

Abstract
A hallmark of imprinted genes in mammals is the occurrence of parent-of-origin-dependent asymmetry of DNA cytosine methylation (5mC) of alleles at CpG islands (CGIs) in their promoter regions. This 5mCpG asymmetry between the parental alleles creates allele-specific imprinted differentially methylated regions (iDMRs). iDMRs are often coupled to the transcriptional repression of the methylated allele and the activation of the unmethylated allele in a tissue-specific, developmental-stage-specific and/or isoform-specific fashion. iDMRs function as regulatory platforms, built through the recruitment of chemical modifications to histones to achieve differential, parent-of-origin-dependent chromatin segmentation states. Here, we used a comparative computational data mining approach to identify 125 novel constitutive candidate iDMRs that integrate the maximal number of allele-specific methylation region records overlapping CGIs in human methylomes. Twenty-nine candidate iDMRs display gametic 5mCpG asymmetry, and another 96 are candidate secondary iDMRs. We established the maternal origin of the 5mCpG imprints of one gametic (PARD6G-AS1) and one secondary (GCSAML) iDMRs. We also found a constitutively hemimethylated, nonimprinted domain at the PWWP2AP1 promoter CGI with oocyte-derived methylation asymmetry. Given that the 5mCpG level at the iDMRs is not a sufficient criterion to predict active or silent locus states and that iDMRs can regulate genes from a distance of more than 1 Mb, we used RNA-Seq experiments from the Genotype-Tissue Expression project and public archives to assess the transcriptional expression profiles of SNPs across 4.6 Mb spans around the novel maternal iDMRs. We showed that PARD6G-AS1 and GCSAML are expressed biallelically in multiple tissues. We found evidence of tissue-specific monoallelic expression of ZNF124 and OR2L13, located 363 kb upstream and 419 kb downstream, respectively, of the GCSAML iDMR. We hypothesize that the GCSAML iDMR regulates the tissue-specific, monoallelic expression of ZNF124 but not of OR2L13. We annotated the non-coding epigenomic marks in the two maternal iDMRs using data from the Roadmap Epigenomics project and showed that the PARD6G-AS1 and GCSAML iDMRs achieve contrasting activation and repression chromatin segmentations. Lastly, we found that the maternal 5mCpG imprints are perturbed in several hematopoietic cancers. We conclude that the maternal 5mCpG imprints at PARD6G-AS1 and GCSAML iDMRs are decoupled from parent-of-origin transcriptional expression effects in multiple tissues.
Keywords: PARD6G-AS1, GCSAML, GRB10, ZNF124, OR2L13, LOC284240, genomic imprinting, imprinting disease
Introduction
Genomic imprinting refers to the epigenetic and epigenomic differentiation of alleles of particular chromosomal loci on the basis of the parent transmitting them (Barlow and Bartolomei, 2014). The process of imprinting entails tagging with chemically stable marks consisting primarily of parent-of-origin-specific (and, therefore, asymmetrical) 5mCpG DNA methylation, enrichment with chemically modified histones, DNA accessibility, and the expression of long non-coding RNAs (lncRNAs) (Skaar et al., 2012). The trademark of genomic imprinting is the transcriptional silencing of the same parental allele in every cell (Chess, 2016). A hallmark of the canonical imprinted genes in mammals is the asymmetrical arrangement of parent-of-origin-dependent methylation at carbon 5 of the cytosine pyrimidine ring in the context of a CpG dinucleotide sequence (5mCpG) in CpG islands (CGIs) in the encompassed promoter regions (Yuen et al., 2011; Docherty et al., 2014; Hannula-Jouppi et al., 2014). The asymmetrical, parental-allele-specific status of 5mCpG at CGIs enables the construction of parent-of-origin-dependent regulatory platforms, which are built through the recruitment of chemical modifications to histones to achieve chromatin segmentations that impose differential repressive versus activation statuses on the parental alleles (Li and Zhang, 2014). Thus, the imprinting of the transmitted alleles from a subset of genes makes them subject to monoallelic transcriptional expression in specific tissues or developmental stages. Parent-of-origin-dependent allele-specific 5mCpG imprints are silencing epigenetic (e.g., heritable) marks that can be set in the germline or somatically acquired and conserved through mitotic divisions (Guo et al., 2014; Smith et al., 2014).
Genomic imprinting occurs in therian mammals (Barlow and Bartolomei, 2014) and angiosperms (Rodrigues and Zilberman, 2015). The exact repertoire of imprinted genes and the precise set of cis-acting regulatory elements, which make up the “imprintome” (Cooper and Constancia, 2010; Skaar et al., 2012), have not been established. In humans, approximately 150 chromosomal loci are known to be imprinted (Morison et al., 2005; Zhang et al., 2010; Jirtle and Murphy, 2012; Da Silva-Santiago et al., 2014; Wei et al., 2014; Cuellar Partida et al., 2017), although no unified, updated information repository is available. Through different imputation strategies, a predicted (candidate) imprinting status has been assigned to over 200 other loci (Luedi et al., 2007; Wei et al., 2014). Dysregulation of the epigenetic marks or mutations of the imprinted loci or domains is associated with 12 congenital diseases (Soellner et al., 2017).
Within the past 5 years, the reanalysis of big-data resource depositories of deep sequencing (methylomes, RNA-Seq and ChIP-Seq) from different tissues in a multitude of individuals or single cells at various developmental stages has substantially increased our ability to discover and characterize imprinted genes. The most informative epigenetic features currently used to detect imprinted loci are the following: (i) the occurrence of allelically methylated regions (AMRs) or genomic intervals exhibiting allele-specific (or allelically skewed) methylation (ASM) at CGIs (Fang et al., 2012; Song et al., 2013; Marzi et al., 2016); (ii) allele-specific expression (ASE) ratios across heterozygous SNPs (Babak et al., 2015; Baran et al., 2015); (iii) asymmetrical allele-specific enrichment of histone modifications associated with activation or repression states at the allele-specific candidate imprinted differentially methylated regions (iDMRs) (Savol et al., 2017); (iv) dysregulation of the intermediate methylation states at candidate iDMRs in diseased versus healthy tissues (Choufani et al., 2011; Docherty et al., 2014; Hannula-Jouppi et al., 2014; Maeda et al., 2014; Kagami et al., 2017); and, most recently, (v) cis methylation quantitative trait loci (cis-meQTLs) that exhibit parent-of-origin effects (POEs), with a physical distance ranging from 0.01 to 743 kb between the effector SNP and the affected 5mCpG site (Cuellar Partida et al., 2017).
Herein, through a comparative computational data mining approach, we report the identification of 125 novel candidate iDMRs in the human genome that integrated the maximal number of AMR records overlapping CGIs in methylomes from several tissues. We show that 29 candidate iDMRs display germline (gametic, primary) 5mCpG asymmetry, whereas 96 are candidate somatic (secondary) iDMRs. We revealed that the maternally inherited 5mCpG imprints for one gametic (PARD6G-AS1) and one somatic (GCSAML) iDMR are decoupled from POEs on the expression of the parental alleles. Lastly, we show that the oocyte-derived 5mCpG imprints are dysregulated in hematopoietic cancers.
Materials and methods
Subjects
Out of 100 trios from the Northern Region of the State of Rio de Janeiro, we included five nuclear families (mother, father, and son or daughter; median ages: 42, 50, and 19 years, respectively) whose genomic DNA (gDNA) had been genotyped as informative for the appropriate SNPs of the target genes. The investigators were blinded to the identities and scores of all trios throughout the genotyping process.
DNA extraction
Human gDNA from freshly drawn peripheral blood samples (2 mL) was extracted using a commercial illustra blood genomicPrep Mini Spin Kit (GE Healthcare, Little Chalfont, UK), essentially as reported earlier (Machado et al., 2014). gDNA was quantified using an ND-NDL-PR NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and kept frozen at −20°C until used. For genotyping, aliquots of 10 ng of gDNA were used in each PCR reaction.
Identification of candidate iDMRs
To identify novel candidate iDMRs, we employed an integrative data mining strategy based on the detection of AMRs, that is, chromosomal regions where the paternal allele is differentially methylated compared with the maternal allele (Fang et al., 2012; Song et al., 2013) and that overlap CGIs in whole-genome methylomes determined by bisulfite sequencing (BS-Seq) experiments. The strategy entailed extracting all fields (whole genome, hg19 assembly) from track data hubs (Raney et al., 2014) for CGIs that have any overlap with the coordinates of the AMR records reported in 39 BS-Seq methylome repositories having a bisulfite conversion rate of at least 0.95 and a minimum depth of 10 reads per CpG site (Dataset S1A: 39 BS-Seq methylomes). For this task, we used the UCSC Genome Browser Data Integrator web tool (Hinrichs et al., 2016), freely available at UCSC Genome Browser (Kent et al., 2002; Raney et al., 2014). We compared the integration results with the 15,282 AMRs identified previously in hg18 (Fang et al., 2012) by annotating them in hg19 using the UCSC Genome Browser LiftOver coordinate conversion tool. The output was used to populate a spreadsheet, and the data were filtered by the number of overlapping AMR records per CGI (Dataset S1B: Novel candidate iDMRs). As an investigative inclusion criterion, we accepted only CGIs that achieved at least 16 overlapping AMR records, which was the minimal number of overlapping records we observed for most of the known gametic (n = 20) and secondary (n = 9) iDMRs (Okae et al., 2014) (Dataset S1C: Known imprinted DMRs). The selected CGI-bearing AMRs were classified as candidate gametic or secondary iDMRs (Dataset S1B) on the basis of asymmetrical hypermethylated versus hypomethylated statuses in BS-Seq methylomes from human oocytes (Okae et al., 2014) (customized in the UCSC Genome Browser as described previously; Alves da Silva et al., 2016) and spermatozoa (Molaro et al., 2011).
Determination of the parental origin of the 5mCpG methylation imprints
To validate the intermediate methylation statuses observed in the candidate iDMRs in public BS-Seq methylomes, we set up methylation-sensitive restriction enzyme PCR (MSRE-PCR) triplex assays (Dataset S1D: Primers and MSRE assay) specific to the candidate iDMRs; these assays were designed following the rationale described previously (Alves da Silva et al., 2016). Each test amplifies three loci: (i) An HpaII-containing X-chromosome-specific monomorphic locus at the predicted promoter region of the PPP2R3B gene, located on the pseudoautosomal region PAR1 in females and the Y chromosome in males. The PPP2R3B promoter region is unmethylated in healthy and cancerous tissues from males and females (Figure S1); thus, it is not subject to gender-related differential methylation or effects of X-chromosome inactivation in women. PPP2R3B is a gonosomal melanoma tumor suppressor gene (van Kempen et al., 2016). The amplimer from this region was used as a positive control for the HpaII restriction enzyme digestion. (ii) The candidate iDMR, encompassing at least one HpaII recognition site and an informative SNP. (iii) A CpG-rich region of the WRB gene, bearing no HpaII sites, used as a reference control for normalization of the ratio of restriction-enzyme-resistant 5mCpG sites in the target candidate iDMR. The methylation statuses were calculated as the proportion of restriction-enzyme-resistant 5mCpG sites using the equation reported previously (Alves da Silva et al., 2016). We genotyped the methylated alleles of informative SNPs (Dataset S1D), e.g., the alleles resistant to digestion with the restriction enzyme, using single-nucleotide primer extension (SNuPE) and SNaPshot technology (Thermo Fisher Scientific).
DNA CpG methylation changes at candidate iDMRs in the placenta
Although there is widespread interindividual polymorphic methylation in the gametic maternal iDMRs that are specific to the human placenta (Hanna et al., 2016), variable maternal methylation in other tissues has been reported only in the somatic maternal iDMR of the tumor suppressor ncRNA VTRNA2-1 (Paliwal et al., 2013; Romanelli et al., 2014; Green et al., 2015). Thus, we annotated the methylation statuses at the selected PARD6G-AS1, PWWP2AP1, and GCSAML iDMRs in 90 public methylome experiments for signs of polymorphic methylation. The inherent statuses are based on methylation profiling by the Illumina Infinium HumanMethylation450K BeadChip array (Hatt et al., 2015, 2016; Hanna et al., 2016). As a quality assurance control, we excluded from the analysis all unspecific cross-reactive probes, probes with p > 0.01, and probes varying among replicates (Chen et al., 2013). The placenta dataset comprises samples from healthy controls (n = 52); cases of preeclampsia (n = 8); and cases of trisomy 13 (Patau syndrome) (n = 6), 18 (Edwards syndrome) (n = 12) and 21 (Down syndrome) (n = 12) (Dataset S1E: Placenta GEO entries). We also included BS-Seq methylomes from three normal placentas (Dataset S1E). We customized tracks at the UCSC Genome Browser and extracted the 5mCpG levels to measure the mean values and standard deviation across the corresponding chromosomal regions.
DNA CpG methylation changes at the candidate iDMRs in BS-Seq methylomes from cancer
Given that aberrant methylation associated with iDMRs is reported in cancer (Barrow et al., 2015), we annotated the variations in the methylation statuses of the candidate iDMRs exhibited in BS-Seq methylomes from hematopoietic cancers in the BLUEPRINT project (August 2016 data release) (Adams et al., 2012) (Dataset S1F: BLUEPRINT malignancies), compared with the intermediate methylation status typically observed in BS-Seq methylomes from healthy tissues. We restricted the analysis to hematopoietic cancers because the GCSAML candidate imprinted gene encodes a putative signaling protein associated with the sites of proliferation and differentiation of mature B lymphocytes (NCBI Resource Coordinators, 2017). The BLUEPRINT project was initially designed to decode the epigenetic signatures of healthy blood and hematopoietic malignancies. The project included samples from bone marrow (acute lymphocytic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, and multiple myeloma) and venous blood (acute lymphocytic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, mantle cell lymphoma, and T-cell prolymphocytic leukemia). We viewed the target regions using the track hub available at the UCSC Genome Browser (Raney et al., 2014), with the hg38 chromosomal coordinates. We extracted the values of the 5mCpG calls, determined the average methylation level across the CpG sites that span the corresponding physical region in healthy tissues, and normalized the data by excluding the CpG sites with no data.
