoimmune disease of the central nervous system characterized by chronic inflammation and progressive demyelination (20). MCs and activated MCs are present in the target tissues of MS patients and correlated with disease severity (21–24). The animal model of MS, experimental autoimmune encephalomyelitis (EAE), can be induced by active immunization of susceptible mouse strains with myelin components such as myelin basic protein and myelin oligodendrocyte glycoprotein (MOG) (25). RA is an autoimmune disease of the joints characterized by chronic inflammation and cartilage destruction (26). Increased MCs and MC-derived inflammatory mediators are found in the inflamed joints of RA patients (27–29). K/BxN mouse serum contains autoantibodies against the glucose-6-phosphate isomerase and, when passively transferred to mice, induces experimental RA (30).
Role of MCs in Experimental MS and Rheumatoid Arthritis
Mast cell-deficient mice have been widely used to determine the role of MCs in various physiological and pathological conditions, including autoimmune diseases. Whether MCs actively participate in the pathogenesis of MS and RA has been extensively debated recently due to controversial results obtained from different MC-deficient mouse strains. For a more comprehensive and in-depth review, please refer to the studies by Yu et al. and Rivellese et al. (15, 31). In MOG-induced EAE, MC-deficient KitW/W-v mice (caused by Kit mutations) developed a significantly reduced disease, and reconstitution of MC-deficient KitW/W-v mice with wild-type bone marrow-derived MCs restored the disease (32). Similarly, MC-deficient KitW/W-v mice were protected from K/BxN serum-induced RA (33). K/BxN serum also failed to induce RA in MgfSl/Sl-d mice, another MC-deficient strain caused by mutations in the gene encoding the Kit ligand SCF (33). Since MC deficiency by Kit or SCF mutations also caused a variety of immunological abnormalities, new Kit-independent MC-specific deletion mouse strains were developed recently. It turned out that MCs were not required in the development of EAE and serum-induced RA (34).
BP: Clinical and Immunohistological Features
Bullous pemphigoid is an autoimmune subepidermal blistering disease induced by autoantibodies against the two components of the hemidesmosome, BP180 and BP230. BP is the most common autoimmune blistering disease and most prevalent in the elderly. BP typically presents with tense, mostly clear blisters, and erythema, frequently in conjunction with urticarial plaques (35). Blisters occur on either a normal or a erythematous base, containing serous or serosanguinous fluid (36). The disease has a symmetric distribution, and the predilection sites include the lower abdomen, flexor surfaces of the limbs, groin, and axillae (37). In almost all patients, severe pruritus is present. About 10–20% of patients show mucosal involvement, with the oral mucosa being the most common mucosal site (38, 39). Two prospective studies showed that up to 20% of patients with BP have no obvious blistering at the time of diagnosis (38–40).
Histopathologically, hematoxylin and eosin staining of early bulla in BP reveals subepidermal blistering with dense inflammatory infiltrate consisting predominantly of eosinophils, but also lymphocytes, neutrophils, and MCs. Eosinophils are seen within the blister and in the edematous papillary dermis (41). In the early non-bullous phase, subepidermal clefts and eosinophilic spongiosis (epidermal spongiosis with eosinophils within the epidermis) can be found (41). Therefore, BP is an autoimmune and inflammatory disease (Figure 1). Direct immunofluorescence staining exhibits linear deposition of IgG and/or complement components (C3 and/or C5) at the dermal–epidermal junction. IgG deposition sometimes is combined with weaker linear IgA or IgE staining. To identify circulating autoantibodies to the DEJ, indirect immunofluorescence (IIF) with normal human skin as the substrate is usually examined. Artificial blisters can be induced by incubating the skin specimen with 1 M NaCl solution. Since BP180 and BP230 are on the epidermal side of the artificial blisters, autoantibodies from BP patients are known to react with the epidermal side of the blisters (42). In contrast, autoantibodies from other autoimmune blistering diseases, including EBA and anti-laminin γ1 pemphigoid, react with the dermal side of the artificial blisters (35). Thus, IIF with the salt-split skin as a substrate is helpful in distinguishing BP from other autoimmune blistering disorders.
Figure 1.

Human bullous pemphigoid (BP). (A) Large, tense bullae, and erythematous patches seen in BP patient. (B) Histology reveals dermal–epidermal junction separation with inflammatory cell infiltration. Immunofluorescence shows linear deposition of IgG (C) and complement C3 (D) at the basement membrane zone (BMZ). d, dermis; e, epidermis. Arrow, the BMZ. Original magnification, 100× for panels (B–D).
BP Autoantigens
Bullous pemphigoid autoantibodies target two hemidesmosomal components BP180 (BPAG2) and BP230 (BPAG1), which are involved in dermal–epidermal cohesion (43–45). BP180 is a type II transmembrane glycoprotein with a globular cytoplasmic domain and a large extracellular region containing 15 collagenous and 16 non-collagenous (NC1-16) domains. The 16th non-collagenous (NC16A) domain is the immunodominant region in BP (46). Anti-NC16A IgG autoantibodies are detected in more than 90% of BP patients (47) and have been shown to be pathogenic in skin organ culture system and in animal models of BP (48–50) (see below). BP230 is a 230-kDa intracellular component of the hemidesmosomal plaque and belongs to the plakin family of proteins. Anti-BP230 autoantibodies are detected in nearly 60% of BP patients (51). In addition to IgG reactivity, anti-BP180/BP230 IgE autoantibodies are present in serum samples from most patients (47, 52, 53).
Genetics of BP
Genetic, environmental, and stochastic factors contribute to susceptibility to most autoimmune diseases. The human MHC encodes many glycoproteins that include the HLA class I and class II molecules, which provide a pivotal role in the recognition of antigenic peptides by T cells. A lot of polymorphisms of HLA-II class alleles have been identified in several populations of patients with BP (54–58). These polymorphisms HLA class II alleles occur likely due to changes in the charge of the active binding site on the HLA molecules for binding of autoantigenic peptides. A common HLA class II allele, HLA-DQB1*03:01, is positively associated with BP in multiple populations (54, 55, 58) and also appears to be associated with distinct clinical pemphigoid variants (59–61). In addition, the activation of BP180-autoreactive T cells from a cohort of BP patients with HLA-DQB1*03:01 was found to be restricted by this BP-associated HLA class II allele (55).
