

Abstract
Environmentally induced phenotypic plasticity is thought to play an important role in the adaption of plant populations to heterogeneous habitat conditions, and yet the importance of epigenetic variation as a mechanism of adaptive plasticity in natural plant populations still merits further research. In this study, we investigated populations of Vitex negundo var. heterophylla (Chinese chastetree) from adjacent habitat types at seven sampling sites. Using several functional traits, we detected a significant differentiation between habitat types. With amplified fragment length polymorphisms (AFLP) and methylation‐sensitive AFLP (MSAP), we found relatively high levels of genetic and epigenetic diversity but very low genetic and epigenetic differences between habitats within sites. Bayesian clustering showed a remarkable habitat‐related differentiation and more genetic loci associated with the habitat type than epigenetic, suggesting that the adaptation to the habitat is genetically based. However, we did not find any significant correlation between genetic or epigenetic variation and habitat using simple and partial Mantel tests. Moreover, we found no correlation between genetic and ecologically relevant phenotypic variation and a significant correlation between epigenetic and phenotypic variation. Although we did not find any direct relationship between epigenetic variation and habitat environment, our findings suggest that epigenetic variation may complement genetic variation as a source of functional phenotypic diversity associated with adaptation to the heterogeneous habitat in natural plant populations.
Keywords: Chinese chastetree, DNA methylation, ecological epigenetics, MSAP, phenotypic plasticity
1. INTRODUCTION
Adaptation of plants to the heterogeneous environment may be achieved through natural selection of fixed traits and plasticity of variable traits (Pigliucci, 2001; Roda et al., 2013; Williams, 2008). Various studies have demonstrated the important role of ecologically relevant phenotypic plasticity in persistence across a range of habitats with spatial heterogeneity and temporal dynamics (Douhovnikoff & Hazelton, 2014; McLean et al., 2014; Nicotra et al., 2010). Although whether intraspecific variation in functional traits, most of which is assumed to be highly plastic, is more due to genetic control or epigenetic regulation remains essentially unexplored to date, emerging evidence has suggested that epigenetic mechanisms could facilitate phenotypic plasticity in response to ecologically relevant stressors under complex habitat conditions (Herrera & Bazaga, 2013; Medrano, Herrera, & Bazaga, 2014; Nicotra et al., 2015; Wilschut, Oplaat, Snoek, Kirschner, & Verhoeven, 2016; Zhang, Fischer, Colot, & Bossdorf, 2013).
Although there are several epigenetic mechanisms, including chemical modification of DNA and histones, position effects and interference by small noncoding RNAs (Richards, 2011), DNA methylation of cytosine is most the extensively studied epigenetic mechanism with important effects on ecologically relevant traits (Herrera & Bazaga, 2008, 2013; Schrey et al., 2013; Wilschut et al., 2016; Xie et al., 2015). While DNA is predominantly methylated at CG sites, cytosine methylations in plants occurs throughout the genome in all sequence contexts (CG, CHG and CHH where H = A, C or T) (Law & Jacobsen, 2010) and could affect whether transposons are silenced and genes are expressed. Biologists have illuminated that the amount and pattern of DNA methylation in model plants is sensitive to various environmental stressors under laboratory conditions, and ecologists have focused on the variation in DNA methylation in wild populations to understand the role of DNA methylation in plant adaptation to real environmental stress in nature (Bossdorf, Richards, & Pigliucci, 2008; Kilvitis et al., 2014). The rapidly increasing number of publications has illustrated that variation in DNA methylation is correlated with herbivory in violets (Herrera & Bazaga, 2010), salinity in marsh perennials (Foust et al., 2016), artificial disturbance in Lavandula latifolia (Herrera & Bazaga, 2016), metals in red maple (Kim, Im, & Nkongolo, 2016), and climate in Quercus lobata (Gugger, Fitz‐Gibbon, PellEgrini, & Sork, 2016; Platt, Gugger, Pellegrini, & Sork, 2015).
There is a complex relationship between genetic and epigenetic variation in the wild. Epigenetic variants can be under genetic control (Slotkin & Martienssen, 2007), but environmental factors can also directly alter the epigenetic variation that may be inherited through meiosis over several generations (Jablonka & Raz, 2009). The pattern of epigenetic variation in wild populations contributing to phenotypic traits which cannot be explained by genetic variation may be a consequence of natural selection on pre‐existing epigenetic variation, environmental induction of variable epigenetic variation, or both (Bossdorf et al., 2008; Verhoeven, vonHoldt, & Sork, 2016).