Assessment of transcriptional allele-specific expression
For the genes within 2.3 Mb of the candidate iDMRs in either direction, we queried RNA-Seq experiments for signs of ASE across SNPs in dbSNP (Sherry et al., 2001) having global minor allele frequency (MAF) ≥ 0.1 as reported by the 1,000 Genome Project (Sudmant et al., 2015). We used both secondary (pre-processed) and primary (unprocessed) sources of RNA-Seq experiments.
Secondary source of RNA-Seq experiments
The secondary source comprised the free track hubs for the Genotype-Tissue Expression (GTEx) project (data release V6, October 2015) (The GTEx Project, 2015), available from the UCSC Genome Browser and the dbGaP Data Browser. The GTEx track hubs, as designed and provided by the Lappalainen lab at the New York Genome Center, part of the GTEx Analysis Working Group (Castel et al., 2015), enable users to access the supporting tables. The GTEx track hubs report ASE measurement values at imputed heterozygous variants identified from transcriptome and genotype data collected from 51 primary tissues in RNA-Seq experiments (8,555 samples from 570 postmortem donors) (Dataset S2A: GTEx sample info V7). The ASE measurement values provided by GTEx refer to the median difference in the number of RNA-Seq read counts between the two alleles in an imputed heterozygous SNP across donors, with a depth ≥8 reads per site per donor. ASE is calculated as [0.5–Ref_allele_read count/(Ref_allele_read count + Alt_allele_read count)] (Castel et al., 2015). We used the UCSC Data Integrator web tool to extract the information about the physical coordinates' overlapping gene names; minimum, median, maximum, and quartiles (Q1 and Q3) for ASE values; number of donors; and read depth at SNPs present in the target genes. Because the ancestral allele of an SNP can occur in the context sequence of a different locus, we filtered out such SNPs from the downstream processing using a Short Match script in R-codes. We note that <5% of all SNP sites across the genome are excluded this way. The expression data were classified per gene locus using a script in R-codes that sorts the evidence into four mutually exclusive ASE categories. The ASE categories and their parameters were as follows: strictly monoallelic (minimal, maximal and Q1 ASE values = 0.5); consistent with monoallelic (minimal ASE values <0.5, Q1 and maximal ASE values ≥ 0.33); strictly biallelic (minimal, maximal and Q1 ASE values = 0), and consistent with biallelic (minimal ASE values <0.5, Q1 ASE values <0.33, and maximal ASE values > 0). The R script was validated using the corresponding data for a reference gene set consisting of the 40 known imprinted genes reported by the GTEx Analysis Working Group (Dataset S2B: 40 known imprinted genes) (Babak et al., 2015; Baran et al., 2015), also accessible from the GTEx portal. For the final expression profile designation (on a per-gene and per-tissue basis), we called only the genes with at least three non-discordant informative SNPs per tissue, and a depth ≥12 reads per SNP.
Primary source of RNA-Seq experiments
The primary source consisted of 2,164 RNA-Seq experiments (Dataset S2C: 2,164 SRA experiment atlas), representing an atlas of 25 tissues, selected from the NCBI RNA sequence read archive (SRA) public data repositories. The unicity (e.g., the certainty of excluding mixed samples and disease tissues) of each SRA experiment was established by querying SNPs in SNURF, H19, and PLAGL1, known imprinted genes expressed monoallelically in multiple tissues (Dataset S1D) using the NCBI SRA search web tool, essentially as reported (Alves da Silva et al., 2016). The query sequence strings were 29 nt in length, and the alleles were represented by the International Union of Pure and Applied Chemistry (IUPAC) substitution codes. The unicity inclusion criteria were as follows: (i) ASE ≥ 0.42 (consistent with monoallelic expression), (ii) occurrence of allele flip (both alleles observed monoallelically in individual tissues) and (iii) absence of strictly biallelic patterns.
Assessment of histone modification enrichment
To compile evidence about the likely involvement of the candidate iDMR in the regulation of imprinting, we examined the enrichment of histone modifications associated with activation or repression chromatin states, including promoter and enhancer functions and DNA accessibility (e.g., inter-nucleosome DNase-hypersensitive site distribution). We used the Grid Visualization web resource (available at the Roadmap Epigenomics Browser from the NIH Roadmap Epigenomics Mapping Consortium Bernstein et al., 2010; Roadmap Epigenomics Consortium et al., 2015) and the WashU Epigenome Browser (Zhou et al., 2011) to analyze uniformly processed datasets. We also annotated the non-coding genome in the candidate iDMRs by computationally exploring integrative large-scale functional- and comparative-genomics datasets on gene expression (RNA-Seq, cDNA mapping, active transcriptional start site (TSS) mapping, 5′-cap mapping) using the FANTOM ZENBU Browser (Severin et al., 2014). In summary, we analyzed secondary data from chromatin immunoprecipitation and DNA sequencing (ChIP-Seq) of 31 histone modification marks in 22 healthy human somatic tissues and cell lines (Dataset S1G: Roadmap Epigenomics samples). We extracted both the observed (Roadmap Epigenomics Consortium et al., 2015) and the imputed data points (Ernst and Kellis, 2015) for histone modifications across the candidate iDMRs from every tissue experiment and plotted them using the software GraphPad Prism. The chromosomal segmentation enrichment was compared at five constitutive oocyte-derived iDMRs (GRB10, INPP5F, ZNF331, KCNQ1, MEST) and three secondary maternal iDMRs (MAGEL2, NDN, MEG8) (Dataset S1H: iDMR reference set). The GRB10 iDMR was included as it regulates monoallelic expression from the paternal or maternal allele in a tissue- and isoform-specific fashion (Blagitko et al., 2000; Yoshihashi et al., 2000; Hitchins et al., 2001; McCann et al., 2001; Mergenthaler et al., 2001; Monk et al., 2009).
Genotype-phenotype associations
We queried the PheGenI (Ramos et al., 2014), e-GRASP (Karim et al., 2016), PhenoScanner (Staley et al., 2016), and HaploReg v4.1 (Ward and Kellis, 2016) web database tools to cross-reference SNPs at the PARD6G-AS1, PWWP2AP1, and GCSAML candidate iDMRs (Dataset S3A: iDMR SNP lookup) with a broad range of phenotypes from large-scale genome-wide association studies (GWAS) to gain insights into possible disease-associated loci and to assign chromatin states to the lead variants. We set the p and r2 cut-off values at 5 × 10−8 and 0.8, respectively, and requested results for 1,000 Genomes SNP proxies at significant gametic disequilibrium. We ran the SNP variants within the GRB10 iDMR (Dataset S3A) as a control set of SNPs that did not cross-reference with disease phenotypes in the above databases.
Results
Novel candidate iDMRs
To identify novel candidate iDMRs, we integrated AMR records that overlap annotated CGIs from 39 BS-Seq methylomes, including 26 primary (uncultured) healthy tissues and 13 cell types and cell lines (Dataset S1A). We limited the analysis to the autosomes because evidence of 5mCpG imprinted marks on the X chromosome in women is only now beginning to emerge. We further restricted the screening to CGIs overlapping AMR records in at least 16 methylomes, which was the minimal number of records observed in 29 out of 67 known iDMRs (Okae et al., 2014; Dataset S1C). The analysis yielded 480 CGIs, 168 of which consistently exhibited methylation levels ranging from 0.35 to 0.65 at the overlapping CpG sites in the esophagus tissue methylome, as the reference methylome because every chosen CGI has at least one AMR record in that tissue methylome (Dataset S1B). The distribution of the 168 CGI-bearing AMRs per autosome is shown in Figure 1. Chromosomes 6 through 9 had no hits. Chromosome 19 bears the highest density of hits per megabase. Eleven CGI-bearing AMRs correspond to known gametic iDMRs (10 maternal and one paternal), six to known secondary iDMRs (Okae et al., 2014), and 26 to known candidate iDMRs for which parent-of-origin-specific methylation is uncertain (Court et al., 2013, 2014; Docherty et al., 2014) (Dataset S1B). The 125 remaining CGI-bearing AMRs appear to be new candidate iDMRs. Of these, 29 exhibit asymmetrical methylation statuses in the methylomes of oocytes and spermatozoa; thus, they were classified as candidate gametic iDMRs (18 maternal and 11 paternal). Ninety-six are candidate secondary iDMRs (Dataset S1B).
Figure 1.
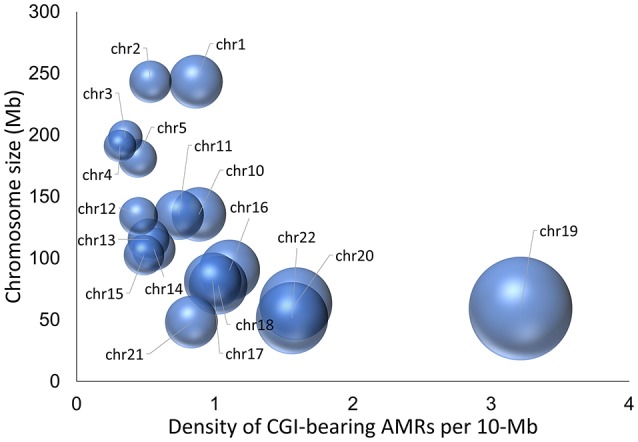
Three-dimensional bubble chart display of the density of novel candidate iDMRs. The diameter of the bubble represents the density of candidate iDMRs per 10 Mb autosomal region, whereas the position of the balloon on the y-axis is related to the chromosomal size in Mb. The highest density of candidate iDMRs is 3.1/Mb on chromosome 19.
Because our laboratory is interested in the discovery of imprinted genes located in the autosomes that are commonly affected by nondisjunction (Alves da Silva et al., 2016), we selected two candidate gametic maternal iDMRs—PARD6G-AS1 (par-6 family cell polarity regulator gamma antisense RNA 1 on chromosome 18; Edwards syndrome) (Figure 2) and PWWP2AP1 (PWWP domain containing 2A pseudogene 1 on chromosome 13; Patau syndrome) (Figure 3)—and the secondary candidate iDMR in the GCSAML (germinal center associated signaling and motility like) gene on chromosome 1 (Figure 4), the autosome with the highest number of candidate iDMR hits (Figure 1). The three candidate iDMRs consistently exhibited intermediate methylation profiles in somatic tissues (levels ranging from 0.35 to 0.65 across all CpG sites) but were hypermethylated (>0.65) in human embryonic stem cell (hESC)-derived CD56+ ectoderm and mesoderm and CD184+ endoderm cell cultures (Figure 5).
Figure 2.

Primary intermediate methylation status at the PARD6G-AS1 predicted promoter region. Chromosome 18 ideogram; physical positions and domain features of the PARD6G-AS1 locus depicting the methylation status at the CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr18:77886816–77926815). The methylation levels are represented on a scale from 0 to 1 (hypomethylated to hypermethylated). The image is centered on the CGI localized in the PARD6G-AS1 predicted promoter region, labeled “PARD6G-AS1 CGI-1.” The light green ticks represent the positions of the CpG sites. The methylation level across the PARD6G-AS1 CGI ranges from 0.35 to 0.65 in adult somatic tissues. In the same region, there is asymmetrical methylation in gametes (hypermethylation in oocytes and hypomethylation in spermatozoa). The PARD6G-AS1 gene partly overlaps the downstream PARD6G gene locus, which is transcribed in the opposite direction from the minus DNA strand. The annotated features are (from top to bottom) the exon-intron organization of the principal and alternative PARD6G-AS1 and PARD6G splice isoforms (variants 1 and 2 in shades of dark blue) and the species-conserved PARD6G-AS1 and PARD6G principal isoform transcripts (ENST00000586421 and ENST00000353265, respectively, in brown) (Rodriguez et al., 2013).
Figure 3.

Primary intermediate methylation status at the PWWP2AP1 processed pseudogene. Chromosome 13 ideogram; physical positions and domain features of the PWWP2AP1 locus showing the methylation status at the CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr13:81210960-81250959). The methylation levels are represented on a scale from 0 to 1 (hypomethylated to hypermethylated). The image is centered on the CGI localized in the PWWP2AP1 predicted promoter-flanking region, labeled “PWWP2AP1 CGI-1.” The light green ticks represent the positions of the CpG sites. The methylation across the CpG sites within the PWWP2AP1 candidate iDMR ranges from 0.35 to 0.65 in adult somatic tissues. In the same region, however, there is asymmetrical methylation in gametes (hypermethylation in oocytes and hypomethylation in spermatozoa). The GENCODE ENST00000429776.2 transcript for the PWWP2AP1 locus is shown. The annotated features (from top to bottom) are as in Figure 2.
Figure 4.

Secondary intermediate methylation status at the GCSAML predicted secondary promoter. Chromosome 1 ideogram; physical positions and domain features of the GCSAML locus displaying the methylation status at the CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr1:247663350-247703349). The methylation levels are represented on a scale from 0 to 1 (hypomethylated to hypermethylated). The image is centered on the intronic CGI (GCSAML CGI-1 track) near the GCSAML predicted secondary promoter-flanking region. The light green ticks represent the positions of the CpG sites. The GCSAML locus overlaps two other annotated genes (GCSAML-AS1 and OR2C3). The annotated features (from top to bottom) are as in Figure 2. The methylation level across the CpG sites within the GCSAML candidate iDMR ranges from 0.35 to 0.65 in adult somatic tissues. In methylomes from male and female gametes and from blastocysts, this region is hypomethylated.
Figure 5.

Intermediate mean methylation level across the PARD6G-AS1, PWWP2AP1, and GCSAML candidate iDMRs. Comparative variation in the average methylation levels across all 5mCpG sites on the candidate iDMRs in the methylomes of healthy tissues depicted in Figures 2–4. In adult somatic tissues, the overall mean levels were 0.39 (PARD6G-AS1 iDMR), 0.42 (PWWP2AP1 iDMR) and 0.42 (GCSAML iDMR). A slightly skewed hypermethylation state (0.70) was observed in the liver for the PARD6G-AS1 iDMR. The three candidate iDMR regions are consistently hypermethylated in hESC-derived CD56+ ectoderm and mesoderm and hESC-derived CD184+ endoderm cell cultures. The light blue zone represents the expected intermediate methylation range (0.35–0.65). Each methylome is represented by a different color of bar.