T Cell Response in BP
CD4+ T helper (Th) cells are thought to participate in early disease development and perpetuation of autoantibody-mediated autoimmune blistering diseases. Th cells, upon proper costimulation, are activated and produce and secrete distinct cytokines that stimulate B cells. This Th–B cell interaction thus fosters plasma cell development and autoantibody production (62). In BP, autoreactive CD4+ T lymphocytes recognize unique epitopes within the extracellular region of BP180 (63). The majority of BP patients examined have both Th1 and Th2 responses against the BP180 ectodomain (55, 64). BP180-reactive Th cells and IgG autoantibodies recognized similar or identical epitopes clustered in distinct regions of the BP180 ectodomain and BP230 (49, 62, 65). Li et al. found that follicular T helper (Tfh) cells and IL-21 were crucial for the secretion of antibodies against BP180NC16A domain in T cell/B cell co-culture system, indicating that these Tfh cells may be involved in the pathogenesis of BP (66).
MCs in Human BP
In 1978, Wintroub et al. found that increased MCs and increased degranulation of MCs at the BP lesional sites are the earliest events in BP lesion formation (67). The evolution of clinical BP lesions is associated with a sequence of histopathologic events, starting with MC alternation and proceeding to immune cell infiltration first with lymphocytes followed by eosinophils and basophils. Electron and light microscopy revealed that MCs are mainly present in the papillary dermis adjacent to the dermal–epidermal junction and demonstrate a unique, focal, irregular loss of granule contents (68).
Various inflammatory mediators have been found in lesional/perilesional skin, blister fluids, and/or blood of patients with BP, including C5a, histamine, LTs, and many cytokines/chemokines (e.g., IL-1, IL-2, IL-5, IL-6, IL-8, TNF-α, eotaxins, and IFN-γ) (69–75). These mediators can recruit and directly activate MCs and leukocytes. Moreover, MCs can influence biological responses through the production of multifunctional cytokines and enzymes (76–78). Evidence suggests that metalloproteinase (MMP9 in particular), leukotrienes (LT), heparin and platelet activating factor (PAF) derived from MCs also play a role in the inflammatory process during blister formation (67). Tryptase is a specific proteolytic enzyme synthesized and stored in MCs and released by MCs when activated by various stimulating factors. Tryptase, therefore, is considered a reliable marker for the presence of MCs (79). A previous study showed that tryptase levels in BP blister fluid were increased compared with the respective sera and significantly correlated with several cytokines/chemokines (IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, and RANTES), VEGF, and sICAM-1. Most importantly, the blister fluid tryptase levels were also positively correlated with titers of autoantibodies against basement membrane zone antigens (80), which relates to the severity of the disease. Increased levels of cytokines (including IL-1β, IL-5, IL-6, IL-10, IL-15, and TNF-α) and chemokines (such as CCL2, CCL5, CCL11, CCL13, and CCL18, and IL-8) were identified in serum samples and blister fluids of patients with BP, and some of these mediators parallel disease activity (81). Bieber et al. investigated serum parameters related to activation of different inflammatory cells and found higher serum concentrations of MCs tryptase during ongoing disease. The serum levels of MCs tryptase significantly decreased at the time of clinical remission of the patients. In addition, serum concentrations of MCs tryptase were significantly associated with levels of circulating anti-BP180 autoantibodies (82). These data suggested that increased concentrations of MCs tryptase in BP blister fluids and/or serum partly correlate with cytokines, autoantibodies, and clinical disease severity in BP patients.
BP180-specific IgG autoantibodies are the most abundant immunoglobulin isotype; however, IgE autoantibodies with the same or similar epitope specificity are also present in about 70–90% of BP patients (83, 84). It has been speculated that IgE autoantibody–mediated activation of MCs in the skin may be involved in the development of certain clinical symptoms typical of BP, such as urticarial plaques, dermal edema, and eosinophilic inflammation. Dimson et al. found IgE-coated MCs in the perilesional skin of the BP patients, and BP180 peptides were co-localized on these MCs, suggesting that BP180-specific IgE that bind to the surface of MCs through IgE receptors, when interacting with BP180 peptides, result in MC degranulation. Moreover, basophils obtained from untreated BP patients stimulated with recombinant BP180NC16A released significantly higher histamine compared to NC16-stimulated basophils from normal control or from treated BP patients (83). In addition, Freire et al. reported that IgE co-localized with MCs in the perilesional skin of BP patients, and IgE-BP180 complexes could activate MCs via the high-affinity IgE receptor (FcεRI), conceivably triggering MC degranulation-mediated events resulting in tissue inflammation (85).
Omalizumab is a recombinant humanized monoclonal antibody that inhibits the binding of IgE to FcεRI on the surface of MCs and basophils. Patients with BP treated with omalizumab showed reduced disease severity including decreased itching and blister count, reduced urticarial plaques, and reduced eosinophilic inflammation (86, 87). Together, these clinical research and clinical trial data suggest that IgE autoantibodies in BP patients are involved in BP development likely through FcεRI-induced degranulation of MCs and basophils. However, pathogenic anti-BP180 IgE autoantibodies could also act on eosinophils in BP tissue injury since eosinophils express IgE receptors and are predominant infiltrating immune cells in BP (41).
Animal Models of BP
Bullous pemphigoid autoantibodies were thought to be responsible for blister formation in BP; however, passive transfer of IgG autoantibodies from BP patients could not induce a BP-like disease in animals (88, 89). It turned out that BP autoantibodies reacting with NC16A domain that harbors immunodominant and potentially pathogenic epitopes fail to cross-react with mouse BP180; therefore, BP IgG autoantibodies cannot be tested for pathogenicity in a conventional passive transfer mouse model. In 1993, Liu et al. (90) subcloned a segment of the murine BP180 protein homologous with the human BP180 NC16A (mBP180 NC14A), generated rabbit polyclonal antibodies against mBP180 NC14A, and administrated the purified rabbit anti-mBP180 IgG intradermally or intraperitoneally into neonatal BALB/c mice. This experimental BP model reproduced all of the key clinical, histological, and immunopathological features of BP, including deposition of rabbit anti-mBP180 IgG and mouse complement C3 at dermal–epidermal junction, infiltration of inflammatory cells, and subepidermal blistering (90) (Figure 2). Anti-BP180 IgG-induced BP blistering required complement activation and neutrophil recruitment (91, 92). Subsequently, BP serum-purified IgG autoantibodies against BP180 or NC16A domain were also demonstrated to be pathogenic in BP180 humanized mouse models (93, 94).