Vitex negundo var. heterophylla (Chinese chastetree, or five‐leaved chaste tree), a native deciduous officinal shrub with a heterophylly leaf and entomophilous flower (Figure 1), is widely distributed in the hilly areas of North China (Hu et al., 2015). Vitex negundo var. heterophylla is a typical species that can survive in a broad range of habitats, including bush and understory, varying in an array of other biotic and abiotic parameters (Li, Yang, & Wu, 2008). Chinese chastetree in roadside and bush habitats is frequently cut by farmers as firewood or to prevent it from occupying farmlands. It can adapt to cardinal environmental factors through modification of a series of morphological and physiological characteristics (Du, Guo, Zhang, & Wang, 2010; Du et al., 2012). As a long long‐lived perennial, V. negundo var. heterophylla may face variable environments through epigenetic processes (Bräutigam et al., 2013). Therefore, it provides an ideal study system to compare genetic and epigenetic differences in response to heterogeneous habitat conditions.
Figure 1.

The study organism Vitex negundo var. heterophylla, a deciduous shrub with entomophilous flowers and digitate leaves containing five lanceolate leaflets, sometimes three
In this study, we measured several plant functional traits for individuals sampled from each plot to determine habitat differentiation in phenotype. We investigated the genetic variation using amplified fragment length polymorphism (AFLP) and epigenetic variation using methylation‐sensitive AFLP (MSAP). These dominant markers provided a considerable number of anonymous loci, which generate powerful data for detecting the genetic and epigenetic structure across several heterogeneous habitats in a nonmodel species without a reference genome. We compared the patterns of genetic and epigenetic variation shaped by the habitat conditions to test the hypothesis that epigenetic variation plays a potential role independent of genetic variation in the adaptation of V. negundo var. heterophylla to the environment.
2. MATERIALS AND METHODS
2.1. Sampling design
We selected plots for this study from seven sites in the south hills of Shandong Province, eastern China, separated by approximately 20–150 km (Figure 2). For each site, we collected samples from two adjacent plots with contrasting habitat conditions at a distance of <2 km (Table 1) in May 2016. At each plot, 13–15 widely spaced individuals were randomly chosen and marked with permanent tags. To avoid developmental epigenetic variation confusing the differences among heterogeneous environments, all leaf samples for molecular analyses were picked at the same position of the plant before the flowering phase. Young expanding leaves were collected from each plant, placed in paper envelopes, and dried immediately with silica gel. This material was used for genomic DNA extraction, and additional expanding leaves were randomly collected to measure the leaf traits.
Figure 2.
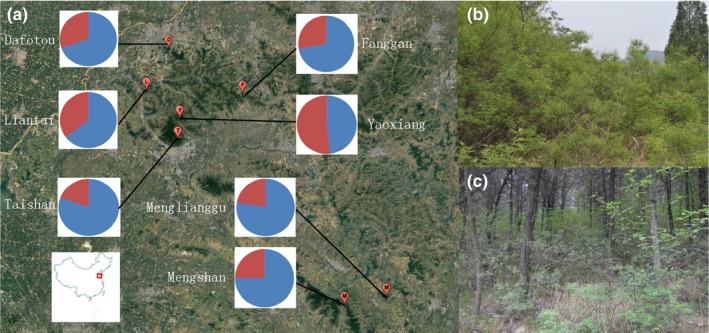
(a) Maps of the seven Vitex negundo var. heterophylla sampling sites in Shandong Province, China, with the results of Bayesian clustering from STRUCTURE. The shaded portion of the circle indicates population assignment to two groups. Examples of (b) the open habitat and (c) the forest understory habitat
Table 1.
Overview of sampled populations of Vitex negundo var. heterophylla
| Population | Sample size | Site | Latitude (°N) | Longitude (°E) | Habitat Type | Dominant tree |
|---|---|---|---|---|---|---|
| D1 | 14 | Dafotou | 36.63179 | 117.0347 | Understory | Platycladus orientalis |
| D2 | 14 | Dafotou | 36.62825 | 117.0507 | Open | |
| F1 | 15 | Fanggan | 36.43172 | 117.4516 | Understory | Quercus acutissima, Robinia pseudoacacia |
| F2 | 14 | Fanggan | 36.43333 | 117.4515 | Open | |
| L1 | 14 | Liantai | 36.44292 | 116.937 | Understory | Cotinus coggygri, Robinia pseudoacacia, Quercus acutissima |
| L2 | 13 | Liantai | 36.44398 | 116.9425 | Open | |
| Y1 | 14 | Yaoxiang | 36.32133 | 117.12 | Understory | Quercus acutissima |
| Y2 | 14 | Yaoxiang | 36.31391 | 117.1388 | Open | |
| M1 | 14 | Mengshan | 35.5376 | 117.9895 | Understory | Pinus armandii, Robinia pseudoacacia |
| M2 | 14 | Mengshan | 35.52588 | 117.987 | Open | |
| N1 | 13 | Menglianggu | 35.57246 | 118.2078 | Understory | Pinus armandii |
| N2 | 14 | Menglianggu | 35.57203 | 118.2214 | Open | |
| T1 | 14 | Taishan | 36.2313 | 117.1125 | Understory | Quercus acutissima, Robinia pseudoacacia, Platycladus orientalis |
| T2 | 14 | Taishan | 36.2257 | 117.1141 | Open |
2.2. Measurement of plant functional traits
For each sampled individual, plant maximum height (H), basal diameter (D), and number of resprouts (NR) were measured, and more than five fully expanded compound leaves were collected at different positions of the stem. Both leaf fresh weight (LFW) and leaf dry weight (LDW) were measured and leaf water content [LWC = (LFW − LDW)/LDW] was obtained. Leaflet area (Area) was determined with image processing program ImageJ (Abràmoff, Magalhães, & Ram, 2004), and the specific leaf area (SLA = LA/LDW) was calculated. The number of resprouts (NR) was divided into three levels to assess the environmental disturbance (Pérez‐Harguindeguy et al., 2013): level 0 for one stem, level 1 for more than one but <5 stems, and level 2 for more than five stems. Finally, the coefficient of height‐diameter allometry (HDA) was calculated for each population using function MSA(D~H, log = “xy”) in R package smart (Warton, Duursma, Falster, & Taskinen, 2012).