We validated the intermediate methylation profiles observed in public methylomes in the selected candidate iDMRs using gDNA from venous blood by DMR-specific MSRE-PCR triplex assays (Figure S2).
We next cross-referenced the physical coordinates of the candidate iDMRs with the overlapping domains in public methylomes of control versus diseased specimens. We found that all three candidate iDMRs had been predicted to be differentially methylated in a maternally dependent manner in the placenta (Hamada et al., 2016; Sanchez-Delgado et al., 2016a), but the parental origin of the methylated alleles had not been established (Figures 6A–C). We observed that the PARD6G-AS1 DMR domain is hypomethylated in three imprinted diseases—Beckwith-Wiedemann syndrome, transient neonatal diabetes (Docherty et al., 2014), and pseudohypoparathyroidism in patients with GNAS cluster imprinting defects (Rochtus et al., 2016)—compared with matched control subjects (Figure 6A). Moreover, we revealed that the GCSAML DMR is differentially hypermethylated in patients with Klinefelter syndrome (47,XXY) compared with matched males but not females (Viana et al., 2014; Wan et al., 2015; Figure 6C).
Figure 6.

Cross-reference overlaps of the PARD6G-AS1, PWWP2AP1, and GCSAML candidate iDMRs with differentially methylated regions reported in control-disease methylomes and healthy placentas. Customized graphics of the intersections between the candidate iDMRs at PARD6G-AS1 (A), PWWP2AP1 (B) and GCSAML (C) and the public records reporting parent-of-origin predictions, abnormal methylation profiles, and haplotype-dependent allele-specific 5mCpG sites. In each panel, the annotated features are (from top to bottom) the exon-intron organization of the principal and alternative isoforms, the evidence of large intergenic non-coding RNAs (lincRNAs) and small non-coding RNAs (sncRNAs) (DASHR RNA [pos] and [neg] tracks), the FANTOM5 transcriptional start sites (TSS peaks track) (Lizio et al., 2017), the Ensembl Regulatory Build predicted promoters (Ensembl Reg Build track) and transcriptional factor binding sites (TFBS summary track) (Cunningham et al., 2015), CpG islands and CGI-bearing AMRs (this study) and the comparative custom Fang_AMR track (Fang et al., 2012). (A) The PARD6G-AS1 CGI-1 overlaps the domain previously predicted to be differentially methylated in a parent-of-origin-dependent manner in blastocysts and placenta (Okae et al., 2014; Hamada et al., 2016; Sanchez-Delgado et al., 2016a) (Hamada_DMRs, Sanchez_DMRs_set:I, and Sanchez_DMRs_set:II tracks, respectively), for which the parental origin of the methylated alleles had not been established. The allelic methylation levels in the aforementioned regions are maternally skewed, varying from 0.69 in first- and second-trimester to 0.82 in full-term placentas (Hamada_1st_trim and Hamada_2nd_trim_full tracks). The PARD6G-AS1 CGI-1 also intersects with the domains reported to be hypomethylated in patients with Beckwith-Wiedemann syndrome and transient neonatal diabetes (Docherty et al., 2014) (Docherty_DMRs track) and in pseudohypoparathyroidism patients with GNAS cluster imprinting defects (Rochtus et al., 2016) (Rochtus_DMRs track), as well as with the haplotype-dependent allele-specific 5mCpG sites reported in placenta, liver, lung, brain and T cells (Do_Hap:ASM track) (Do et al., 2016). The genes that are known to be biallelically expressed in placenta (Hamada et al., 2016) are depicted in the Hamada_biallelic track. (B) The PWWP2AP1 CGI-1 overlaps the domain previously predicted to be differentially methylated in a parent-of-origin-dependent manner in blastocysts and placenta (Hamada et al., 2016; Sanchez-Delgado et al., 2016a) (Hamada_DMRs, and Sanchez_DMRs_set:II tracks, respectively), for which the parental origin of the methylated alleles had not been established. The methylation levels in the aforementioned regions varied from 0.38 in first- and second-trimester placentas to 0.24 in full-term placentas (Hamada_1st_trim and Hamada_2nd_trim_full tracks). (C) The GCSAML CGI-1 overlaps the domain reported to be skewed toward hypermethylation in Klinefelter syndrome (47,XXY karyotype) versus control males (Viana et al., 2014; Wan et al., 2015) (Viana-hyperDMR-in-KS and Wan-hyperDMR-in-KS), for which the parental origin of the allele methylation had not been established.
We also searched for evidence of haplotype-dependent allele-specific 5mCpG sites within the selected candidate iDMRs by integrating the corresponding records reported in methylomes (Do et al., 2016). Within the PARD6G-AS1 candidate iDMR, we noted evidence of haplotype-dependent allele-specific 5mCpG sites in placenta, liver, lung, brain and T cells methylomes (Figure 6A).
The 5mCpG sites at the PARD6G-AS1 and GCSAML DMRs are maternally inherited
The methylation statuses of the transmitted alleles were determined by genotyping the methylation-sensitive HpaII-resistant alleles amplified from venous blood gDNA samples from nuclear trios (mother, father, and son or daughter) informative for SNPs within the candidate iDMRs for the PARD6G-AS1 (upstream gene indel variant rs11281142: -/CTGTGGTGC), PWWP2AP1 (rs1176323), and GCSAML (intron variant, upstream gene variant rs6700954) loci (Dataset S1D). The 5mCpG sites at the PARD6G-AS1 (Figure 7A) and GCSAML (Figure 7B) DMRs are maternally inherited. By contrast, at the PWWP2AP1 candidate iDMR, both parental alleles are methylated (Figure 7C). Given the asymmetrical methylation statuses observed in the BS-Seq methylomes of oocytes and spermatozoa at the PARD6G-AS1 iDMR (Figure 2), we inferred that the 5mCpG imprints are primary (e.g., oocyte-derived). Thus, we classified this domain as a gametic maternal iDMR. On the other hand, the 5mCpG imprints at the GCSAML iDMR are not oocyte-derived but somatically acquired) (Figure 4), making the domain a secondary maternal iDMR. The primary versus secondary designations above are concordant with the emerging view that, in blastocysts, the primary iDMRs (Figures 2, 4) exhibit intermediate methylation levels while the secondary iDMRs are hypomethylated (Okae et al., 2014; Hamada et al., 2016; Sanchez-Delgado et al., 2016a).
Figure 7.
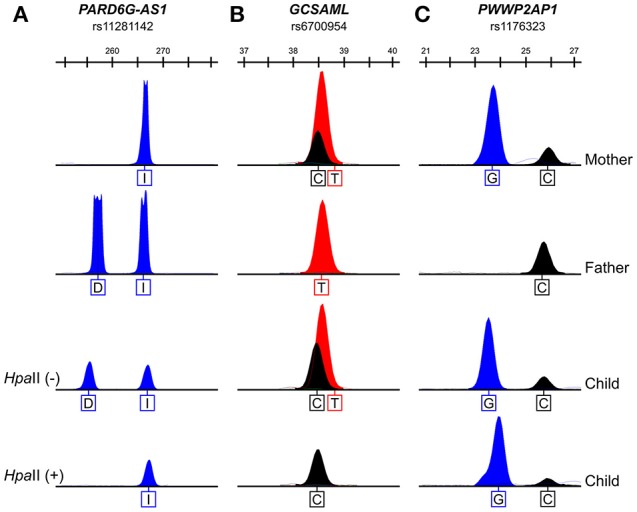
The 5mCpG sites at the PARD6G-AS1 and GCSAML iDMRs are on the maternally transmitted alleles. The maternally transmitted alleles and not the paternally transmitted alleles at the PARD6G-AS1 (A) and GCSAML (B) DMRs are consistently methylated, as supported by the resistance of the maternally inherited alleles to digestion with the methylation-sensitive HpaII restriction enzyme and the lack of amplification of the paternally transmitted alleles from the digested genomic DNA samples for the informative variant SNPs (the PARD6G-AS1 upstream gene indel variant rs11281142 (I = insertion of CTGTGGTGC; D = deletion of CTGTGGTGC) and the GCSAML intron variant/upstream gene variant rs6700954, respectively) in three representative nuclear families that were informative for at least one SNP. Therefore, the 5mCpG marks in the PARD6G-AS1 and GCSAML DMRs are maternal imprints. On the other hand, both parental alleles of the PWWP2AP1 rs1176323 variant are resistant to digestion (C). Thus, the 5mCpG marks in the PWWP2AP1 CGI-1 occur in a parent-of-origin-independent manner. Given the intermediate rate of methylation in PWWP2AP1 CGI-1 (see Figure 5), the occurrence of 5mCpG sites in both parental alleles indicates that the PWWP2AP1 CGI-1 alleles are methylated in a stochastic fashion.
The parent-of-origin-independent methylation at the PWWP2AP1 candidate iDMR is not consistent with imprinting, despite the occurrence of the asymmetrical methylation observed in gametes (Figure 3). The biallelic methylation and the intermediate methylation seen at the PWWP2AP1 DMR in various BS-Seq somatic methylomes, including those of blastocysts (Figure 3), support the scenario of random (e.g., switchable) allelic methylation of this domain. Therefore, the gametic 5mCpG asymmetry observed at the PWWP2AP1 promoter CGI is not maintained in the somatic tissues investigated. The PWWP2AP1 CGI appears to be the first example of a constitutively hemimethylated, nonimprinted domain that is hemimethylated in blastocysts.
The maternal 5mCpG sites are not polymorphic in the placenta
The methylation statuses at the PARD6G-AS1 and GCSAML iDMRs in BS-Seq and 450K array placenta methylomes were coherent with the intermediate statuses observed in methylomes from diverse tissues (Figure 8). Overall, the intermediate methylation level was undisturbed in normal, preeclampsia, and trisomy 13, 18, and 21 placentas. However, we noted hypomethylation (observed lower limit 0.26) for the PARD6G-AS1 iDMR in some samples from trisomy 18 (Figure 8A). The hypomethylated status of some but not all trisomy 18 placentas suggests a paternal origin of the nondisjunction of the supernumerary chromosome 18 in some placentas (e.g., two unmethylated paternal allele copies versus one maternal methylated allele; expected methylation rate ≤ 0.33). We reported a similar nondisjunction parent-of-origin effect for the known maternal gametic iDMR in the WRB gene in chromosome 21 in cases of paternal trisomy 21 (Alves da Silva et al., 2016). Interestingly, the methylation level in the WRB iDMR appears to be perturbed in trisomy 18 placentas (Figure 8D).
Figure 8.
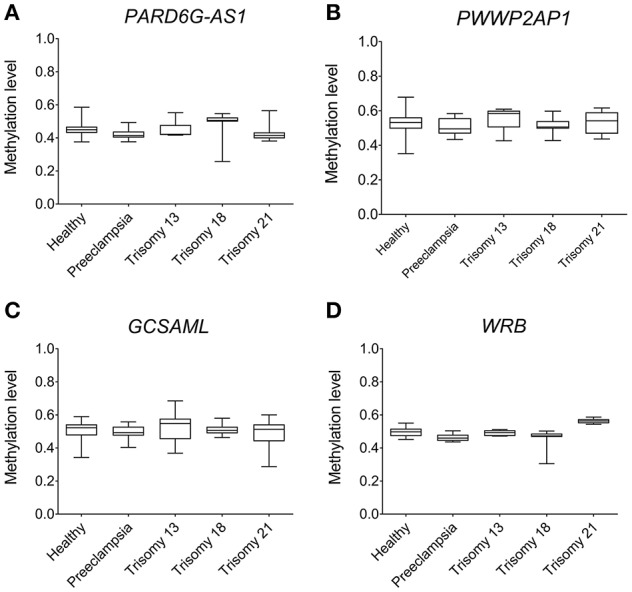
Absence of epipolymorphism at the PARD6G-AS1 and GCSAML maternal iDMRs in placentas from healthy and diseased pregnancies. The new maternal iDMRs are consistently hemimethylated in 450 K array (n = 90) methylomes from healthy; preeclampsia; and trisomy 13, 18, and 21 placentas. Comparison of the observed methylation levels in (A) the PARD6G-AS1 iDMR, (B) the PWWP2AP1 DMR, (C) the GCSAML iDMR, and (D) the WRB iDMR. Note that none of the conditions led to an unmethylated or hypermethylated status at the iDMRs. The hypomethylated status at the PARD6G-AS1 iDMR in some but not all trisomy 18 placentas suggests a paternal origin of the nondisjunction of the extra copy of chr18 in some placentas. The supernumerary chromosome 18 also perturbs the methylation status of the WRB iDMR in chromosome 21.
5mCpG DNA methylation dysregulation in the novel iDMRs in hematopoietic cancers
The methylation statuses at the PARD6G-AS1 and GCSAML iDMRs were perturbed in the methylomes of hematopoietic cancers, with both hypo- and hypermethylated profiles observed in different types of malignancies (Figure 9). Similar patterns of dysregulation were found in the known PPIEL and DIRAS3 iDMRs located on the same or different chromosomes than the novel PARD6G-AS1 and GCSAML iDMRs, respectively (Figure 9). Thus, the new PARD6G-AS1 and GCSAML imprinted loci undergo cancer-associated epigenetic changes in hematopoietic malignancies.
Figure 9.

Aberrant 5mCpG DNA methylation at the new maternal iDMRs in hematopoietic cancers. The 5mCpG DNA methylation statuses at the PARD6G-AS1 and GCSAML iDMRs are perturbed in methylomes of hematopoietic cancers, displaying either hypo- or hypermethylated profiles. Abnormal patterns are also observed at the known PPIEL and DIRAS3 iDMRs, located on chromosome 1, which were used as a reference set. The light blue zone represents the expected intermediate methylation range (0.35–0.65). Each methylome is represented by a different color of bar.