Figure 2.

Mouse bullous pemphigoid. The anti-BP180 IgG induce extensive blistering disease in neonatal B6 mice clinically (A) and histologically (B). The skin of these animals shows linear deposition of anti-BP180 IgG (C) and murine C3 (D) at the BMZ, as determined by direct IF. Toluidine blue staining shows resting and degranulating mast cells in the dermis (E). d, dermis; e, epidermis; v, vesicle; arrow, the BMZ. Original magnification, 200× for panels (B–D), 400× for panel (E). (E) Arrows for degranulating mast cells, and arrow heads for normal resting mast cells.
BP180-specific IgE autoantibodies purified from serum of BP patients when passively transferred into human skin grafted onto athymic nude mice induced skin lesions that recapitulated the initial phase of disease. The features of the early phase of the disease are characterized by increased plaques and MC degranulation in comparison with injection of normal control human IgE (95). Lesional skin of the anti-BP180 IgE-injected mice also exhibited infiltration of neutrophils and eosinophils (95). However, it remains to be determined whether the pathogenic activity of anti-BP180 IgE depends on eosinophils, MCs, or both.
Role of MCs in Experimental BP
To determine whether MCs were involved in experimental BP, Chen et al. (19) demonstrated that wild-type MC-sufficient mice administrated intradermally with pathogenic anti-mBP180 IgG developed BP disease with extensive MC degranulation in the upper dermis, which preceded infiltration of neutrophils and subsequent dermal–epidermal separation. In contrast, MC-deficient KitW/W-v and MgfSl/Sl-d mice failed to develop BP (19). Moreover, these MC-deficient mice reconstituted with wild-type bone marrow-derived MCs, and polymorphonuclear leukocytes from these MC-deficient mice or by intradermal injection of IL-8 (a neutrophil chemoattractant) became susceptible to experimental BP (19). Blocking MC degranulation by treating MC-sufficient mice with an MC degranulation inhibitor also significantly reduced disease phenotype (19).
To determine the functional relationship between MCs and neutrophils, Chen et al. found that anti-BP180 antibody-induced neutrophil infiltration depends mainly on MCs in experimental BP (19). Without MCs, KitW/W-v and MgfSl/Sl-d mice injected with pathogenic IgG show about 70% reduction of infiltrating neutrophils in the skin (96). Further examination of the experimental BP model also implicated macrophages in anti-BP180 IgG-triggered neutrophil infiltration in mice, and that macrophage-mediated neutrophil infiltration depends on MC activation (96). The findings that neutrophil recruitment is not completely impaired in MC-deficient mice in experimental BP suggest that at least two neutrophil recruitment pathways could exist: MC-dependent and MC-independent (96). Nevertheless, these data suggest a major role of MCs in infiltration of neutrophils into the dermis in this animal model setting.
Mast cells express surface receptors that directly bind the cleaved products of the activated complement cascade (97). Skin MCs express the C5aR (98), and upon the molecular interaction of C5a and C5aR, MCs degranulate, releasing several pro-inflammatory cytokines including TNF-α, IL-1, IL-6, and GM-CSF (99). Moreover, human C3a and C5a could degranulate MCs in vitro to release histamine and tryptase. Heimbach et al. (100) demonstrated that interaction of C5a–C5aR on MCs activated the p38 MAPK pathway that trigger MC degranulation and subsequent tissue injury and blister formation.
Mast cells store proteases in large quantities in the secretory granules, and these fully functional enzymes are a major class of inflammatory mediators (101, 102). Human cutaneous MCs contain a single chymase, and mouse MC protease-4 (mMCP-4) has been generally recognized as the likely homolog of the human chymase (103–105). Importantly, mMCP-4 can activate MMP-9, a key proteolytic enzyme for tissue injury in experimental BP (106). Interestingly, mMCP-4−/− mice are resistant to anti-BP180 IgG-induced experimental BP (107). In experimental BP, mMCP-4 activates MMP-9 and directly cleaves BP180. mMCP-4, MMP-9, and other proteolytic enzymes work together to degrade BP180 and other hemidesmosomal proteins and proteins in extracellular matrix of the BMZ (107), leading to clinical and histological BP-like blistering.
Taken together, results of these studies using MC-deficient and C5aR and mMCP-4 knockout mice implicate a pathogenic role of MCs in BP (Figure 3). However, since the studies on the role of MCs in anti-BP180 IgG-induced experimental BP have been performed only in MC-deficient KitW/W-v and MgfSl/Sl-d mice, KIT-independent MC-specific deletion mouse strains need to be tested to confirm or clarify the involvement of MCs in experimental BP.
Figure 3.