2.3. AFLP and MS‐AFLP protocol
We investigated a total of 195 individuals for genetic and epigenetic variation with AFLP and MSAP using the same DNA sample for each individual. Total genomic DNA was extracted from dried leaf tissue according to the cetyl trimethylammonium bromide (CTAB) method (Doyle & Doyle, 1987) with some modifications. PVP 40,000 was used to improve DNA yield and quality. DNA was quantified with both 0.8% agarose gels and microscopic spectrophotometry.
The protocol for MSAP was adapted from a standard AFLP (Vos et al. 1995), replacing the MseI enzyme in two separate runs with the methylation‐sensitive enzymes HpaII and MspI using appropriate adaptors and primers. For restriction digests, 2 μl genomic DNA (ca. 150 ng) was combined with 10 μl double digestion mix containing 1 μl 10 × CutSmart Buffer (New England Biolabs, NEB), 2.5 μEcoRI (NEB), 0.5 μMseI or 2.5 μHpaII or 2.5 μMspI (NEB) in parallel reactions. The reaction was incubated at 37°C for 2 hr and inactivated at 80°C for 20 min. Then, the product was combined with 48 μT4 DNA ligase (NEB), 1.4 μl 10 × T4 DNA ligase buffer, 1 μl EcoRI adapter (5 μM), and 1 μl MseI adapter (50 μM) or 1 μl HpaII/MspI adapter (50 μM). The reaction was incubated at 16°C for 2 hr, inactivated at 65°C for 10 min, and diluted 1:5. For the preselective amplification (PCR1), 2 μl ligation product was combined with 13 μl PCR1 reaction mix containing 0.7 μl preselective primers (5 μM) each, 0.6 μl dNTPs (TIANGEN, 2.5 mM each), 1.5 μl 10× buffer (TIAGEN), 0.75 U polymerase (TIAGEN), and 9.2 μl H2O. The thermocycler protocol was 72.0°C (2 min) followed by 20 cycles of 94.0°C (20 s), 56.0°C (30 s), and 72.0°C (2 min) and a final extension at 60.0°C (30 min), performed on a Biometra TGradient. The PCR1 product was diluted 1:5. For the selective amplification (PCR2), 2 μl PCR1 product was combined with 13 μl PCR2 reaction mix containing 0.7 μl selective primers (5 μM) each, 0.6 μl dNTPs (TIANGEN, 2.4 mM each), 1.5 μl 10 × buffer (TIAGEN), 0.75 U polymerase (TIAGEN), and 9.2 μl H2O. The thermocycler protocol was 94.0°C (2 min) followed by 10 cycles of 94.0°C (20 s), 66.0°C (30 s, decreasing 1°C per cycle) and 72.0°C (2 min) and 20 cycles of 94.0°C (20 s), 56.0°C (30 s) and 72.0°C (2 min), and a final extension at 60.0°C (30 min). Six selective primer combinations were chosen for the AFLP (EcoRI + ACA and MseI + CTA, EcoRI + ACT and MseI + CAT, EcoRI + ACC and MseI + CTC) and MSAP (EcoRI + AAC and HpaII/MspI + TC, EcoRI + ACG and HpaII/MspI + TC, EcoRI + ACT and HpaII/MspI + TA) analyses (Table 2), of which the EcoRI + 3 primers were 5′‐end labeled with FAM dye.
Table 2.