The 5mCpG epigenetic imprints are decoupled from parent-of-origin expression effects in multiple human tissues
Given that an iDMR can control the parent-of-origin-dependent monoallelic expression of a cluster of genes, we searched for evidence of ASE of genes located within 2.3 Mb of the PARD6G-AS1 and GCSAML iDMRs in either direction. The 2.3 Mb range was selected because it matches the distance between the genes controlled by the SNURF iDMR (Eggermann et al., 2015). It is worth noting that certain known iDMRs are dissociated from POEs (Alves da Silva et al., 2016). We used two large sets of RNA-Seq experiments from 52 primary tissues to search for ASE across SNPs within 4.6 Mb chromosomal region spans, centered at the PARD6G-AS1 and GCSAML iDMRs and the PWWP2AP1 pseudogene. In the secondary GTEx set of RNA-Seq experiments, we designated a gene expression profile only if the evidence included at least three non-discordant SNPs with ≥12 reads each per tissue. Overall, we observed no signs of monoallelic expression for PARD6G-AS1 or GCSAML and no transcriptional activity of the PWWP2AP1 pseudogene in either the secondary (Dataset S2A) or the primary (Dataset S2C) source of RNA-Seq experiments. PARD6G-AS1 was expressed biallelically in six tissues: brain amygdala (4 SNPs), brain putamen (5 SNPs), heart atrial appendage (6 SNPs), pituitary (4 SNPs), testis (3 SNPs) and thyroid (7 SNPs) (Dataset S2D: 4.6 Mb screen analysis). GCSAML was expressed biallelically in five tissues: visceral adipose tissue (omentum) (4 SNPs), brain - cerebellar hemisphere (6 SNPs), brain cerebellum (4 SNPs), prostate (6 SNPs), and testis (6 SNPs).
In humans, certain iDMRs can control the expression of genes that are up to 2.3 Mb away in either direction (Eggermann et al., 2015). Therefore, we searched the GTEx dataset for signs of monoallelic expression of genes neighboring the novel iDMRs (Dataset S2D). On chromosome 1, we found evidence of tissue-specific monoallelic expression of ZNF124 (lung; 5 SNPs) and OR2L13 (testis; 6 SNPs). ZNF124 and OR2L13 are located 363 kb upstream and 419 kb downstream, respectively, of the GCSAML iDMR (Dataset S2D). The ZNF124 locus has no overlapping hemimethylated CGIs in normal tissues, including lung tissue (Figure S4A). In the placenta (Hamada et al., 2016), there is evidence of biallelic expression of ZNF124 (Figure S4B, Dataset S2D).
On the other hand, the OR2L13 locus has a predicted CGI-bearing AMR overlapping the promoter region (Figure S5A) in blood, spleen, lung, liver, esophagus, and brain methylomes, as well as two intergenic DMRs, predicted to be maternal in the placenta (Figure S5B). Nevertheless, the CGI-bearing AMR is hypomethylated in gamete, blastocyst, placenta, thymus, and spinal cord methylomes and hypermethylated in hESC-derived CD56+ ectoderm and mesoderm and in hESC-derived CD184+ endoderm cell cultures (Figure S5A). Unfortunately, there are no BS-Seq or 450 K methylomes available from testis tissue; thus, we cannot establish the methylation status in that tissue.
On chromosome 18, we found evidence of pituitary-specific expression of the RP11-567M16.1-LOC284240 locus (8 SNPs) (Dataset S2D). The RP11-567M16.1-LOC284240 locus encodes a lincRNA. At loci 18.6 and 4.9 kb upstream, we annotated two predicted paternal DMRs, neither of which exhibited a constitutively or a tissue-specific hemimethylated profile (Figure S6). Because no BS-Seq or 450 K methylomes are available for the pituitary, we cannot rule out the possibility of a pituitary-specific iDMR. Importantly, a CGI (LOC284240 CGI-2) located 3.2 kb upstream from the LOC284240 locus shows an intermediate methylation status with paternal, rather than maternal, methylation asymmetry in gametes (Figure S7) in BS-Seq methylomes from various tissues. The hemimethylated status at LOC284240 CGI-2 is not constitutive because CGI-2 is hypermethylated in spleen, liver, thymus, and hESC-derived cells (Figure S7). Querying the primary RNA-Seq experiments from lung (ZNF124), testis (OR2L13) and pituitary (LOC284240) tissues yielded >10 reads only for the ZNF124 SNPs, with no evidence of biallelic expression (Dataset S2E).
From the 4.6 Mb screen described above, we selected the SNPs with MAF ≥0.1 that exhibited ≥80 reads in the GTEx subset (n = 47; Dataset S2F: Heatmap of top SNPs) and queried them against the primary set of 2,164 RNA-Seq experiments. We confirmed the evidence of biallelic expression with ≥20 reads for 46 SNPs, including the four SNPs in PARD6G-AS1 (Dataset S2G: ASE top biallelic SNPs). The only discordant profile found was for the SNP rs75287701 in the HSBP1L1 gene, which showed monoallelic expression in 10 tissues. The discordance is apparently due to the low allele G frequency (0.144) in the general population. Lastly, we observed no evidence of RNA-Seq expression for the PWWP2AP1 pseudogene in either the secondary or the primary subset of RNA-Seq experiments. Nevertheless, the neighboring loci are biallelically expressed in multiple tissues (Dataset S2D).
Epigenomic chromatin segmentation at the PARD6G-AS1 and GCSAML iDMRs
Constitutive iDMRs (e.g., those that are stably maintained through mitosis in many tissues and cell lines) can regulate ASE of the underlying gene(s) in an isoform-specific and tissue-specific manner. For example, the oocyte-derived GRB10 intronic iDMR (Arnaud et al., 2003), depicted in Figure S3, regulates the imprinting of the paternal and maternal GRB10 alleles in a highly tissue- and isoform-specific manner (Blagitko et al., 2000; Yoshihashi et al., 2000; Mergenthaler et al., 2001). GRB10 is biallelically expressed in multiple tissues (Blagitko et al., 2000; Monk et al., 2009; Babak et al., 2015; Baran et al., 2015), including the growth plate cartilage (McCann et al., 2001), which is the tissue most important for linear growth. Importantly, there is alternate monoallelic expression in brain (transcribed from the paternally transmitted allele), skeletal muscle (from the maternal gamma 1 and gamma 2 isoform alleles) (Blagitko et al., 2000; Yoshihashi et al., 2000), human lymphocyte/rodent somatic hybrid cells (from the maternal allele) (Yoshihashi et al., 2000), and placental villous trophoblasts (from the maternal allele) (Monk et al., 2009). To gain insight into the epigenomic architecture regulating this class of POEs and to explore the possibility of the PARD6G-AS1 and GCSAML iDMRs similarly controlling ASE, we compared the chromatin segmentation enrichment states achievable by these two constitutive maternal iDMRs through chemically modified histones. We used data from the integrative analysis of 111 reference epigenomes (Roadmap Epigenomics Consortium et al., 2015). We cross-referenced those data with the combinatorial histone signatures associated with five known maternal gametic iDMRs (Dataset S1G, Figure 10A) and three maternal secondary iDMRs (Figure 10B) in tissues where the corresponding genes are expressed either monoallelically or biallelically (Babak et al., 2015; Baran et al., 2015; Dataset S2B). We identified differential enrichment signatures for the iDMRs of INPP5F, ZNF331, GRB10, KCNQ1, MAGEL2, and MEG8 in tissues with monoallelic expression (Figure 10). For the PARD6G-AS1 and GCSAML iDMRs, genes for which monoallelic expression was absent from several tissues, we did not identify a tissue-specific enrichment signature (Figure 10). The PARD6G-AS1 iDMR showed a chromatin segmentation enrichment with the H3K4me1, H3K4me3, and H3K27ac activation marks (Figure 10). The GCSAML iDMR showed enrichment of the H3K9me3 repressive mark. For the GRB10 maternal gametic iDMR, we observed a brain-specific differential enrichment of the H3K27ac activation tag (Figure 11), which we propose to be associated with the differential reading of the 5mCpG imprints in the brain.
Figure 10.

Absence of a tissue-specific histone modification enrichment signature at the PARD6G-AS1 and GCSAML iDMRs. Hierarchical clustering analysis of the histone modification enrichment profiles associated with maternal (A) gametic and (B) secondary iDMRs. The color intensity is correlated with the magnitude of the histone modification enrichment observed in tissues in which the genes are expressed either monoallelically or biallelically. Differential enrichment signatures in tissues with monoallelic expression are revealed for the GRB10, MEG8, and MAGEL2 iDMRs. The PARD6G-AS1 and GCSAML iDMRs were associated with contrasting chromatin segmentation marked by enrichment of activation and repression marks, respectively.
Figure 11.

H3K27ac is differentially enriched at the GRB10 maternal iDMR in the brain. (A) Distribution of the H3K27ac activation mark across the GRB10 maternal iDMR in 22 tissues and one stem cell line. (B) A constitutively hemimethylated status is observed in all tissue methylomes. The line traces in (A) correspond in color to the tissue methylation profiles in (B). Despite the constitutive hemimethylated status, there is a brain-specific enrichment of H3K27ac, an activation mark, at the GRB10 iDMR; this enrichment is taken to be a differential signature of the active paternal allele in the brain.
iDMR genotype-phenotype associations
By cross-referencing SNPs in a broad range of phenotypes from large-scale GWAS repositories, we found the lead variant rs1050919 in the PARD6G-AS1 iDMR to exert cis methylation QTL effects 113,318 bp away, on the TXNL4A gene at the chr18:77793182 hg19 coordinate cg04727522 probe, in frontal cortex and caudal pons (genome-wide p-value 3.39 × 10−8) (Dataset S3B: iDMR effect SNPs; and Dataset S3C: Effect and proxy SNP MAF). We note that H3K4me1, H3K4me3, and H3K27ac all contribute to the promoter-enhancer chromatin active state assignment at the lead variant rs1050919 in multiple tissues (Dataset S3D: rs1050919 chromatin states). We also found 38 proxies (linkage disequilibrium r2 ≥ 0.8) of two other lead SNPs (rs56357216 and rs1106086) in the PARD6G-AS1 iDMR exerting the same cis-meQTL effect in temporal cortex (p < 1.76 × 10−8) (Datasets S3B,C). These two lead variants are associated with an active TSS regulatory chromatin state in >20 epigenomes (Dataset S3E: rs56357216 chromatin states and Dataset S3F: rs1106086 chromatin states). On the other hand, the rs7550918 proxy of the lead variant rs10925069 at the GCSAML iDMR showed a negative association with platelet counts (p = 3.00 × 10−11) (Dataset S3B). H3K4me1, H3K4me3, H3K27ac, and H3K9ac contribute to the chromatin active state assignment at the lead SNP rs10925069 in blood cells and only a few other tissues (Dataset S3G: rs10925069 chromatin states).
Discussion
We demonstrated the occurrence of maternally inherited 5mCpG imprints at the DMRs linked to the non-coding RNA PARD6G-AS1 and the protein-coding GCSAML gene. The 5mCpG imprints are oocyte-derived (PARD6G-AS1 iDMR) or somatic (GCSAML iDMR), and their inheritance is dissociated from POEs in at least 10 adult somatic tissues, as determined by RNA-Seq profiles that were robustly consistent with biallelic expression across multiple informative SNPs. In the multitissue transcriptomes investigated, we did not see evidence for monoallelic expression from the parental alleles at SNPs that are isoform-specific or those that overlap isoforms or are in overlapping genes. Our study has a critical limitation regarding the number of informative SNPs covering the entire set of known or predicted genes included in each of three 4.6 Mb genomic regions that were screened for signs of allele expression profiles. The coverage rate of genes with informative SNPs ranged from 30% (PWWAP2AP1) to 59% (PARD6G-AS1 and GCSAML) (Dataset S2H: Effective gene coverage). The limitation derives from the conservative and stringent criteria used to call the allele transcriptional profiles of genes. Specifically, we reported only unambiguous evidence supported by at least three informative SNPs per gene per body tissue. The findings do not rule out the possibility that, in other datasets with considerably higher read depths, the encompassed genes that our analysis found uninformative may exhibit profiles consistent with monoallelic expression. Moreover, our approach cannot rule out the possibility of monoallelic expression in tissues in which the levels of expression are below the limit of detection of 12 reads per SNP accepted in our experimental design, or in tissues that are not covered by the atlas set of 2,164 SRA RNA-Seq experiments. Similarly, we cannot exclude the possibility that the iDMRs are associated with the expression from the paternal allele of coding and non-coding genes that are over 2.3 Mb away in either direction, which is the greatest physical distance between any known iDMR and the underlying controlled gene(s) (e.g., the SNRPN/SNURF iDMR) (Eggermann et al., 2015).
A second significant limitation of the present study is that the experimental setting does not permit us to assess cell-specific differences in 5mCpG methylation levels, RNA-Seq allele expression, or enrichment of histone modifications within the same homogeneous cells, which may add considerable noise to the tissue-specificity analysis reported here. We recognize that the majority of experiments on 5mCpG DNA methylation, RNA-Seq and ChIP histone modifications from public repositories are conducted with tissue samples (e.g., a heterogeneous mixture of cell types), which may confound our findings. It is possible, for example, that a given gene is monoallelically expressed in a parent-of-origin-dependent manner in just one cell type (e.g., platelets, lymphocytes or monocytes in peripheral blood) while the expression profile in whole blood is consistently biallelic. Moreover, we are aware that metabolic differences between cultured tissues and tissues collected by biopsy from living or necropsy donors (e.g., the GTEx study) must be kept in mind when trying to replicate our results with other biosamples.
The maternal origin of the 5mCpG imprints at the PARD6G-AS1 gametic iDMR enables a direct interrogation of the parental origin of the nondisjunction in trisomy 18 index cases without the need to test gDNA from the progenitors, since the methylation levels at the iDMR will be differential in cases of paternally (skewed hypomethylation; ≤0.33) versus maternally inherited (skewed hypermethylation; ≥0.66) supernumerary chromosome 18. This parent-of-origin-dependent epigenetic feature of iDMRs was recently explored in the identification of the parental origin of trisomy 21 nondisjunction on the basis of the WRB gametic iDMR (Alves da Silva et al., 2016).