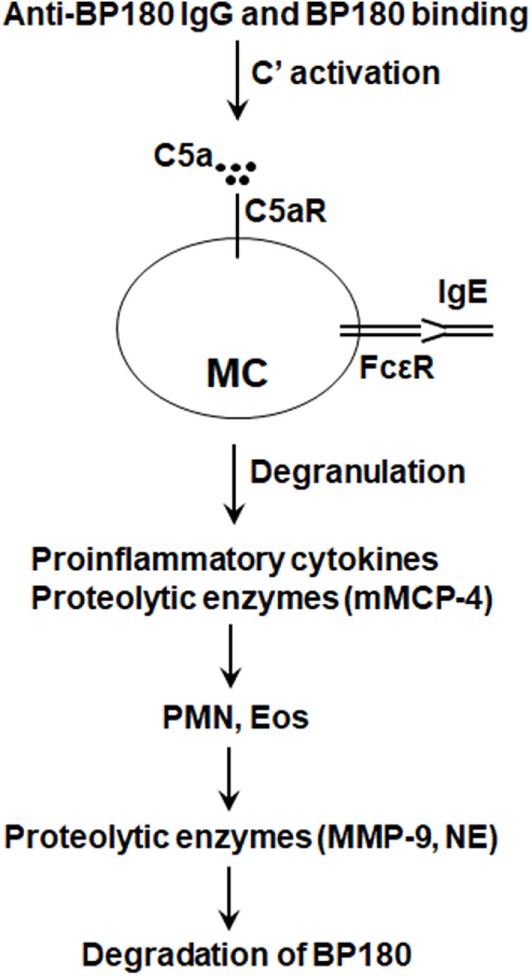
Proposed role of mast cells (MCs) in bullous pemphigoid (BP). Anti-BP180 IgG binding to BP180 on the surface of basal keratinocytes activates the complement (C), generating C5a. C5a acts on C5a receptor (C5aR) to cause MCs to degranulate and release pro-inflammatory cytokines/chemokines (e.g., TNFα) and proteolytic enzymes including mouse MC protease-4 (mMCP-4). Anti-BP180 IgE could also activate MCs. The released pro-inflammatory mediators interact with local cells to recruit neutrophils (PMN) and eosinophils (Eos). Infiltrating PMN and Eos, upon activation through interactions between immobilized anti-BP180 IgG/IgE and FcγR/FcεR, release neutrophil elastase (NE), MMP-9, and other proteolytic enzymes. mMCP-4 activates MMP-9 and also directly cleaves BP180 and other BP180-associated proteins in concert with MMP-9 and NE, resulting in subepidermal blistering.
Role of MCs in Epidermal Bullosa Acquisita (EBA)
Epidermal bullosa acquisita is another autoimmune subepidermal blistering skin disease caused by autoantibodies against collagen VII (108). Experimental EBA can be induced by passive transfer of anticollagen VII IgG (109, 110). Immunopathogenically, experimental EBA shares many key features with experimental BP such as their dependency on complement, C5a-C5aR signaling, and neutrophils (109, 111). However, anticollagen VII IgG causes similar disease severity in both wild-type control and MC-deficient KitW/W-v mice (112). KIT-independent MC-specific deletion mice are also not protected from experimental EBA (112). These studies demonstrate that MCs do not contribute to experimental EBA, further emphasizing a need to revisit the role of MCs in experimental BP using KIT-independent MC-specific deletion strains.
Concluding Remarks
We presented several lines of BP animal model evidence, together with clinical observations, implicating that MCs are likely to be involved in the immunopathogenesis of BP. The role of MCs in experimental BP, however, was investigated exclusively in KIT-dependent MC-deficient mice. Based on the observed discrepancies in different MC-deficient models of EAE, RA, and EBA, it is necessary to perform anti-BP180 IgG-induced BP studies in KIT-independent MC-specific deletion strains to clarify whether MCs play a role in BP.
Bullous pemphigoid patients also have anti-BP180 IgE autoantibodies, which are involved in tissue injury (95); therefore, a potential role of MCs in anti-BP180 IgE-induced BP should be determined in both KIT-dependent and KIT-independent MC-deficient strains. Future studies could also investigate whether and how MCs interact with anti-BP180 IgG, anti-BP180 IgE, and eosinophils during disease development. Giving the fact that MCs have a variety of immunomodulatory activities (14), MC contribution to BP could be multifaceted. Advanced tools need to be developed to clarify and fully appreciate the contribution of MCs to BP and help uncover new therapeutic targets for this potentially fatal skin autoimmune disease.
Author Contributions
HF, YZ, NL, GW, and ZL wrote the manuscript; HF, YZ, and NL prepared the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our colleagues and collaborator for their support. We apologize for not mentioning and citing some related studies due to space limitations. This work was supported by the National Institutes of Health (grants R01 AI40768 and R01 AR070276 to ZL) and National Natural Science Foundation of China (no. 81220108016 to GW).
References
- 1.Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S73–80. 10.1016/j.jaci.2009.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maurer M. Urticaria and angioedema. Chem Immunol Allergy (2014) 100:101–4. 10.1159/000358614 [DOI] [PubMed] [Google Scholar]
- 3.Sayed BA, Christy A, Quirion MR, Brown MA. The master switch: the role of mast cells in autoimmunity and tolerance. Annu Rev Immunol (2008) 26:705–39. 10.1146/annurev.immunol.26.021607.090320 [DOI] [PubMed] [Google Scholar]
- 4.Rivera J, Fierro NA, Olivera A, Suzuki R. New insights on mast cell activation via the high affinity receptor for IgE. Adv Immunol (2008) 98:85–120. 10.1016/S0065-2776(08)00403-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arthur G, Bradding P. New developments in mast cell biology: clinical implications. Chest (2016) 150(3):680–93. 10.1016/j.chest.2016.06.009 [DOI] [PubMed] [Google Scholar]
- 6.Young JD, Liu CC, Butler G, Cohn ZA, Galli SJ. Identification, purification, and characterization of a mast cell-associated cytolytic factor related to tumor necrosis factor. Proc Natl Acad Sci U S A (1987) 84(24):9175–9. 10.1073/pnas.84.24.9175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol (2008) 9(11):1215–23. 10.1038/ni.f.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakae S, Suto H, Iikura M, Kakurai M, Sedgwick JD, Tsai M, et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J Immunol (2006) 176(4):2238–48. 10.4049/jimmunol.176.4.2238 [DOI] [PubMed] [Google Scholar]
- 9.Harmon B, Chylek LA, Liu Y, Mitra ED, Mahajan A, Saada EA, et al. Timescale separation of positive and negative signaling creates history-dependent responses to IgE receptor stimulation. Sci Rep (2017) 7(1):15586. 10.1038/s41598-017-15568-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muto Y, Wang Z, Vanderberghe M, Two A, Gallo RL, Di Nardo A. Mast cells are key mediators of cathelicidin-initiated skin inflammation in rosacea. J Invest Dermatol (2014) 134(11):2728–36. 10.1038/jid.2014.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovanen PT. Mast cells and degradation of pericellular and extracellular matrices: potential contributions to erosion, rupture and intraplaque haemorrhage of atherosclerotic plaques. Biochem Soc Trans (2007) 35(Pt 5):857–61. 10.1042/BST0350857 [DOI] [PubMed] [Google Scholar]