Adapter and primer sequences for AFLP and MSAP amplification
| Primer | Sequence(from 5'to 3’) |
|---|---|
| Adapters | |
| EcoRI_adapter top | CTCGTAGACTGCGTACC |
| EcoRI_adapter bottom | AATTGGTACGCAGTCTAC |
| MseI_adapter top | GAGCGATGAGTCCTGAG |
| MseI_adapter bottom | TACTCAGGACTCAT |
| HpaII/MspI_adapter top | GATCATGAGTCCTGCT |
| HpaII/MspI_adapter bottom | CGAGCAGGACTCATGA |
| Preselective primers | |
| EcoRI+A | GACTGCGTACCAATTCA |
| MseI+C | GATGAGTCCTGAGTAAC |
| HpaII/MspI | ATCATGAGTCCTGCTCGG |
| Selective primers | |
| EcoRI+AAC4 | GACTGCGTACCAATTCAAC |
| EcoRI+ACA1 | GACTGCGTACCAATTCACA |
| EcoRI+ACT2,6 | GACTGCGTACCAATTCACT |
| EcoRI+ACC3 | GACTGCGTACCAATTCACC |
| EcoRI+ACG5 | GACTGCGTACCAATTCACG |
| MseI+CAT2 | GATGAGTCCTGAGTAACAT |
| MseI+CTA1 | GATGAGTCCTGAGTAACTA |
| MseI+CTC3 | GATGAGTCCTGAGTAACTC |
| HpaII/MspI+TA6 | ATCATGAGTCCTGCTCGGTA |
| HpaII/MspI+TC4,5 | ATCATGAGTCCTGCTCGGTC |
Superscript numbers indicate primer combinations used for the selective amplification, and every primer combination is tagged with same number.
The final selective PCR productions were separated and visualized on an ABI3730XL DNA capillary sequencer (Applied Biosystems, Foster City, USA) with Rox‐500 internal size standard (Applied Biosystems) at the Shandong Academy of Agricultural Sciences. We used PEAKSCANNER v1.0 (Applied Biosystems) to analyze the AFLP and MSAP profiles. Binning of fragments was performed using a peak height threshold of 50 relative fluorescence units and a minimal size of 50 base pairs. Peak height data were scored with a binary code, zero for band absent, and one for band present. For every polymorphic locus, each allele must exist in more than two individuals (>1% of all samples).
2.4. Data analysis
To identify how many different genetic groups are represented in our collection regardless of the geographic sampling location, we performed Bayesian clustering of genetic data using STRUCTURE v2.3.4 (Falush, Stephens, & Pritchard, 2007; Pritchard, Stephens, & Donnelly, 2000). Structure estimates the number of groups (K) present among individuals and assigns individuals to each K using Bayesian modeling. We tested 20 populations (k = 1–20), which are more than the maximum anticipated based on sampling location, with 20 runs at each k. We used both the log probability of observing the data (Ln Pr(x|k)) method of Structure and Delta K (Evanno, Regnaut, & Goudet, 2005) with the online program STRUCTURE HARVESTER for visualizing STRUCTURE output and implementing (Earl & vonHoldt, 2012), which determines the number of populations that best fit the data. We incorporated sampling locations in our model to assist in detecting sampling weak differentiation. We performed clustering with the admixture model, 30,000 burn‐in steps, 100,000 postburn‐in steps, and allowed correlated allele frequencies. We assigned individuals to groups based on the highest q‐value.
All basic statistical analyses were carried out using the R environment. The MSAP profiles were analyzed with the R script MSAP_calc (Schulz, Eckstein, & Durka, 2013) using the function Extract_MSAP_epigenotypes with parameters Epicode = “Mix1,” delete.monomorphic.loci = TRUE, and MinPoly = 2. Under the scoring scheme “Mix1,” M‐MSAP makers were obtained with a methylation scoring approach scoring the presence of both EcoRI–HpaII and EcoRI–MspI products as “1” and scoring other conditions as “0,” while u‐MSAP makers were transformed with a nonmethylation scoring approach treating the presence of only the EcoRI–MspI fragment (hemi‐ or fully methylated at the internal cytosine) or only the EcoRI–MspI fragment (hemi‐methylated at the external cytosine) as “1” and treating other conditions as “0.”
To estimate the amount of epigenetic and genetic variation, Shannon's diversity index (H) and percentage of polymorphisms (PPL) for each population were calculated with GENALEX 6.5 (Peakall & Smouse, 2006). We also used GENALEX to estimate genetic and epigenetic differentiation using hierarchical analysis of molecular variation (AMOVA) to determine whether spatial location structured genetic or epigenetic differences by comparing the genetic and epigenetic variation among sites (ФRT), among populations (i.e., habitats) within sites (ФPR), and within populations (ФPT). We used 9,999 permutations to estimate statistical significance and an initial alpha of 0.05. Moreover, we used generalized linear models (GLM) for each loci to determine the specific AFLP and MSAP loci correlated with the habitat type using function glm in R.