We note that the maternally derived 5mCpG imprints at the PARD6G-AS1 and GCSAML iDMRs displayed concordant methylation levels in 90 human placentas, with no evidence for epipolymorphism in 5mCpG DNA methylation level. The finding is consistent with the rapidly evolving concept that only placenta-specific maternal iDMRs represent stochastic (e.g., switchable) epipolymorphic traits (Yuen et al., 2009; Hamada et al., 2016; Hanna et al., 2016; Sanchez-Delgado et al., 2016a). The epipolymorphism in placenta-specific maternal iDMRs is interindividual (e.g., across individual placentas), and it is not associated with distinct profiles generated by sampling different sections of the same placenta (Yuen et al., 2009).
Regarding other possible biological relevance of the 5mCpG imprints, we note that significant abnormal hypomethylation has been reported for individual CpG sites located within the PARD6G-AS1 gametic iDMR in some patients with one of three imprinted defects. Between 3 and 9 CpG sites are affected in Beckwith-Wiedemann syndrome (the lowest p = 1.01 × 10−19) and transient neonatal diabetes (4.38 × 10−71) patients, respectively, compared with matched control subjects (Docherty et al., 2014), while two encompassing CpG sites are impacted in pseudohypoparathyroidism patients with GNAS cluster imprinting defects (Rochtus et al., 2016).
An epigenome-wide association study of aggressive behavior showed a significant positive relationship (p = 9.0 × 10−7, FDR = 0.18) between the increase in methylation (over the average intermediate level; mean 0.35, SD = 0.096) in venous blood at the cg06092953 site (located 135 bp downstream from the newly validated PARD6G-AS1 maternal iDMR) and the increase in residual aggression scores across the entire Netherlands Twin Register (van Dongen et al., 2015). Thus, we hypothesize that heritability influences the aforementioned epigenetic association with aggression.
Similarly, the methylation status at the cg18973878 site (located 179 bp upstream from the non-coding PARD6G-AS1 maternal iDMR) was recently identified as a cis-meQTL, significantly affected when the rs11659843 SNP minor allele T is transmitted from the father (genome-wide p-values of 1.95 × 10−7−5.32 × 10−21) (Cuellar Partida et al., 2017). The SNP exerting the cis-meQTL effect is intronic to the protein-coding PARD6G gene, and it maps 79,598 bp (hg19) away from the PARD6G-AS1 maternal iDMR, which evidently does not overlap the PARD6G gene.
We also note that within the PARD6G-AS1 maternal iDMR, evidence for haplotype-dependent allele-specific 5mCpG sites has been reported in several methylomes (Do et al., 2016). We propose that the epi-haplotypes above occur in a parent-of-origin-dependent manner and, thus, that they would be useful in studying susceptibility to complex imprinted genetic diseases (Sanchez-Delgado et al., 2016b).
We notice that a significant change toward a hypermethylated status at four CpG sites (β differences ranged from +0.15 to +0.22) within the chromosomal region corresponding to the GCSAML secondary maternal iDMR has been reported in postmortem prefrontal cortex in one case of Klinefelter syndrome (47,XXY karyotype) versus control males (Viana et al., 2014), and the perturbation was subsequently confirmed in a second study using venous blood samples from five patients with Klinefelter syndrome (Wan et al., 2015). The perturbed 5mCpG sites are cg05639522, cg18198730, cg11166453, and cg07313626 (genome-wide p-values ranged from 3.85 × 10−5−5.07 × 10−7). The first Klinefelter case had comorbid schizophrenia along with a notably reduced cerebellum mass, and the β difference in methylation levels between the index case and sex-matched control samples was 0.16 (p = 1.35 × 10−6) (Viana et al., 2014). In both studies, the difference from female controls was nonsignificant. Thus, the supernumerary X chromosome in the index patients may be the direct cause of the disturbance in the imprinted intermediate methylation at this autosomal locus. It follows that the epigenetic perturbance may be linked to the development and evolution of psychotic spectrum conditions, as has been suggested for differential methylation of the candidate imprinted GNAL gene with possible maternal effects in the linkage to psychosis (reviewed in Crespi, 2008). On the other hand, we showed herein that the supernumerary autosomes 13, 18, and 21 cause significant changes in the asymmetrical methylation statuses at this iDMR in the placenta.
We hypothesize that the GCSAML maternal iDMR regulates the lung-specific, monoallelic expression of ZNF124 from the paternal allele. Addressing this possibility will require establishing the parental origin of the expressed allele(s) and carrying out functional genetics studies to determine the effects of, for example, permanently perturbing the distant GCSAML iDMR. ZNF124 encodes a potential transcriptional factor, and no variants have been flagged in GWAS. ZNF124 is a candidate gene for the Dandy-Walker complex at 1q44 (Poot et al., 2007). Interestingly, the reported Dandy-Walker complex propositus had a 5 Mb segmental aneuploidy (spanning the ZNF124, GCSAML, and OR2L13 genes) of the 1q44 → qter region due to a paternal t(1;20) (q44;q13.33). Thus, in the scenario of maternal genomic imprinting, the monoallelic expression of the predicted paternal allele will be precluded in the haploinsufficient patient, who would manifest the presumed imprinting defect as a pathogenic null mutation.
We view the testis-specific monoallelic expression of OR2L13 as indicative of allele exclusion rather than genomic imprinting coupled to the distant GCSAML iDMR. The reasons are as follows: the OR2L13 gene encodes an olfactory receptor, and the monoallelic expression of olfactory receptors is mediated by epigenetic mechanisms leading to allelic exclusion (Monahan and Lomvardas, 2015). It is worth noting that the abovementioned 5 Mb segmental aneuploidy of the 1q44 → qter region also encompasses the cluster of olfactory receptor genes that includes OR2L13 (Poot et al., 2007).
To date, the placenta is the only confirmed source of tissue-specific iDMRs (Hanna et al., 2016). The candidate LOC284240 paternal iDMR occurs in the skin, lung, fetal brain cortex, fetal spinal cord, and placenta; therefore, it exemplifies a new variable type of iDMR. We propose that the observed pituitary-specific monoallelic expression of the RP11-567M16.1-LOC284240 locus is coupled to the novel candidate paternal iDMR represented by LOC284240 CGI-2, rather than to the distant PARD6G-AS1 maternal iDMR.
Finally, we view as unusual the finding that the maternal secondary GCSAML iDMR is not associated with a primary iDMR within a 4.6 Mb genomic span. Canonical secondary iDMRs are found most frequently near functional gametic iDMRs. For example, in the case of the gametic IG-DMR and the secondary MEG3-DMR (15.9 kb apart), there is evidence of hierarchical regulation of imprinting in 14q32. The IG-DMR regulates the methylation state of the MEG3-DMR, but not vice versa (Beygo et al., 2015). Although our analysis does not rule out the possibility of the occurrence of a primary iDMR beyond the 2.3 Mb limits in either direction, it appears that the GCSAML iDMR is the second example in humans of a secondary iDMR that lacks a primary iDMR. The first reported iDMR lacking a known constitutive, primary iDMR was the maternal ZDBF2 iDMR (Kobayashi et al., 2013). In contrast to the GCSAML iDMR, the orphan ZDBF2 iDMR regulates the monoallelic expression of the ZDBF2 gene in at least 46 tissues (Dataset S2B; Baran et al., 2015).
Concluding remarks
We identified 125 constitutively hemimethylated domains in the human genome. Twenty-nine domains displayed asymmetrical 5mCpG imprints in gametes, including 18 oocyte-derived and 11 spermatozoon-derived epigenetic marks, and those 29 domains were classified as candidate gametic (primary) iDMRs. The remaining 96 hemimethylated domains were classified as candidate secondary iDMRs. We established the maternal inheritance of the 5mCpG constitutive imprints at the PARD6G-AS1 and GCSAML iDMRs and their disassociation from POEs in the allelic transcriptional expression profiles across a 4.6 Mb domain around each iDMR in multiple human primary tissues. Our results also revealed the occurrence of a constitutively hemimethylated, nonimprinted domain at PWWP2AP1 CGI-1 with oocyte-derived methylation asymmetry. The maternally inherited 5mCpG imprints at the PARD6G-AS1 and GCSAML iDMRs are perturbed in hematopoietic cancers.
Web resources
The URLs for data presented herein are as follows:
UCSC Genome Browser, https://genome.ucsc.edu/
BLUEPRINT project, http://www.blueprint-epigenome.eu/
GTEx Portal, https://www.gtexportal.org/
NCBI SRA, https://www.ncbi.nlm.nih.gov/sra/
NCBI GEO, http://www.ncbi.nlm.nih.gov/geo/
Roadmap Epigenomics Browser, http://www.roadmapepigenomics.org/
WashU Epigenome Browser, http://epigenomegateway.wustl.edu/
FANTOM ZENBU Browser, http://fantom.gsc.riken.jp/zenbu/
dbGaP, http://www.ncbi.nlm.nih.gov/gap
PhenoScanner. http://www.phenoscanner.medschl.cam.ac.uk/phenoscanner
e-GRASP, http://www.mypeg.info/egrasp
HaploReg, http://compbio.mit.edu/HaploReg
PheGenI; https://www.ncbi.nlm.nih.gov/gap/phegeni
Geneimprint, http://www.geneimprint.com/
OMIM, http://www.omim.org
R software package, http://www.R-project.org
Ethics statement
This study complied with the recommendations of the Ethics Committee of the Faculdade de Medicina de Campos, Brazil (approval code FR-278769). The goal of the study was to identify imprinted differentially methylated regions for the development of molecular genetic tests. The study did not involve physical or history examinations of subjects or laboratory testing for a genetic disease, as the subjects' DNA was used exclusively for DNA marker validation purposes. Healthy volunteer subjects were sampled by convenience or accessibility. Peripheral blood samples from participating adult subjects were collected with the written informed consent in compliance with the Declaration of Helsinki. For the youth, written consent was given by the legally authorized next of kin on behalf of the participants.
Author contributions
EM-A, FBM, GdSMA, RdSFJ, PTMR, CdSF, DTM, JTdS: conceived and designed experiments. GdSMA, RdSFJ, PTMR, FMB, CCFA, AFAdS, CdSF, EM-A: performed comparative screening of candidate iDMR in methylomes. GdSMA and TLdS: performed MSRE-PCR and SNuPE genotyping. RdSFJ, CdSF, VR, EM-A: developed R and Excel scripts. RdSFJ, CdSF, EM-A: carried out the GTEx analysis. DTM, ATdS, CFOdF, ABG: performed SRA RNA-Seq analysis. PTMR: executed the placenta methylome analysis. JTdS, RdSFJ, CFOdF, EM-A: searched chromatin segmentation enrichment signatures. DTM, CdSF, PTMR, RdSFJ, JTdS, CFOdF, TLdS, EM-A: prepared figures. EM-A: carried out comprehensive computational analyses; contributed biological samples, reagents, and materials; and wrote the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank the members of the Medina-Acosta laboratory for their insights and productive brainstorm lab meetings.
Footnotes
Funding. This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ (BR) (http://www.faperj.br/) [grant number E26/010.001036/2015 to EM-A] and from the Conselho Nacional de Desenvolvimento Científico e Tecnológico–CNPq (BR) (http://cnpq.br/) [grant numbers 301034/2012-5 and 308780/2015-9 to EM-A]. RdSFJ and DTM received undergraduate PIBIC/CNPq fellowships from the Universidade Estadual do Norte Fluminense Darcy Ribeiro UENF (BR) (http://www.uenf.br/). CdSF and PTMR are recipients of graduate fellowships from the Fundação Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–CAPES (http://www.capes.gov.br/). ATdS received a graduate fellowship from FAPERJ/UENF, and FBM received a postdoctoral fellowship from CAPES/PADCT. The agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00036/full#supplementary-material
Information about the novel candidate imprinted DMRs. Seven spreadsheets: (A) List of the 39 BS-Seq methylomes used to extract the AMR records; (B) List of the new candidate iDMR reported in this study; (C) List of known constitutive imprinted DMRs (Okae et al., 2014) used to set the investigative inclusion criterion of ≥16 overlapping AMR records in each candidate iDMR; (D) Information about the primers, MSRE-PCR triplex assays, and SNPs used to determine the parent-of-origin of the 5mCpG imprints in the PARD6G-AS1, PWWP2AP1, and GCSAML candidate iDMRs; (E) List of 90 GEO entries for 450 K methylation arrays from healthy; preeclampsia; and trisomy 13 (Patau syndrome), 18 (Edwards syndrome) and 21 (Down syndrome) placentas and list of BS-Seq methylomes from healthy (n = 3) placentas; (F) List of BS-Seq methylomes from hematopoietic cancers from the BLUEPRINT project (Adams et al., 2012); (G) Roadmap Epigenomics samples; (H) Reference set of iDMRs used in the hierarchical clustering analysis of histone modification enrichment.
Overall evidence of allele-specific expression (ASE) from RNA-Seq experiments. Six spreadsheets: (A) Information about the 8,555 RNA-Seq experiments in 51 tissues from the GTEx project (release V7; n = 9,931 RNA-Seq entries extracted with the dbGaP run selector tool), used as a secondary source; (B) Sorting and classification of 40 known imprinted genes (Babak et al., 2015; Baran et al., 2015) by the patterns of ASE across informative SNPs using the GTEx data and classification criteria described in the present study; (C) Information about the 2,164 RNA-Seq experiments in 25 tissues used as the primary source. Jointly, the sets of RNA-Seq experiments listed in (A,C) represent an atlas of 52 tissues; (D) Sorting and classification of the genes located up to 2.3Mb in either direction from the PARD6G-AS1, PWWP2AP1, and GCSAML genes by the patterns of ASE across informative SNPs using the GTEx data; (E) Allele-specific expression of ZNF124 yielding at least 10 reads per SNP in primary RNA-Seq experiments from lung tissue; (F) List of the top lead SNPs (MAF > 0.1) selected from the results in (D) and heatmap of the distribution of the informative RNA-Seq experiments yielding at least 20 reads per SNP; (G) Evidence of biallelic expression across the lead SNPs listed in (F); (H) Effective gene coverage according to the GTEx expression analysis.