- 12.Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci U S A (2007) 104(50):19977–82. 10.1073/pnas.0704620104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wasiuk A, de Vries VC, Hartmann K, Roers A, Noelle RJ. Mast cells as regulators of adaptive immunity to tumours. Clin Exp Immunol (2009) 155(2):140–6. 10.1111/j.1365-2249.2008.03840.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galli SJ, Grimbaldeston M, Tsai M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev Immunol (2008) 8(6):478–86. 10.1038/nri2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu X, Kasprick A, Petersen F. Revisiting the role of mast cells in autoimmunity. Autoimmun Rev (2015) 14(9):751–9. 10.1016/j.autrev.2015.04.008 [DOI] [PubMed] [Google Scholar]
- 16.Steinman L. Multiple sclerosis: a two-stage disease. Nat Immunol (2001) 2(9):762–4. 10.1038/ni0901-762 [DOI] [PubMed] [Google Scholar]
- 17.Brown MA, Tanzola MB, Robbie-Ryan M. Mechanisms underlying mast cell influence on EAE disease course. Mol Immunol (2002) 38(16–18):1373–8. 10.1016/S0161-5890(02)00091-3 [DOI] [PubMed] [Google Scholar]
- 18.Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FMA, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity (2002) 16(2):157–68. 10.1016/S1074-7613(02)00275-3 [DOI] [PubMed] [Google Scholar]
- 19.Chen R, Ning G, Zhao ML, Fleming MG, Diaz LA, Werb Z, et al. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest (2001) 108(8):1151–8. 10.1172/JCI11494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol (2007) 8(9):913–9. 10.1038/ni1507 [DOI] [PubMed] [Google Scholar]
- 21.Couturier N, Zappulla JP, Lauwers-Cances V, Uro-Coste E, Delisle MB, Clanet M, et al. Mast cell transcripts are increased within and outside multiple sclerosis lesions. J Neuroimmunol (2008) 195(1–2):176–85. 10.1016/j.jneuroim.2008.01.017 [DOI] [PubMed] [Google Scholar]
- 22.Kruger PG, Bo L, Myhr KM, Karlsen AE, Taule A, Nyland HI, et al. Mast cells and multiple sclerosis: a light and electron microscopic study of mast cells in multiple sclerosis emphasizing staining procedures. Acta Neurol Scand (1990) 81(1):31–6. 10.1111/j.1600-0404.1990.tb00927.x [DOI] [PubMed] [Google Scholar]
- 23.Toms R, Weiner HL, Johnson D. Identification of IgE-positive cells and mast cells in frozen sections of multiple sclerosis brains. J Neuroimmunol (1990) 30(2–3):169–77. 10.1016/0165-5728(90)90101-R [DOI] [PubMed] [Google Scholar]
- 24.Rozniecki JJ, Hauser SL, Stein M, Lincoln R, Theoharides TC. Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann Neurol (1995) 37(1):63–6. 10.1002/ana.410370112 [DOI] [PubMed] [Google Scholar]
- 25.Rangachari M, Kuchroo VK. Using EAE to better understand principles of immune function and autoimmune pathology. J Autoimmun (2013) 45:31–9. 10.1016/j.jaut.2013.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, III, et al. 2010 rheumatoid arthritis classification criteria: an American college of rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum (2010) 62(9):2569–81. 10.1002/art.27584 [DOI] [PubMed] [Google Scholar]
- 27.Bridges AJ, Malone DG, Jicinsky J, Chen M, Ory P, Engber W, et al. Human synovial mast cell involvement in rheumatoid arthritis and osteoarthritis. Relationship to disease type, clinical activity, and antirheumatic therapy. Arthritis Rheum (1991) 34(9):1116–24. 10.1002/art.1780340907 [DOI] [PubMed] [Google Scholar]
- 28.Gotis-Graham I, Smith MD, Parker A, McNeil HP. Synovial mast cell responses during clinical improvement in early rheumatoid arthritis. Ann Rheum Dis (1998) 57(11):664–71. 10.1136/ard.57.11.664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano S, Mishiro T, Takahara S, Yokoi H, Hamada D, Yukata K, et al. Distinct expression of mast cell tryptase and protease activated receptor-2 in synovia of rheumatoid arthritis and osteoarthritis. Clin Rheumatol (2007) 26(8):1284–92. 10.1007/s10067-006-0495-8 [DOI] [PubMed] [Google Scholar]
- 30.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell (1996) 87(5):811–22. 10.1016/S0092-8674(00)81989-3 [DOI] [PubMed] [Google Scholar]
- 31.Rivellese F, Nerviani A, Rossi FW, Marone G, Matucci-Cerinic M, de Paulis A, et al. Mast cells in rheumatoid arthritis: friends or foes? Autoimmun Rev (2017) 16(6):557–63. 10.1016/j.autrev.2017.04.001 [DOI] [PubMed] [Google Scholar]
- 32.Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med (2000) 191(5):813–22. 10.1084/jem.191.5.813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science (2002) 297(5587):1689–92. 10.1126/science.1073176 [DOI] [PubMed] [Google Scholar]
- 34.Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity (2011) 35(5):832–44. 10.1016/j.immuni.2011.09.015 [DOI] [PubMed] [Google Scholar]
- 35.Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (2013) 381(9863):320–32. 10.1016/S0140-6736(12)61140-4 [DOI] [PubMed] [Google Scholar]
- 36.Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol (2017) 54(1):26–51. 10.1007/s12016-017-8633-4 [DOI] [PubMed] [Google Scholar]
- 37.Walsh SRA, Hogg D, Mydlarski PR. Bullous pemphigoid: from bench to bedside. Drugs (2005) 65(7):905–26. 10.2165/00003495-200565070-00002 [DOI] [PubMed] [Google Scholar]