We analyzed the correlation between genetic variation, epigenetic variation, and habitat by performing Mantel and partial Mantel tests using zt software (Bonnet & Van de Peer, 2002). Using a simple Mantel, we compared the genetic distance matrix to the epigenetic distance matrix to test for a relationship between genetic and epigenetic variation. While a Mantel test determines correlations between two distance matrices, the partial Mantel test determines correlations between two distance matrices while controlling for correlations with a third matrix. In this case, we used a partial Mantel to test for an independent relationship between genetic variation and habitat while controlling for epigenetic variation. Likewise, we tested for a relationship between epigenetic variation and habitat while controlling for correlations with genetic variation. To create the habitat distance matrix, two different strategies were adopted. For the first habitat distance matrix, we used zero to indicate understory habitat and one to indicate open habitat. The second one was based on the fitness‐related traits, including SLA, LWC, NR, and HDA, performed using function dist (method = “euclidean”) in R software. Both strategies make the assumption that differences between habitats will be essentially the same magnitude regardless of individual population differences. In all cases, we used the Euclidean genetic and epigenetic distance matrices generated by GENALEX. We also constructed Euclidian geographic distances and Nei unbiased genetic and epigenetic distances to test the roles of major role for population differentiation. As simple and partial Mantel tests have been questioned for a number of drawbacks (Bradburd, Ralph, & Coop, 2013; Guillot & Rousset, 2013; Legendre, Fortin, & Borcard, 2015), we applied multiple matrix regression with randomization (MMRR) (Wang, 2013) as an alternative approach to Mantel procedures. Computations were implemented in with the MMRR function using 9,999 permutations.
3. RESULTS
3.1. Differences in plant functional traits
At the population level, we found significant variation for D [paired t‐test: t (6) = −2.51, p = .046], LWC [paired t‐test: t(6) = 3.19, p = .019] and SLA [paired t‐ test: t(6) = 2.96, p = .025], and similar patterns of disturbance between two populations were revealed among sites other than Dafotou and Yaoxiang (Figure 3a). At the individual level, significant differences between two habitats were detected for LWC and SLA (Figure 3b) at four sites: Dafotou, Yaoxiang, Liantai, and Taishan (Table 3). Principal coordinates analyses (PCoA) displayed different phenotypic divergences among sites, indicating complex heterogeneous habitats. The cord. 2 in PCoA may suggest the phenotypic variation between two habitat types (Figure 4).
Figure 3.
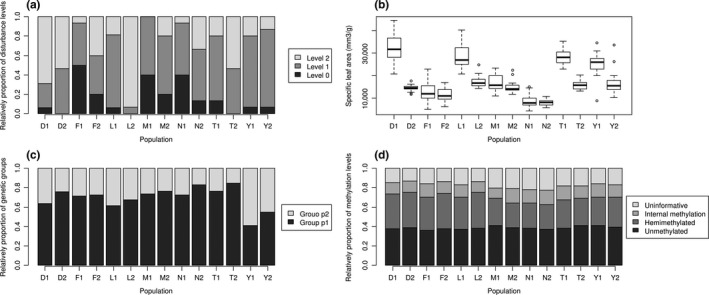
Differences among population in disturbance (a), specific leaf area (b), genetic structure (c), and DNA methylation (d). (a) The individual with only one resprout indicates a minor disturbance (level 0), the individual with 2–5 resprouts indicates a medium disturbance (level 1), and the individual with more than five resprouts indicates a recent severe disturbance (level 2). (b) Specific leaf area is significantly different between habitats in most sites and varies among sites. (c) The genetic assignment of group p1 in open habitat is slightly higher than that in understory habitat within every site. (d) There is no significant difference in DNA methylation level between habitats or among sites
Table 3.
T‐value for individuals between two populations for each site
| Dafotou | Fanggan | Yaoxiang | Liantai | Mengshan | Menglianggu | Taishan | |
|---|---|---|---|---|---|---|---|
| Height | −1.732 | 1.531 | −0.806 | −1.625 | 1.638 | 0.231 | −1.852 |
| Diameter | −3.187 | −0.702 | −1.663 | −3.218* | −0.310 | 0.087 | −0.4030* |
| LWC | 3.390* | 2.014 | 2.903* | 3.651** | −0.736 | 3.967** | 3.703** |
| SLA | 10.13** | 0.658 | 3.604* | 6.778** | 1.278 | 0.655 | 11.85** |
*p < .01; **p < .001.
Figure 4.