PhenoScanner, e-GRASP, PheGenI, and HaploReg lookup of genotype-phenotype genome-wide associations for SNPs in the PARD6G-AS1 and GCSAML iDMRs. Seven spreadsheets: (A) Complete set of SNPs in each iDMR; (B) The list of SNPs and proxies found in association with traits at genome-wide significance; (C) Gene positions, minor alleles and global MAFs for the SNPs and proxies with genome-wide significance in GWAS; regulatory chromatin states at the lead variants rs1050919 (D), rs56357216 (E), rs1106086 (F), and rs10925069 (G).
Constitutive unmethylated status at the PPP2R3B predicted promoter region. X- chromosome ideogram; physical positions and domain features of the PPP2R3B locus depicting the methylation status at CpG sites across a 40 kb long-span view (hg19; 329240–369239). The methylation levels are represented on a scale from 0 to 1 (hypomethylated to hypermethylated). The image is centered on CGI-6, localized in the PPP2R3B predicted promoter region. The light green ticks represent the positions of the CpG sites. The PPP2R3B gene is transcribed from the minus DNA strand. The annotated features are (from top to bottom) the segmental duplication in chrY (chrY:231384 track), the exon-intron organization of the principal isoform, the evidence of small non-coding RNAs (sncRNAs) (DASHR RNA [neg] track), the FANTOM5 transcriptional start sites (TSS peaks track) (Lizio et al., 2017), the Ensembl Regulatory Build predicted promoters (Ensembl Reg Build track) (Cunningham et al., 2015), CpG islands and CGI-bearing AMRs (this study) and the comparative custom Fang_AMR track (Fang et al., 2012). A constitutive unmethylated status is observed across PPP2R3B CGI-6 in gametes, as well as in healthy (golden ticks) and cancerous (orange ticks) adult tissues. The chromosomal position of the region corresponding to the HpaII-sensitive amplimer (PCR results track) is highlighted in light blue within PPP2R3B CGI-6.
Validation of the intermediate methylation statuses at the PARD6G-AS1, PWWP2AP1, and GCSAML DMRs by MSRE-PCR triplex assays. Representative methylation profiles before and after digestion with the methylation-sensitive HpaII restriction enzyme. (A) PARD6G-AS1 DMR, (B) PWWP2AP1 DMR, and (C) GCSAML DMR. Each profile generated from uncut DNA consists of two control products (left and right peaks) and a test product (peak in the middle). The control products correspond to the amplimers of a chromosomal region known to be 100% unmethylated in the human genome (left peak) (see Materials and Methods for details) and a chromosomal region bearing no HpaII sites (right peak). The PARD6G-AS1 DMR amplimer comprises the common indel variant rs11281142 (MAF > 0.4), and thus, a heterozygous sample yields two peaks representing the two variant alleles, one with and the other without the CTGTGGTGC insertion. The GCSAML DMR amplimer comprises two peaks (255 and 257 bp), consistent with the addition of a 3′-end A residue overhang to the PCR product, since the two-peak product occurs in 20 genomic DNA samples tested, and the two annotated, encompassing, one-base-pair indel variants rs533592199 (-/G), and rs773445240 (-/T) reported in dbSNP150 have not been detected in over 841,883 public exomes (NHLBI GO Exome Sequencing Project, 2012; Sulem et al., 2015; Chen et al., 2016; Lek et al., 2016; Narasimhan et al., 2016). Each panel corresponds to a different individual from one out of three representative nuclear families that were informative for at least one SNP. The biallelic methylation profile at the PWWP2AP1 hemimethylated CGI DMR was confirmed using a second MSRE (HhaI) in two informative individuals (data not shown).
Constitutive hemimethylation statuses across the known GRB10 maternal iDMR. (A) Chromosome 7 ideogram; physical positions and domain features of the GRB10 locus showing the methylation status at CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr7:50832014-50872013). The image is centered on the CGI localized in the GRB10 predicted promoter region, labeled “GRB10 maternal iDMR” (GRB10 CGI-1), which maps to a predicted secondary promoter region upstream of the transcriptional start site of the short GRB10 isoform. The GRB10 locus is transcribed from the minus DNA strand. The light green ticks represent the positions of the CpG sites. The methylation levels are represented on a scale from 0 to 1 (hypomethylated to hypermethylated). The methylation levels across the CpG sites within the GRB10 maternal iDMR range from 0.35 to 0.65 in adult somatic tissues. In the same region, however, there is asymmetrical methylation in gametes (hypermethylation in oocytes and hypomethylation in spermatozoa). (B) Cross-reference overlaps of the GRB10 maternal iDMR with differentially methylated regions reported in control-disease methylomes and healthy placentas. The annotated features are (from top to bottom) the exon-intron organization of the principal and alternative isoforms, the evidence of large intergenic non-coding RNAs (lincRNA) and small non-coding RNAs (sncRNAs) (DASHR RNA [neg] track), the FANTOM5 transcriptional start sites (TSS peaks track) (Lizio et al., 2017), the Ensembl Regulatory Build predicted promoters (Ensembl Reg Build track) and transcriptional factor binding sites (TFBS summary track) (Cunningham et al., 2015), CpG islands and the comparative custom Fang_AMR track (Fang et al., 2012). The GRB10 maternal iDMR overlaps the domain known be differentially methylated in a maternal-origin-dependent manner in blastocysts and the placenta (Okae et al., 2014; Hamada et al., 2016; Sanchez-Delgado et al., 2016a) (Okae_DMRs, Hamada_DMRs, Sanchez_DMRs_set:I and Sanchez_DMRs_set:II tracks, respectively). GRB10 CGI-1 also intersects the domains reported to be hypomethylated in pseudohypoparathyroidism patients with GNAS cluster imprinting defects (Rochtus et al., 2016) (Rochtus_DMR track) and the maternally derived 5mCpG sites in placenta from (Yuen et al., 2011) (Yuen_5mCpG sites track). In the brain, the short GRB10 isoform is expressed from the paternal allele from the promoter that overlaps the GRB10 maternal iDMR. In the placenta, the expression of the long GRB10 isoform is biased (0.79) toward the maternal allele (Hamada_biased_maternal_PL track) (Hamada et al., 2016), and it starts from the promoter region that overlaps GRB10 CGI-2, which is constitutively hypomethylated.
The ZNF124 locus has no overlapping hemimethylated CGIs. (A) Chromosome 1 ideogram; physical positions and domain features of the ZNF124 locus showing the methylation status at CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr1:247315500–247355499). The image is centered on the CGI located in the ZNF124 predicted promoter region. The ZNF124 locus is transcribed from the minus DNA strand. The light green ticks represent the positions of the CpG sites. The ZNF124 CGI is hypomethylated in all shown BS-Seq methylomes. (B) Cross-reference overlaps of the predicted differentially methylated regions reported in healthy placentas. The annotated features are as in Figure S3. In the placenta, there is evidence of biallelic expression (Hamada et al., 2016).
The OR2L13 locus has no overlapping hemimethylated CGIs. (A) Chromosome 1 ideogram; physical positions and domain features of the OR2L13 locus showing the methylation status at the CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr1:248098501–248138500). The light green ticks represent the positions of the CpG sites. The OR2L13 CGI, localized in the OR2L13 predicted promoter region, is hypomethylated in all shown BS-Seq methylomes analyzed (average level of 0.24). (B) Cross-reference overlaps of the predicted DMR reported in healthy placentas across a 70kb long-span view (hg19; chr1:248098401–248168400). In the placenta (Hamada-DMR track), there are two intergenic predicted maternal DMRs 56.3 and 65.6 kb downstream, respectively, of the OR2L13 CGI. The annotated features are as in Figure S3.
The RP11-567M16.1-LOC284240 locus exhibits a hemimethylated CGI. (A) Chromosome 18 ideogram; physical positions and domain features of the RP11-567M16.1-LOC284240 locus showing the methylation status at the CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr18:77317230–77357229). The image is centered at the RP11-567M16.1-LOC284240 CGI-2 predicted promoter region. The light green ticks represent the position of the CpG sites. RP11-567M16.1-LOC284240 CGI-2 is hemimethylated (average level of 0.53) in several BS-Seq methylomes, including those of the epidermis, esophagus, lung, fetal brain cortex, fetal spinal cord, and placenta tissues, with paternal rather than maternal methylation asymmetry in gametes (this study). The occurrence of a paternal primary candidate iDMR is consistent with the observation of monoallelic expression of the locus in the pituitary (this study). (B) Cross-reference for two predicted paternal DMRs (Hamada_DMRs, hamada_1st_trim, Sanchez_pDMR_PL, and Sanchez_pDMR_embryo tracks), none of which exhibit either a constitutive or a tissue-specific hemimethylation profile.
Predicted paternally-derived intermediate methylation at LOC284240 CGI-2. Methylation levels across CGI-2 at the predicted promoter-flanking region of the LOC284240 locus. Note the paternally-derived 5mCpG asymmetry in gametes and the intermediate methylation levels in BS-Seq methylomes from epidermis, lung, esophagus, gastric muscle, brain cortex, fetal spinal cord, and placenta. The hemimethylated status is not constitutive, since, in spleen, liver, blood, thymus, and the hESC-derived endodermal, mesodermal and endodermal cell lines, there is a bias toward hypermethylation.
References
- Adams D., Altucci L., Antonarakis S. E., Ballesteros J., Beck S., Bird A., et al. (2012). BLUEPRINT to decode the epigenetic signature written in blood. Nat. Biotechnol. 30, 224–226. 10.1038/nbt.2153 [DOI] [PubMed] [Google Scholar]
- Alves da Silva A. F., Machado F. B., Pavarino E. C., Biselli-Perico J. M., Zampieri B. L., Da Silva Francisco Junior R., et al. (2016). Trisomy 21 Alters DNA methylation in parent-of-origin-dependent and -independent manners. PLoS ONE 11:e0154108. 10.1371/journal.pone.0154108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud P., Monk D., Hitchins M., Gordon E., Dean W., Beechey C. V., et al. (2003). Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum. Mol. Genet. 12, 1005–1019. 10.1093/hmg/ddg110 [DOI] [PubMed] [Google Scholar]
- Babak T., Deveale B., Tsang E. K., Zhou Y., Li X., Smith K. S., et al. (2015). Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet. 47, 544–549. 10.1038/ng.3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran Y., Subramaniam M., Biton A., Tukiainen T., Tsang E. K., Rivas M. A., et al. (2015). The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 25, 927–936. 10.1101/gr.192278.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow D. P., Bartolomei M. S. (2014). Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 6, 1–19. 10.1101/cshperspect.a018382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow T. M., Barault L., Ellsworth R. E., Harris H. R., Binder A. M., Valente A. L., et al. (2015). Aberrant methylation of imprinted genes is associated with negative hormone receptor status in invasive breast cancer. Int. J. Cancer 137, 537–547. 10.1002/ijc.29419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B. E., Stamatoyannopoulos J. A., Costello J. F., Ren B., Milosavljevic A., Meissner A., et al. (2010). The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 28, 1045–1048. 10.1038/nbt1010-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beygo J., Elbracht M., De Groot K., Begemann M., Kanber D., Platzer K., et al. (2015). Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur. J. Hum. Genet. 23, 180–188. 10.1038/ejhg.2014.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagitko N., Mergenthaler S., Schulz U., Wollmann H. A., Craigen W., Eggermann T., et al. (2000). Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion. Hum. Mol. Genet. 9, 1587–1595. 10.1093/hmg/9.11.1587 [DOI] [PubMed] [Google Scholar]
- Castel S. E., Levy-Moonshine A., Mohammadi P., Banks E., Lappalainen T. (2015). Tools and best practices for data processing in allelic expression analysis. Genome Biol. 16:195. 10.1186/s13059-015-0762-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Shi L., Hakenberg J., Naughton B., Sklar P., Zhang J., et al. (2016). Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 34, 531–538. 10.1038/nbt.3514 [DOI] [PubMed] [Google Scholar]
- Chen Y. A., Lemire M., Choufani S., Butcher D. T., Grafodatskaya D., Zanke B. W., et al. (2013). Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209. 10.4161/epi.23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess A. (2016). Monoallelic Gene Expression in Mammals. Annu. Rev. Genet. 50, 317–327. 10.1146/annurev-genet-120215-035120 [DOI] [PubMed] [Google Scholar]
- Choufani S., Shapiro J. S., Susiarjo M., Butcher D. T., Grafodatskaya D., Lou Y., et al. (2011). A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 21, 465–476. 10.1101/gr.111922.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper W. N., Constancia M. (2010). How genome-wide approaches can be used to unravel the remaining secrets of the imprintome. Brief. Funct. Genomics 9, 315–328. 10.1093/bfgp/elq018 [DOI] [PubMed] [Google Scholar]
- Court F., Martin-Trujillo A., Romanelli V., Garin I., Iglesias-Platas I., Salafsky I., et al. (2013). Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum. Mutat. 34, 595–602. 10.1002/humu.22276 [DOI] [PubMed] [Google Scholar]
- Court F., Tayama C., Romanelli V., Martin-Trujillo A., Iglesias-Platas I., Okamura K., et al. (2014). Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 24, 554–569. 10.1101/gr.164913.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi B. (2008). Genomic imprinting in the development and evolution of psychotic spectrum conditions. Biol. Rev. Camb. Philos. Soc. 83, 441–493. 10.1111/j.1469-185X.2008.00050.x [DOI] [PubMed] [Google Scholar]
- Cuellar Partida G., Laurin C., Ring S., Gaunt T. R., Relton C. L., Davey Smith G., et al. (2017). Imprinted loci may be more widespread in humans than previously appreciated and enable limited assignment of parental allelic transmissions in unrelated individuals. bioRxiv. 10.1101/161471 [DOI] [Google Scholar]
- Cunningham F., Amode M. R., Barrell D., Beal K., Billis K., Brent S., et al. (2015). Ensembl 2015. Nucleic Acids Res. 43, D662–D669. 10.1093/nar/gku1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva-Santiago S. C., Pacheco C., Rocha T. C., Brasil S. M., Pacheco A. C., Silva M. M., et al. (2014). The linked human imprintome v1.0: over 120 genes confirmed as imprinted impose a major review on previous censuses. Int. J. Data Min. Bioinform. 10, 329–356. 10.1504/IJDMB.2014.064547 [DOI] [PubMed] [Google Scholar]
- Do C., Lang C. F., Lin J., Darbary H., Krupska I., Gaba A., et al. (2016). Mechanisms and disease associations of haplotype-dependent allele-specific DNA methylation. Am. J. Hum. Genet. 98, 934–955. 10.1016/j.ajhg.2016.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty L. E., Rezwan F. I., Poole R. L., Jagoe H., Lake H., Lockett G. A., et al. (2014). Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J. Med. Genet. 51, 229–238. 10.1136/jmedgenet-2013-102116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann T., Perez De Nanclares G., Maher E. R., Temple I. K., Tumer Z., Monk D., et al. (2015). Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics 7:123. 10.1186/s13148-015-0143-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. (2015). Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol. 33, 364–376. 10.1038/nbt.3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F., Hodges E., Molaro A., Dean M., Hannon G. J., Smith A. D. (2012). Genomic landscape of human allele-specific DNA methylation. Proc. Natl. Acad. Sci. U.S.A. 109, 7332–7337. 10.1073/pnas.1201310109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green B. B., Kappil M., Lambertini L., Armstrong D. A., Guerin D. J., Sharp A. J., et al. (2015). Expression of imprinted genes in placenta is associated with infant neurobehavioral development. Epigenetics 10, 834–841. 10.1080/15592294.2015.1073880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Zhu P., Yan L., Li R., Hu B., Lian Y., et al. (2014). The DNA methylation landscape of human early embryos. Nature 511, 606–610. 10.1038/nature13544 [DOI] [PubMed] [Google Scholar]
- Hamada H., Okae H., Toh H., Chiba H., Hiura H., Shirane K., et al. (2016). Allele-specific methylome and transcriptome analysis reveals widespread imprinting in the human placenta. Am. J. Hum. Genet. 99, 1045–1058. 10.1016/j.ajhg.2016.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna C. W., Peñaherrera M. S., Saadeh H., Andrews S., Mcfadden D. E., Kelsey G., et al. (2016). Pervasive polymorphic imprinted methylation in the human placenta. Genome Res. 26, 756–767. 10.1101/gr.196139.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannula-Jouppi K., Muurinen M., Lipsanen-Nyman M., Reinius L. E., Ezer S., Greco D., et al. (2014). Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics 9, 351–365. 10.4161/epi.27160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatt L., Aagaard M. M., Bach C., Graakjaer J., Sommer S., Agerholm I. E., et al. (2016). Microarray-based analysis of methylation of 1st trimester trisomic placentas from down syndrome, edwards syndrome and patau syndrome. PLoS ONE 11:e0160319. 10.1371/journal.pone.0160319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatt L., Aagaard M. M., Graakjaer J., Bach C., Sommer S., Agerholm I. E., et al. (2015). Microarray-Based Analysis of Methylation Status of CpGs in Placental DNA and Maternal Blood DNA–potential new epigenetic biomarkers for cell free Fetal DNA-based diagnosis. PLoS ONE 10:e0128918. 10.1371/journal.pone.0128918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs A. S., Raney B. J., Speir M. L., Rhead B., Casper J., Karolchik D., et al. (2016). UCSC data integrator and variant annotation integrator. Bioinformatics 32, 1430–1432. 10.1093/bioinformatics/btv766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchins M. P., Monk D., Bell G. M., Ali Z., Preece M. A., Stanier P., et al. (2001). Maternal repression of the human GRB10 gene in the developing central nervous system; evaluation of the role for GRB10 in Silver-Russell syndrome. Eur. J. Hum. Genet. 9, 82–90. 10.1038/sj.ejhg.5200583 [DOI] [PubMed] [Google Scholar]
- Jirtle J., Murphy C. (2012). Geneimprint database [Online]. Available online at: http://www.geneimprint.com (Accessed September 10, 2017).