- 38.Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol (2012) 30(1):3–16. 10.1016/j.clindermatol.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 39.della Torre R, Combescure C, Cortes B, Marazza G, Beltraminelli H, Naldi L, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol (2012) 167(5):1111–7. 10.1111/j.1365-2133.2012.11108.x [DOI] [PubMed] [Google Scholar]
- 40.Joly P, Baricault S, Sparsa A, Bernard P, Bedane C, Duvert-Lehembre S, et al. Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol (2012) 132(8):1998–2004. 10.1038/jid.2012.35 [DOI] [PubMed] [Google Scholar]
- 41.Kneisel A, Hertl M. Autoimmune bullous skin diseases. Part 2: diagnosis and therapy. J Dtsch Dermatol Ges (2011) 9(11):927–47. 10.1111/j.1610-0387.2011.07809.x [DOI] [PubMed] [Google Scholar]
- 42.Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Sci (2014) 73(3):179–86. 10.1016/j.jdermsci.2013.12.001 [DOI] [PubMed] [Google Scholar]
- 43.Stanley JR, Hawley-Nelson P, Yuspa SH, Shevach EM, Katz SI. Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell (1981) 24(3):897–903. 10.1016/0092-8674(81)90115-X [DOI] [PubMed] [Google Scholar]
- 44.Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol (1986) 136(4):1231–5. [PubMed] [Google Scholar]
- 45.Diaz LA, Ratrie H, III, Saunders WS, Futamura S, Squiquera HL, Anhalt GJ, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. Immunolocalization of this protein to the hemidesmosome. J Clin Invest (1990) 86(4):1088–94. 10.1172/JCI114812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA. Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol (1993) 151(10):5742–50. 10.1016/0923-1811(93)90940-Q [DOI] [PubMed] [Google Scholar]
- 47.Ishiura N, Fujimoto M, Watanabe R, Nakashima H, Kuwano Y, Yazawa N, et al. Serum levels of IgE anti-BP180 and anti-BP230 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci (2008) 49(2):153–61. 10.1016/j.jdermsci.2007.08.008 [DOI] [PubMed] [Google Scholar]
- 48.Perriard J, Jaunin F, Favre B, Budinger L, Hertl M, Saurat JH, et al. IgG autoantibodies from bullous pemphigoid (BP) patients bind antigenic sites on both the extracellular and the intracellular domains of the BP antigen 180. J Invest Dermatol (1999) 112(2):141–7. 10.1046/j.1523-1747.1999.00497.x [DOI] [PubMed] [Google Scholar]
- 49.Hofmann S, Thoma-Uszynski S, Hunziker T, Bernard P, Koebnick C, Stauber A, et al. Severity and phenotype of bullous pemphigoid relate to autoantibody profile against the NH2- and COOH-terminal regions of the BP180 ectodomain. J Invest Dermatol (2002) 119(5):1065–73. 10.1046/j.1523-1747.2002.19529.x [DOI] [PubMed] [Google Scholar]
- 50.Di Zenzo G, Thoma-Uszynski S, Fontao L, Calabresi V, Hofmann SC, Hellmark T, et al. Multicenter prospective study of the humoral autoimmune response in bullous pemphigoid. Clin Immunol (2008) 128(3):415–26. 10.1016/j.clim.2008.04.012 [DOI] [PubMed] [Google Scholar]
- 51.Kromminga A, Sitaru C, Hagel C, Herzog S, Zillikens D. Development of an ELISA for the detection of autoantibodies to BP230. Clin Immunol (2004) 111(1):146–52. 10.1016/j.clim.2003.12.007 [DOI] [PubMed] [Google Scholar]
- 52.Messingham KN, Srikantha R, DeGueme AM, Fairley JA. FcR-independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol (2011) 187(1):553–60. 10.4049/jimmunol.1001753 [DOI] [PubMed] [Google Scholar]
- 53.Delaporte E, Dubost-Brama A, Ghohestani R, Nicolas JF, Neyrinck JL, Bergoend H, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol (1996) 157(8):3642–7. [PubMed] [Google Scholar]
- 54.Banfield CC, Wojnarowska F, Allen J, George S, Venning VA, Welsh KI. The association of HLA-DQ7 with bullous pemphigoid is restricted to men. Br J Dermatol (1998) 138(6):1085–90. 10.1046/j.1365-2133.1998.02350.x [DOI] [PubMed] [Google Scholar]
- 55.Budinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest (1998) 102(12):2082–9. 10.1172/JCI3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okazaki A, Miyagawa S, Yamashina Y, Kitamura W, Shirai T. Polymorphisms of HLA-DR and -DQ genes in Japanese patients with bullous pemphigoid. J Dermatol (2000) 27(3):149–56. 10.1111/j.1346-8138.2000.tb02141.x [DOI] [PubMed] [Google Scholar]
- 57.Gao XH, Winsey S, Li G, Barnardo M, Zhu XJ, Chen HD, et al. HLA-DR and DQ polymorphisms in bullous pemphigoid from northern China. Clin Exp Dermatol (2002) 27(4):319–21. 10.1046/j.1365-2230.2002.01037.x [DOI] [PubMed] [Google Scholar]
- 58.Esmaili N, Mortazavi H, Chams-Davatchi C, Daneshpazhooh M, Damavandi MR, Aryanian Z, et al. Association between HLA-DQB1*03:01 and bullous pemphigoid in Iranian patients. Iran J Immunol (2013) 10(1):1–9. [PubMed] [Google Scholar]
- 59.Delgado JC, Turbay D, Yunis EJ, Yunis JJ, Morton ED, Bhol K, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A (1996) 93(16):8569–71. 10.1073/pnas.93.16.8569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan LS, Hammerberg C, Cooper KD. Significantly increased occurrence of HLA-DQB1*0301 allele in patients with ocular cicatricial pemphigoid. J Invest Dermatol (1997) 108(2):129–32. 10.1111/1523-1747.ep12332352 [DOI] [PubMed] [Google Scholar]
- 61.Setterfield J, Theron J, Vaughan RW, Welsh KI, Mallon E, Wojnarowska F, et al. Mucous membrane pemphigoid: HLA-DQB1*0301 is associated with all clinical sites of involvement and may be linked to antibasement membrane IgG production. Br J Dermatol (2001) 145(3):406–14. 10.1046/j.1365-2133.2001.04380.x [DOI] [PubMed] [Google Scholar]
- 62.Thoma-Uszynski S, Uter W, Schwietzke S, Schuler G, Borradori L, Hertl M. Autoreactive T and B cells from bullous pemphigoid (BP) patients recognize epitopes clustered in distinct regions of BP180 and BP230. J Immunol (2006) 176(3):2015–23. 10.4049/jimmunol.176.3.2015 [DOI] [PubMed] [Google Scholar]