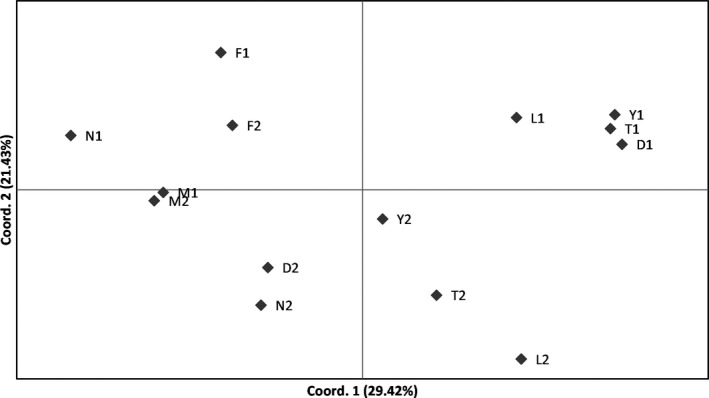
Principal coordinates analyses (PCoA) of distances for functional traits of 14 populations of Vitex negundo var. heterophylla
3.2. Genetic diversity and structure
The AFLP analysis resulted in 142 polymorphic loci across which every individual displayed a unique genotype. Values of diversity and percent polymorphisms across populations are given in Table 4. These loci identified that genetic diversity across all populations was high (h‐AFLP ranged from 0.408 to 0.498). There was no difference in genetic diversity between understory and open habitats (paired t‐test: t = 0.60723, df = 6, p‐value = .566 for H; t = 0.85722, df = 6, p‐value = .4242 for PPL). We detected population structure at every level of hierarchy and found significant variation among regions (explaining 7% of the genetic variance) and among populations within region (2%), and the most (91%) among individuals within population (Table 5).
Table 4.
Values of diversity (h) and percent polymorphisms (%P) of populations
| Pop | Sample size | h‐AFLP | %P‐AFLP | h‐u‐MSAP | %P‐u‐MSAP | h‐m‐MSAP | %P‐m‐MSAP |
|---|---|---|---|---|---|---|---|
| D1 | 14 | 0.54 | 84.51 | 0.471 | 95.89 | 0.563 | 100.00 |
| D2 | 14 | 0.463 | 73.24 | 0.477 | 97.26 | 0.562 | 99.31 |
| F1 | 15 | 0.496 | 78.17 | 0.449 | 95.89 | 0.566 | 100.00 |
| F2 | 14 | 0.537 | 85.21 | 0.467 | 95.89 | 0.566 | 99.31 |
| L1 | 14 | 0.436 | 71.83 | 0.460 | 96.58 | 0.551 | 100.00 |
| L2 | 13 | 0.495 | 78.87 | 0.471 | 95.21 | 0.545 | 98.61 |
| M1 | 14 | 0.506 | 81.69 | 0.485 | 94.52 | 0.495 | 100.00 |
| M2 | 14 | 0.501 | 80.28 | 0.453 | 92.47 | 0.509 | 98.61 |
| N1 | 14 | 0.487 | 78.87 | 0.469 | 95.89 | 0.503 | 98.61 |
| N2 | 14 | 0.459 | 73.24 | 0.447 | 94.52 | 0.502 | 98.61 |
| T1 | 13 | 0.462 | 73.94 | 0.463 | 92.47 | 0.538 | 99.31 |
| T2 | 14 | 0.408 | 65.49 | 0.481 | 94.52 | 0.515 | 98.61 |
| Y1 | 14 | 0.600 | 97.18 | 0.493 | 100.00 | 0.535 | 99.31 |
| Y2 | 14 | 0.586 | 93.66 | 0.476 | 95.89 | 0.531 | 100.00 |
| Mean | 13.929 | 0.498 | 79.73 | 0.469 | 95.50 | 0.534 | 99.31 |
| SE | 0.010 | 0.006 | 2.28 | 0.004 | 0.51 | 0.003 | 0.16 |
Table 5.
Three‐level hierarchical analysis of molecular variance (AMOVA) for AFLP, u‐MSAP and m‐MSAP data sets among sites, among populations within sites (among Pops,) and within populations (within Pops)
| Source | df | % variation | Ф‐statistics |
|---|---|---|---|
| Genetic variation based on AFLP | |||
| Among sites | 6 | 7 | 0.067** |
| Among Pops | 7 | 2 | 0.086** |
| Within Pops | 181 | 91 | 0.020* |
| Epigenetic variations based on u‐MSAP | |||
| Among Sites | 6 | 2 | 0.020** |
| Among Pops | 7 | 1 | 0.011* |
| Within Pops | 181 | 97 | 0.030** |
| Epigenetic variations based on m‐MSAP | |||
| Among Sites | 6 | 2 | 0.023** |
| Among Pops | 7 | 0 | 0.0005 |
| Within Pops | 181 | 98 | 0.024** |
df, degrees of freedom.
Ф‐statistics were calculated using 9,999 permutations.
*p ≤ .01, **p ≤ .0001.