- Kagami M., Matsubara K., Nakabayashi K., Nakamura A., Sano S., Okamura K., et al. (2017). Genome-wide multilocus imprinting disturbance analysis in Temple syndrome and Kagami-Ogata syndrome. Genet. Med. 19, 476–482. 10.1038/gim.2016.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim S., Noureldin H. F., Abusamra H., Salem N., Alhathli E., Dudley J., et al. (2016). e-GRASP: an integrated evolutionary and GRASP resource for exploring disease associations. BMC Genomics 17:770. 10.1186/s12864-016-3088-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., et al. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H., Yanagisawa E., Sakashita A., Sugawara N., Kumakura S., Ogawa H., et al. (2013). Epigenetic and transcriptional features of the novel human imprinted lncRNA GPR1AS suggest it is a functional ortholog to mouse Zdbf2linc. Epigenetics 8, 635–645. 10.4161/epi.24887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E., Zhang Y. (2014). DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 6:a019133. 10.1101/cshperspect.a019133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizio M., Harshbarger J., Abugessaisa I., Noguchi S., Kondo A., Severin J., et al. (2017). Update of the FANTOM web resource: high resolution transcriptome of diverse cell types in mammals. Nucleic Acids Res. 45, D737–D743. 10.1093/nar/gkw995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedi P. P., Dietrich F. S., Weidman J. R., Bosko J. M., Jirtle R. L., Hartemink A. J. (2007). Computational and experimental identification of novel human imprinted genes. Genome Res. 17, 1723–1730. 10.1101/gr.6584707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado F. B., Machado F. B., Faria M. A., Lovatel V. L., Alves Da Silva A. F., Radic C. P., et al. (2014). 5meCpG epigenetic marks neighboring a primate-conserved core promoter short tandem repeat indicate X-chromosome inactivation. PLoS ONE 9:e103714. 10.1371/journal.pone.0103714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T., Higashimoto K., Jozaki K., Yatsuki H., Nakabayashi K., Makita Y., et al. (2014). Comprehensive and quantitative multilocus methylation analysis reveals the susceptibility of specific imprinted differentially methylated regions to aberrant methylation in Beckwith-Wiedemann syndrome with epimutations. Genet. Med. 16, 903–912. 10.1038/gim.2014.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi S. J., Meaburn E. L., Dempster E. L., Lunnon K., Paya-Cano J. L., Smith R. G., et al. (2016). Tissue-specific patterns of allelically-skewed DNA methylation. Epigenetics 11, 24–35. 10.1080/15592294.2015.1127479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann J. A., Zheng H., Islam A., Goodyer C. G., Polychronakos C. (2001). Evidence against GRB10 as the gene responsible for Silver-Russell syndrome. Biochem. Biophys. Res. Commun. 286, 943–948. 10.1006/bbrc.2001.5500 [DOI] [PubMed] [Google Scholar]
- Mergenthaler S., Hitchins M. P., Blagitko-Dorfs N., Monk D., Wollmann H. A., Ranke M. B., et al. (2001). Conflicting reports of imprinting status of human GRB10 in developing brain: how reliable are somatic cell hybrids for predicting allelic origin of expression? Am. J. Hum. Genet. 68, 543–545. 10.1086/318192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A., Hodges E., Fang F., Song Q., Mccombie W. R., Hannon G. J., et al. (2011). Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146, 1029–1041. 10.1016/j.cell.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan K., Lomvardas S. (2015). Monoallelic expression of olfactory receptors. Annu. Rev. Cell Dev. Biol. 31, 721–740. 10.1146/annurev-cellbio-100814-125308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk D., Arnaud P., Frost J., Hills F. A., Stanier P., Feil R., et al. (2009). Reciprocal imprinting of human GRB10 in placental trophoblast and brain: evolutionary conservation of reversed allelic expression. Hum. Mol. Genet. 18, 3066–3074. 10.1093/hmg/ddp248 [DOI] [PubMed] [Google Scholar]
- Morison I. M., Ramsay J. P., Spencer H. G. (2005). A census of mammalian imprinting. Trends Genet. 21, 457–465. 10.1016/j.tig.2005.06.008 [DOI] [PubMed] [Google Scholar]
- Narasimhan V. M., Hunt K. A., Mason D., Baker C. L., Karczewski K. J., Barnes M. R., et al. (2016). Health and population effects of rare gene knockouts in adult humans with related parents. Science 352, 474–477. 10.1126/science.aac8624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI Resource Coordinators (2017). Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 45, D12–D17. 10.1093/nar/gkw1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NHLBI GO Exome Sequencing Project (2012). Exome Variant Server [Online]. Seattle, WA: Available online: http://evs.gs.washington.edu/EVS/ (Accessed April 29, 2016). [Google Scholar]
- Okae H., Chiba H., Hiura H., Hamada H., Sato A., Utsunomiya T., et al. (2014). Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet. 10:e1004868. 10.1371/journal.pgen.1004868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal A., Temkin A. M., Kerkel K., Yale A., Yotova I., Drost N., et al. (2013). Comparative anatomy of chromosomal domains with imprinted and non-imprinted allele-specific DNA methylation. PLoS Genet. 9:e1003622. 10.1371/journal.pgen.1003622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot M., Kroes H. Y., Se V. D. W., Eleveld M. J., Rooms L., Nievelstein R. A., et al. (2007). Dandy-Walker complex in a boy with a 5 Mb deletion of region 1q44 due to a paternal t(1;20)(q44;q13.33). Am. J. Med. Genet. A 143A, 1038–1044. 10.1002/ajmg.a.31690 [DOI] [PubMed] [Google Scholar]
- Ramos E. M., Hoffman D., Junkins H. A., Maglott D., Phan L., Sherry S. T., et al. (2014). Phenotype-Genotype Integrator (PheGenI): synthesizing genome-wide association study (GWAS) data with existing genomic resources. Eur. J. Hum. Genet. 22, 144–147. 10.1038/ejhg.2013.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney B. J., Dreszer T. R., Barber G. P., Clawson H., Fujita P. A., Wang T., et al. (2014). Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 30, 1003–1005. 10.1093/bioinformatics/btt637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochtus A., Martin-Trujillo A., Izzi B., Elli F., Garin I., Linglart A., et al. (2016). Genome-wide DNA methylation analysis of pseudohypoparathyroidism patients with GNAS imprinting defects. Clin. Epigenetics 8:10. 10.1186/s13148-016-0175-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues J. A., Zilberman D. (2015). Evolution and function of genomic imprinting in plants. Genes Dev. 29, 2517–2531. 10.1101/gad.269902.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J. M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J. J., Lopez G., et al. (2013). APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 41, D110–D117. 10.1093/nar/gks1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanelli V., Nakabayashi K., Vizoso M., Moran S., Iglesias-Platas I., Sugahara N., et al. (2014). Variable maternal methylation overlapping the nc886/vtRNA2-1 locus is locked between hypermethylated repeats and is frequently altered in cancer. Epigenetics 9, 783–790. 10.4161/epi.28323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Delgado M., Court F., Vidal E., Medrano J., Monteagudo-Sanchez A., Martin-Trujillo A., et al. (2016a). Human Oocyte-Derived Methylation Differences Persist in the Placenta Revealing Widespread Transient Imprinting. PLoS Genet. 12:e1006427. 10.1371/journal.pgen.1006427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Delgado M., Riccio A., Eggermann T., Maher E. R., Lapunzina P., Mackay D., et al. (2016b). Causes and Consequences of Multi-Locus Imprinting Disturbances in Humans. Trends Genet. 32, 444–455. 10.1016/j.tig.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Savol A. J., Wang P. I., Jeon Y., Colognori D., Yildirim E., Pinter S. F., et al. (2017). Genome-wide identification of autosomal genes with allelic imbalance of chromatin state. PLoS ONE 12:e0182568. 10.1371/journal.pone.0182568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin J., Lizio M., Harshbarger J., Kawaji H., Daub C. O., Hayashizaki Y., et al. (2014). Interactive visualization and analysis of large-scale sequencing datasets using ZENBU. Nat. Biotechnol. 32, 217–219. 10.1038/nbt.2840 [DOI] [PubMed] [Google Scholar]
- Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar D. A., Li Y., Bernal A. J., Hoyo C., Murphy S. K., Jirtle R. L. (2012). The human imprintome: regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. ILAR J. 53, 341–358. 10.1093/ilar.53.3-4.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z. D., Chan M. M., Humm K. C., Karnik R., Mekhoubad S., Regev A., et al. (2014). DNA methylation dynamics of the human preimplantation embryo. Nature 511, 611–615. 10.1038/nature13581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soellner L., Begemann M., Mackay D. J., Gronskov K., Tumer Z., Maher E. R., et al. (2017). Recent advances in imprinting disorders. Clin. Genet. 91, 3–13. 10.1111/cge.12827 [DOI] [PubMed] [Google Scholar]
- Song Q., Decato B., Hong E. E., Zhou M., Fang F., Qu J., et al. (2013). A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 8:e81148. 10.1371/journal.pone.0081148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J. R., Blackshaw J., Kamat M. A., Ellis S., Surendran P., Sun B. B., et al. (2016). PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics 32, 3207–3209. 10.1093/bioinformatics/btw373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant P. H., Rausch T., Gardner E. J., Handsaker R. E., Abyzov A., Huddleston J., et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulem P., Helgason H., Oddson A., Stefansson H., Gudjonsson S. A., Zink F., et al. (2015). Identification of a large set of rare complete human knockouts. Nat. Genet. 47, 448–452. 10.1038/ng.3243 [DOI] [PubMed] [Google Scholar]
- The GTEx Project C. (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. 10.1126/science.1262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen J., Nivard M. G., Baselmans B. M., Zilhao N. R., Ligthart L., Consortium B., et al. (2015). Epigenome-wide association study of aggressive behavior. Twin Res. Hum. Genet. 18, 686–698. 10.1017/thg.2015.74 [DOI] [PubMed] [Google Scholar]
- van Kempen L. C., Redpath M., Elchebly M., Klein K. O., Papadakis A. I., Wilmott J. S., et al. (2016). The protein phosphatase 2A regulatory subunit PR70 is a gonosomal melanoma tumor suppressor gene. Sci. Transl. Med. 8, 369ra177. 10.1126/scitranslmed.aai9188 [DOI] [PubMed] [Google Scholar]
- Viana J., Pidsley R., Troakes C., Spiers H., Wong C. C., Al-Sarraj S., et al. (2014). Epigenomic and transcriptomic signatures of a Klinefelter syndrome (47,XXY) karyotype in the brain. Epigenetics 9, 587–599. 10.4161/epi.27806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan E. S., Qiu W., Morrow J., Beaty T. H., Hetmanski J., Make B. J., et al. (2015). Genome-wide site-specific differential methylation in the blood of individuals with Klinefelter syndrome. Mol. Reprod. Dev. 82, 377–386. 10.1002/mrd.22483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward L. D., Kellis M. (2016). HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881. 10.1093/nar/gkv1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y., Su J., Liu H., Lv J., Wang F., Yan H., et al. (2014). MetaImprint: an information repository of mammalian imprinted genes. Development 141, 2516–2523. 10.1242/dev.105320 [DOI] [PubMed] [Google Scholar]
- Yoshihashi H., Maeyama K., Kosaki R., Ogata T., Tsukahara M., Goto Y., et al. (2000). Imprinting of human GRB10 and its mutations in two patients with Russell-Silver syndrome. Am. J. Hum. Genet. 67, 476–482. 10.1086/302997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen R. K., Avila L., Penaherrera M. S., Von Dadelszen P., Lefebvre L., Kobor M. S., et al. (2009). Human placental-specific epipolymorphism and its association with adverse pregnancy outcomes. PLoS ONE 4:e7389. 10.1371/journal.pone.0007389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen R. K., Jiang R., Penaherrera M. S., Mcfadden D. E., Robinson W. P. (2011). Genome-wide mapping of imprinted differentially methylated regions by DNA methylation profiling of human placentas from triploidies. Epigenetics Chromatin 4:10. 10.1186/1756-8935-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Guan D. G., Yang J. H., Shao P., Zhou H., Qu L. H. (2010). ncRNAimprint: a comprehensive database of mammalian imprinted noncoding RNAs. RNA 16, 1889–1901. 10.1261/rna.2226910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Maricque B., Xie M., Li D., Sundaram V., Martin E. A., et al. (2011). The human epigenome browser at Washington University. Nat. Methods 8, 989–990. 10.1038/nmeth.1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Information about the novel candidate imprinted DMRs. Seven spreadsheets: (A) List of the 39 BS-Seq methylomes used to extract the AMR records; (B) List of the new candidate iDMR reported in this study; (C) List of known constitutive imprinted DMRs (Okae et al., 2014) used to set the investigative inclusion criterion of ≥16 overlapping AMR records in each candidate iDMR; (D) Information about the primers, MSRE-PCR triplex assays, and SNPs used to determine the parent-of-origin of the 5mCpG imprints in the PARD6G-AS1, PWWP2AP1, and GCSAML candidate iDMRs; (E) List of 90 GEO entries for 450 K methylation arrays from healthy; preeclampsia; and trisomy 13 (Patau syndrome), 18 (Edwards syndrome) and 21 (Down syndrome) placentas and list of BS-Seq methylomes from healthy (n = 3) placentas; (F) List of BS-Seq methylomes from hematopoietic cancers from the BLUEPRINT project (Adams et al., 2012); (G) Roadmap Epigenomics samples; (H) Reference set of iDMRs used in the hierarchical clustering analysis of histone modification enrichment.