- 63.Hertl M, Eming R, Veldman C. T cell control in autoimmune bullous skin disorders. J Clin Invest (2006) 116(5):1159–66. 10.1172/JCI28547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin MS, Fu CL, Giudice GJ, Olague-Marchan M, Lazaro AM, Stastny P, et al. Epitopes targeted by bullous pemphigoid T lymphocytes and autoantibodies map to the same sites on the bullous pemphigoid 180 ectodomain. J Invest Dermatol (2000) 115(6):955–61. 10.1046/j.1523-1747.2000.00153.x [DOI] [PubMed] [Google Scholar]
- 65.Thoma-Uszynski S, Uter W, Schwietzke S, Hofmann SC, Hunziker T, Bernard P, et al. BP230- and BP180-specific auto-antibodies in bullous pemphigoid. J Invest Dermatol (2004) 122(6):1413–22. 10.1111/j.0022-202X.2004.22603.x [DOI] [PubMed] [Google Scholar]
- 66.Li Q, Liu Z, Dang E, Jin L, He Z, Yang L, et al. Follicular helper T cells (Tfh) and IL-21 involvement in the pathogenesis of bullous pemphigoid. PLoS One (2013) 8(7):e68145. 10.1371/journal.pone.0068145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wintroub BU, Mihm MCJ, Goetzl EJ, Soter NA, Austen KF. Morphologic and functional evidence for release of mast-cell products in bullous pemphigoid. N Engl J Med (1978) 298(8):417–21. 10.1056/NEJM197802232980803 [DOI] [PubMed] [Google Scholar]
- 68.Dvorak AM, Mihm MCJ, Osage JE, Kwan TH, Austen KF, Wintroub BU. Bullous pemphigoid, an ultrastructural study of the inflammatory response: eosinophil, basophil and mast cell granule changes in multiple biopsies from one patient. J Invest Dermatol (1982) 78(2):91–101. 10.1111/1523-1747.ep12505711 [DOI] [PubMed] [Google Scholar]
- 69.Baba T, Sonozaki H, Seki K, Uchiyama M, Ikesawa Y, Toriisu M. An eosinophil chemotactic factor present in blister fluids of bullous pemphigoid patients. J Immunol (1976) 116(1):112–6. [PubMed] [Google Scholar]
- 70.Endo H, Iwamoto I, Fujita M, Okamoto S, Yoshida S. Increased immunoreactive interleukin-5 levels in blister fluids of bullous pemphigoid. Arch Dermatol Res (1992) 284(5):312–4. 10.1007/BF00372588 [DOI] [PubMed] [Google Scholar]
- 71.Takiguchi Y, Kamiyama O, Saito E, Nagao S, Kaneko F, Minagawa T. Cell-mediated immune reaction in the mechanism of blister formation in bullous pemphigoid. Dermatologica (1989) 179(Suppl):137. 10.1159/000248478 [DOI] [PubMed] [Google Scholar]
- 72.Katayama I, Doi T, Nishioka K. High histamine level in the blister fluid of bullous pemphigoid. Arch Dermatol Res (1984) 276(2):126–7. 10.1007/BF00511070 [DOI] [PubMed] [Google Scholar]
- 73.Kawana S, Ueno A, Nishiyama S. Increased levels of immunoreactive leukotriene B4 in blister fluids of bullous pemphigoid patients and effects of a selective 5-lipoxygenase inhibitor on experimental skin lesions. Acta Derm Venereol (1990) 70(4):281–5. [PubMed] [Google Scholar]
- 74.Schmidt E, Ambach A, Bastian B, Brocker EB, Zillikens D. Elevated levels of interleukin-8 in blister fluid of bullous pemphigoid compared with suction blisters of healthy control subjects. J Am Acad Dermatol (1996) 34(2 Pt 1):310–2. 10.1016/S0190-9622(96)80146-0 [DOI] [PubMed] [Google Scholar]
- 75.Grando SA, Glukhenky BT, Drannik GN, Epshtein EV, Kostromin AP, Korostash TA. Mediators of inflammation in blister fluids from patients with pemphigus vulgaris and bullous pemphigoid. Arch Dermatol (1989) 125(7):925–30. 10.1001/archderm.125.7.925 [DOI] [PubMed] [Google Scholar]
- 76.Galli SJ. New concepts about the mast cell. N Engl J Med (1993) 328(4):257–65. 10.1056/NEJM199301283280408 [DOI] [PubMed] [Google Scholar]
- 77.Galli SJ, Gordon JR, Wershil BK. Cytokine production by mast cells and basophils. Curr Opin Immunol (1991) 3(6):865–72. 10.1016/S0952-7915(05)80005-6 [DOI] [PubMed] [Google Scholar]
- 78.Zebrowska A, Wagrowska-Danilewicz M, Danilewicz M, Stasikowska-Kanicka O, Kulczycka-Siennicka L, Wozniacka A, et al. Mediators of mast cells in bullous pemphigoid and dermatitis herpetiformis. Mediators Inflamm (2014) 2014:936545. 10.1155/2014/936545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Castells MC, Irani AM, Schwartz LB. Evaluation of human peripheral blood leukocytes for mast cell tryptase. J Immunol (1987) 138(7):2184–9. [PubMed] [Google Scholar]
- 80.D’Auria L, Pietravalle M, Cordiali-Fei P, Ameglio F. Increased tryptase and myeloperoxidase levels in blister fluids of patients with bullous pemphigoid: correlations with cytokines, adhesion molecules and anti-basement membrane zone antibodies. Exp Dermatol (2000) 9(2):131–7. 10.1034/j.1600-0625.2000.009002131.x [DOI] [PubMed] [Google Scholar]
- 81.Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity (2012) 45(1):55–70. 10.3109/08916934.2011.606447 [DOI] [PubMed] [Google Scholar]
- 82.Bieber K, Ernst AL, Tukaj S, Holtsche MM, Schmidt E, Zillikens D, et al. Analysis of serum markers of cellular immune activation in patients with bullous pemphigoid. Exp Dermatol (2017) 26(12):1248–52. 10.1111/exd.13382 [DOI] [PubMed] [Google Scholar]
- 83.Dimson OG, Giudice GJ, Fu CL, Van den Bergh F, Warren SJ, Janson MM, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol (2003) 120(5):784–8. 10.1046/j.1523-1747.2003.12146.x [DOI] [PubMed] [Google Scholar]
- 84.Messingham KAN, Holahan HM, Fairley JA. Unraveling the significance of IgE autoantibodies in organ-specific autoimmunity: lessons learned from bullous pemphigoid. Immunol Res (2014) 59(1–3):273–8. 10.1007/s12026-014-8547-7 [DOI] [PubMed] [Google Scholar]