Bayesian clustering identified only two genetic groups (Delta K = 184.07), which indicated a high degree of intermixing between populations. These groups did not clearly reflect geographically based differences among seven sites (Figure 2a), but significant differentiation between understory and open habitats was detected in the amount of each of the two groups [paired t‐test: t(6) = −4.39, p = .005] (Figure 3c). We found 15 AFLP loci correlated with habitat type in replicate populations using GLM.
3.3. Epigenetic diversity and structure
A total of 146 MSAP loci were analyzed, which were transformed into 146 u‐MSAP loci and 146 M‐MSAP. All 195 individuals displayed unique u‐MSAP and M‐MSAP genotypes. The epigenetic diversity was high based on both u‐MSAP (ranged from 0.449 to 0.493) and m‐MSAP (ranged from 0.495 to 0.566). There was also no significant difference in epigenetic diversity between understory and open habitats. We found higher levels of epigenetic than genetic diversity at the population level using index PPL [t(13) = −8.799, p < .001 for m‐MSAP; t(13) = −7.591, p < .001 for u‐MSAP], but two MSAP scoring approaches drew different conclusions comparing index H. Genetic H was lower than epigenetic H calculated with m‐MSAP data [paired t‐test: t(13) = −2.423, p = .031], but higher with u‐MSAP data [paired t‐test: t(13) = 2.193, p = .047]. However, we failed to detect a significant correlation between genetic and epigenetic diversity. For u‐MSAP, hierarchical AMOVA detected little variation (1%) among populations within a site, but failed for m‐MSAP, and most variation existed within population. Moreover, MSAP datasets did not infer significant differences in genome‐wide cytosine methylation level (Figure 3d) between heterogeneous habitats [paired t‐test: t(6) = −0.2443, p = .8151]. Only one MSAP locus was determined with habitat type through analysis with GLM.
3.4. Comparison between genetic and epigenetic variation
Simple Mantel tests showed significant correlations between genetic and epigenetic variation across all populations (Table 6). Both simple Mantel tests (Table 6) and MMRR (Table 7) showed that phenotypic variation was correlated with epigenetic variation, but not with genetic variation. Simple Mantel tests found no relationship between habitat type and genetic or epigenetic variation, but MMRR showed a possible association between genetic differentiation and habitat types (r = .081, p = .0485). We also detected the geographic structure of genetic and epigenetic variation. At the level of population, we found a lower geographic regression slope for epigenetic structure than genetic structure using the Mantel method (βAFLP = 0.0234, βMSAP‐u = 0.0042, βMSAP‐m = 0.0062; Figure 5). The above results obtained from u‐MSAP and m‐MSAP markers separately showed the same trends (Table 6). MMRR also revealed the analogous genetic and epigenetic spatial structure (Table 8).
Table 6.
Correlation coefficients using simple Mantel tests across all sites
| Gen | Epi_u | Epi_m | |
|---|---|---|---|
| Epi_u | 0.110* | – | – |
| Epi_m | 0.055* | 0.344** | – |
| Env | −0.004 | 0.0005 | −0.012 |
| Phe | 0.044 | 0.046* | 0.071** |
| Geo | 0.063** | 0.051** | 0.086** |
Gen, genetic variation; Epi_u, epigenetic variation using u‐MSAP; Epi_m, epigenetic variation using m‐MSAP; Env, environment; Phe, phenotype; Geo, geographical distance.
*p ≤ .05; **p ≤ .001.
Table 7.
Summary of multiple matrix regression analysis with randomization (MMRR) relating the phenotypic distance matrix with genetic and epigenetic distance matrices
| Differentiation matrix | Epigenetic marker used | Overall regression | Linear predictor matrices | ||||
|---|---|---|---|---|---|---|---|
| Genetic distance | Epigenetic distance | ||||||
| F | p | Coefficient | p | Coefficient | p | ||
| Phenotype | u‐AFLP | 35.91 | .0484 | 0.0032 | .1537 | 0.0057 | .0525 |
| Phenotype | m‐AFLP | 63.81 | .005 | 0.0034 | .1484 | 0.0118 | .0001 |
p values were calculated with 9,999 permutations.
Figure 5.
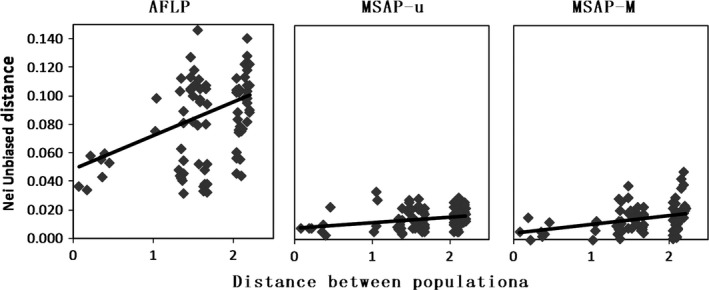
Fitted linear regressions for the genetic (AFLP) and epigenetic (u‐MSAP and m‐MSAP) markers, depicting the relationship between pairwise Nei unbiased distance and spatial separation (note logarithmic scale) for the N = 14 populations
Table 8.