Overall evidence of allele-specific expression (ASE) from RNA-Seq experiments. Six spreadsheets: (A) Information about the 8,555 RNA-Seq experiments in 51 tissues from the GTEx project (release V7; n = 9,931 RNA-Seq entries extracted with the dbGaP run selector tool), used as a secondary source; (B) Sorting and classification of 40 known imprinted genes (Babak et al., 2015; Baran et al., 2015) by the patterns of ASE across informative SNPs using the GTEx data and classification criteria described in the present study; (C) Information about the 2,164 RNA-Seq experiments in 25 tissues used as the primary source. Jointly, the sets of RNA-Seq experiments listed in (A,C) represent an atlas of 52 tissues; (D) Sorting and classification of the genes located up to 2.3Mb in either direction from the PARD6G-AS1, PWWP2AP1, and GCSAML genes by the patterns of ASE across informative SNPs using the GTEx data; (E) Allele-specific expression of ZNF124 yielding at least 10 reads per SNP in primary RNA-Seq experiments from lung tissue; (F) List of the top lead SNPs (MAF > 0.1) selected from the results in (D) and heatmap of the distribution of the informative RNA-Seq experiments yielding at least 20 reads per SNP; (G) Evidence of biallelic expression across the lead SNPs listed in (F); (H) Effective gene coverage according to the GTEx expression analysis.
PhenoScanner, e-GRASP, PheGenI, and HaploReg lookup of genotype-phenotype genome-wide associations for SNPs in the PARD6G-AS1 and GCSAML iDMRs. Seven spreadsheets: (A) Complete set of SNPs in each iDMR; (B) The list of SNPs and proxies found in association with traits at genome-wide significance; (C) Gene positions, minor alleles and global MAFs for the SNPs and proxies with genome-wide significance in GWAS; regulatory chromatin states at the lead variants rs1050919 (D), rs56357216 (E), rs1106086 (F), and rs10925069 (G).
Constitutive unmethylated status at the PPP2R3B predicted promoter region. X- chromosome ideogram; physical positions and domain features of the PPP2R3B locus depicting the methylation status at CpG sites across a 40 kb long-span view (hg19; 329240–369239). The methylation levels are represented on a scale from 0 to 1 (hypomethylated to hypermethylated). The image is centered on CGI-6, localized in the PPP2R3B predicted promoter region. The light green ticks represent the positions of the CpG sites. The PPP2R3B gene is transcribed from the minus DNA strand. The annotated features are (from top to bottom) the segmental duplication in chrY (chrY:231384 track), the exon-intron organization of the principal isoform, the evidence of small non-coding RNAs (sncRNAs) (DASHR RNA [neg] track), the FANTOM5 transcriptional start sites (TSS peaks track) (Lizio et al., 2017), the Ensembl Regulatory Build predicted promoters (Ensembl Reg Build track) (Cunningham et al., 2015), CpG islands and CGI-bearing AMRs (this study) and the comparative custom Fang_AMR track (Fang et al., 2012). A constitutive unmethylated status is observed across PPP2R3B CGI-6 in gametes, as well as in healthy (golden ticks) and cancerous (orange ticks) adult tissues. The chromosomal position of the region corresponding to the HpaII-sensitive amplimer (PCR results track) is highlighted in light blue within PPP2R3B CGI-6.
Validation of the intermediate methylation statuses at the PARD6G-AS1, PWWP2AP1, and GCSAML DMRs by MSRE-PCR triplex assays. Representative methylation profiles before and after digestion with the methylation-sensitive HpaII restriction enzyme. (A) PARD6G-AS1 DMR, (B) PWWP2AP1 DMR, and (C) GCSAML DMR. Each profile generated from uncut DNA consists of two control products (left and right peaks) and a test product (peak in the middle). The control products correspond to the amplimers of a chromosomal region known to be 100% unmethylated in the human genome (left peak) (see Materials and Methods for details) and a chromosomal region bearing no HpaII sites (right peak). The PARD6G-AS1 DMR amplimer comprises the common indel variant rs11281142 (MAF > 0.4), and thus, a heterozygous sample yields two peaks representing the two variant alleles, one with and the other without the CTGTGGTGC insertion. The GCSAML DMR amplimer comprises two peaks (255 and 257 bp), consistent with the addition of a 3′-end A residue overhang to the PCR product, since the two-peak product occurs in 20 genomic DNA samples tested, and the two annotated, encompassing, one-base-pair indel variants rs533592199 (-/G), and rs773445240 (-/T) reported in dbSNP150 have not been detected in over 841,883 public exomes (NHLBI GO Exome Sequencing Project, 2012; Sulem et al., 2015; Chen et al., 2016; Lek et al., 2016; Narasimhan et al., 2016). Each panel corresponds to a different individual from one out of three representative nuclear families that were informative for at least one SNP. The biallelic methylation profile at the PWWP2AP1 hemimethylated CGI DMR was confirmed using a second MSRE (HhaI) in two informative individuals (data not shown).
Constitutive hemimethylation statuses across the known GRB10 maternal iDMR. (A) Chromosome 7 ideogram; physical positions and domain features of the GRB10 locus showing the methylation status at CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr7:50832014-50872013). The image is centered on the CGI localized in the GRB10 predicted promoter region, labeled “GRB10 maternal iDMR” (GRB10 CGI-1), which maps to a predicted secondary promoter region upstream of the transcriptional start site of the short GRB10 isoform. The GRB10 locus is transcribed from the minus DNA strand. The light green ticks represent the positions of the CpG sites. The methylation levels are represented on a scale from 0 to 1 (hypomethylated to hypermethylated). The methylation levels across the CpG sites within the GRB10 maternal iDMR range from 0.35 to 0.65 in adult somatic tissues. In the same region, however, there is asymmetrical methylation in gametes (hypermethylation in oocytes and hypomethylation in spermatozoa). (B) Cross-reference overlaps of the GRB10 maternal iDMR with differentially methylated regions reported in control-disease methylomes and healthy placentas. The annotated features are (from top to bottom) the exon-intron organization of the principal and alternative isoforms, the evidence of large intergenic non-coding RNAs (lincRNA) and small non-coding RNAs (sncRNAs) (DASHR RNA [neg] track), the FANTOM5 transcriptional start sites (TSS peaks track) (Lizio et al., 2017), the Ensembl Regulatory Build predicted promoters (Ensembl Reg Build track) and transcriptional factor binding sites (TFBS summary track) (Cunningham et al., 2015), CpG islands and the comparative custom Fang_AMR track (Fang et al., 2012). The GRB10 maternal iDMR overlaps the domain known be differentially methylated in a maternal-origin-dependent manner in blastocysts and the placenta (Okae et al., 2014; Hamada et al., 2016; Sanchez-Delgado et al., 2016a) (Okae_DMRs, Hamada_DMRs, Sanchez_DMRs_set:I and Sanchez_DMRs_set:II tracks, respectively). GRB10 CGI-1 also intersects the domains reported to be hypomethylated in pseudohypoparathyroidism patients with GNAS cluster imprinting defects (Rochtus et al., 2016) (Rochtus_DMR track) and the maternally derived 5mCpG sites in placenta from (Yuen et al., 2011) (Yuen_5mCpG sites track). In the brain, the short GRB10 isoform is expressed from the paternal allele from the promoter that overlaps the GRB10 maternal iDMR. In the placenta, the expression of the long GRB10 isoform is biased (0.79) toward the maternal allele (Hamada_biased_maternal_PL track) (Hamada et al., 2016), and it starts from the promoter region that overlaps GRB10 CGI-2, which is constitutively hypomethylated.
The ZNF124 locus has no overlapping hemimethylated CGIs. (A) Chromosome 1 ideogram; physical positions and domain features of the ZNF124 locus showing the methylation status at CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr1:247315500–247355499). The image is centered on the CGI located in the ZNF124 predicted promoter region. The ZNF124 locus is transcribed from the minus DNA strand. The light green ticks represent the positions of the CpG sites. The ZNF124 CGI is hypomethylated in all shown BS-Seq methylomes. (B) Cross-reference overlaps of the predicted differentially methylated regions reported in healthy placentas. The annotated features are as in Figure S3. In the placenta, there is evidence of biallelic expression (Hamada et al., 2016).
The OR2L13 locus has no overlapping hemimethylated CGIs. (A) Chromosome 1 ideogram; physical positions and domain features of the OR2L13 locus showing the methylation status at the CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr1:248098501–248138500). The light green ticks represent the positions of the CpG sites. The OR2L13 CGI, localized in the OR2L13 predicted promoter region, is hypomethylated in all shown BS-Seq methylomes analyzed (average level of 0.24). (B) Cross-reference overlaps of the predicted DMR reported in healthy placentas across a 70kb long-span view (hg19; chr1:248098401–248168400). In the placenta (Hamada-DMR track), there are two intergenic predicted maternal DMRs 56.3 and 65.6 kb downstream, respectively, of the OR2L13 CGI. The annotated features are as in Figure S3.
The RP11-567M16.1-LOC284240 locus exhibits a hemimethylated CGI. (A) Chromosome 18 ideogram; physical positions and domain features of the RP11-567M16.1-LOC284240 locus showing the methylation status at the CpG sites (golden ticks) across a 40 kb long-span view (hg19; chr18:77317230–77357229). The image is centered at the RP11-567M16.1-LOC284240 CGI-2 predicted promoter region. The light green ticks represent the position of the CpG sites. RP11-567M16.1-LOC284240 CGI-2 is hemimethylated (average level of 0.53) in several BS-Seq methylomes, including those of the epidermis, esophagus, lung, fetal brain cortex, fetal spinal cord, and placenta tissues, with paternal rather than maternal methylation asymmetry in gametes (this study). The occurrence of a paternal primary candidate iDMR is consistent with the observation of monoallelic expression of the locus in the pituitary (this study). (B) Cross-reference for two predicted paternal DMRs (Hamada_DMRs, hamada_1st_trim, Sanchez_pDMR_PL, and Sanchez_pDMR_embryo tracks), none of which exhibit either a constitutive or a tissue-specific hemimethylation profile.
Predicted paternally-derived intermediate methylation at LOC284240 CGI-2. Methylation levels across CGI-2 at the predicted promoter-flanking region of the LOC284240 locus. Note the paternally-derived 5mCpG asymmetry in gametes and the intermediate methylation levels in BS-Seq methylomes from epidermis, lung, esophagus, gastric muscle, brain cortex, fetal spinal cord, and placenta. The hemimethylated status is not constitutive, since, in spleen, liver, blood, thymus, and the hESC-derived endodermal, mesodermal and endodermal cell lines, there is a bias toward hypermethylation.