- 85.Freire PC, Munoz CH, Stingl G. IgE auto-reactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol (2017) 177(6):1644–53. 10.1111/bjd.15924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fairley JA, Baum CL, Brandt DS, Messingham KAN. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J Allergy Clin Immunol (2009) 123(3):704–5. 10.1016/j.jaci.2008.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu KK, Crew AB, Messingham KAN, Fairley JA, Woodley DT. Omalizumab therapy for bullous pemphigoid. J Am Acad Dermatol (2014) 71(3):468–74. 10.1016/j.jaad.2014.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anhalt GJ, Diaz LA. Animal models for bullous pemphigoid. Clin Dermatol (1987) 5(1):117–25. 10.1016/0738-081X(87)90056-3 [DOI] [PubMed] [Google Scholar]
- 89.Sams WMJ, Gleich GJ. Failure to transfer bullous pemphigoid with serum from patients. Proc Soc Exp Biol Med (1971) 136(4):1027–31. 10.3181/00379727-136-35421 [DOI] [PubMed] [Google Scholar]
- 90.Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest (1993) 92(5):2480–8. 10.1172/JCI116856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest (1995) 95(4):1539–44. 10.1172/JCI117826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest (1997) 100(5):1256–63. 10.1172/JCI119639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nishie W, Sawamura D, Goto M, Ito K, Shibaki A, McMillan JR, et al. Humanization of autoantigen. Nat Med (2007) 13(3):378–83. 10.1038/nm1496 [DOI] [PubMed] [Google Scholar]
- 94.Liu Z, Sui W, Zhao M, Li Z, Li N, Thresher R, et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun (2008) 31(4):331–8. 10.1016/j.jaut.2008.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fairley JA, Burnett CT, Fu CL, Larson DL, Fleming MG, Giudice GJ. A pathogenic role for IgE in autoimmunity: bullous pemphigoid IgE reproduces the early phase of lesion development in human skin grafted to nu/nu mice. J Invest Dermatol (2007) 127(11):2605–11. 10.1038/sj.jid.5700958 [DOI] [PubMed] [Google Scholar]
- 96.Chen R, Fairley JA, Zhao ML, Giudice GJ, Zillikens D, Diaz LA, et al. Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol (2002) 169(7):3987–92. 10.4049/jimmunol.169.7.3987 [DOI] [PubMed] [Google Scholar]
- 97.Brown MA, Hatfield JK. Mast cells are important modifiers of autoimmune disease: with so much evidence, why is there still controversy? Front Immunol (2012) 3:147. 10.3389/fimmu.2012.00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fureder W, Agis H, Willheim M, Bankl HC, Maier U, Kishi K, et al. Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J Immunol (1995) 155(6):3152–60. [PubMed] [Google Scholar]
- 99.Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol (2004) 4(10):787–99. 10.1038/nri1460 [DOI] [PubMed] [Google Scholar]
- 100.Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem (2011) 286(17):15003–9. 10.1074/jbc.M111.221036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schwartz LB, Irani AM, Roller K, Castells MC, Schechter NM. Quantitation of histamine, tryptase, and chymase in dispersed human T and TC mast cells. J Immunol (1987) 138(8):2611–5. [PubMed] [Google Scholar]
- 102.Schwartz LB, Lewis RA, Austen KF. Tryptase from human pulmonary mast cells. Purification and characterization. J Biol Chem (1981) 256(22):11939–43. [PubMed] [Google Scholar]
- 103.Reynolds DS, Stevens RL, Lane WS, Carr MH, Austen KF, Serafin WE. Different mouse mast cell populations express various combinations of at least six distinct mast cell serine proteases. Proc Natl Acad Sci U S A (1990) 87(8):3230–4. 10.1073/pnas.87.8.3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tchougounova E, Pejler G, Abrink M. The chymase, mouse mast cell protease 4, constitutes the major chymotrypsin-like activity in peritoneum and ear tissue. A role for mouse mast cell protease 4 in thrombin regulation and fibronectin turnover. J Exp Med (2003) 198(3):423–31. 10.1084/jem.20030671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Andersson MK, Karlson U, Hellman L. The extended cleavage specificity of the rodent beta-chymases rMCP-1 and mMCP-4 reveal major functional similarities to the human mast cell chymase. Mol Immunol (2008) 45(3):766–75. 10.1016/j.molimm.2007.06.360 [DOI] [PubMed] [Google Scholar]
- 106.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem (2005) 280(10):9291–6. 10.1074/jbc.M410396200 [DOI] [PubMed] [Google Scholar]
- 107.Lin L, Bankaitis E, Heimbach L, Li N, Abrink M, Pejler G, et al. Dual targets for mouse mast cell protease-4 in mediating tissue damage in experimental bullous pemphigoid. J Biol Chem (2011) 286(43):37358–67. 10.1074/jbc.M111.272401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity (2012) 45(1):91–101. 10.3109/08916934.2011.606450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest (2005) 115(4):870–8. 10.1172/JCI21386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Woodley DT, Ram R, Doostan A, Bandyopadhyay P, Huang Y, Remington J, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. J Invest Dermatol (2006) 126(6):1323–30. 10.1038/sj.jid.5700254 [DOI] [PubMed] [Google Scholar]
- 111.Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol (2007) 178(10):6514–21. 10.4049/jimmunol.178.10.6514 [DOI] [PubMed] [Google Scholar]
- 112.Kasprick A, Yu X, Scholten J, Hartmann K, Pas HH, Zillikens D, et al. Conditional depletion of mast cells has no impact on the severity of experimental epidermolysis bullosa acquisita. Eur J Immunol (2015) 45(5):1462–70. 10.1002/eji.201444769 [DOI] [PubMed] [Google Scholar]