Summary of multiple matrix regression analyses with randomization (MMRR) relating genetic, epigenetic, and phenotypic distance matrices with geographical and environmental distance matrices
| Differentiation matrix | Overall regression | Linear predictor matrices | ||||
|---|---|---|---|---|---|---|
| Geographical distance | Environmental distance | |||||
| F | p | Coefficient | p | Coefficient | p | |
| Gen | 37.91 | .0009 | 1.300 | .0008 | 0.181 | .0485 |
| Epi‐u | 25.38 | .0011 | 0.650 | .0011 | −0.185 | .7992 |
| Epi‐m | 70.44 | .0001 | 0.843 | .0001 | −0.940 | .3175 |
| Phe | 8055 | .0001 | 109.0 | .0001 | 61.18 | .0001 |
Gen, genetic variation using AFLP markers; Epi_u, epigenetic variation using u‐MSAP; Epi_m, epigenetic variation using m‐MSAP; Phe, phenotype.
p values were calculated with 9,999 permutations.
Partial Mantel tests always displayed a significant correlation (r > .10, p < .05 for u‐MSAP; r ≥ .05, p < .05 for m‐MSAP) between genetic and epigenetic variation when we controlled for the additional factor (Figure 6). However, we detected no relationship between habitat type and genetic variation when we controlled for epigenetic variation and no relationship between habitat type and genetic variation when we controlled for epigenetic variation. Moreover, we did not find a significant correlation between phenotypic and genetic variation when we controlled for epigenetic variation, but we found a significant correlation (r = .041, p = .033 for u‐MSAP; r = .059, p < .001 for m‐MSAP) between phenotypic and epigenetic variation when we controlled for genetic variation. Additionally, we detected the spatial structure of genetic (r = .058, p = .002 with controlled u‐MSAP; r = .058, p = .005 with controlled m‐MSAP) and epigenetic (r = .045, p = .003 for u‐MSAP; r = .082, p < .001 for m‐MSAP) diversity.
Figure 6.
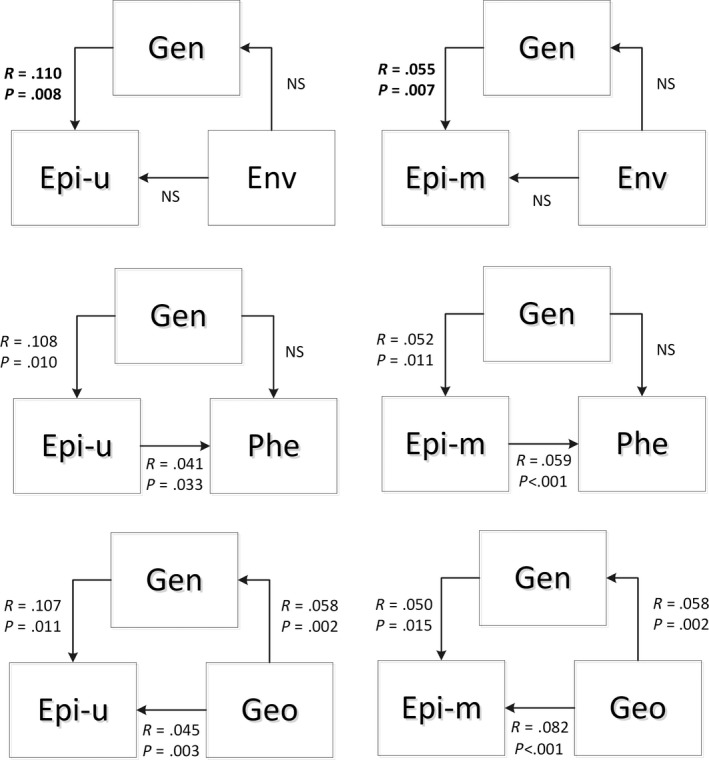
Genetic and epigenetic correlations to variation in habitat environment, functional phenotype, and geographic distance using partial Mantel tests with the Euclidean genetic and epigenetic distance matrices, habitat distance matrices, and trait distance and geographical distance matrices for all individuals across all populations. The correlations between genetic and epigenetic variation were calculated in separate partial Mantel tests. Gen = genetic variation, Epi‐u = Epigenetic variation based on MSAP‐u, Epi‐m = Epigenetic variation based on MSAP‐M, Env = environmental variation, Phe = phenotypic variation, Geo = geographical distance, NS = not significant, r = correlation coefficient when significant and p‐value
4. DISCUSSION
Consistent with the previous studies of V. negundo (Su et al., 2003; Zhang, Zheng, & Ge, 2007), we found high levels of genetic diversity and low population genetic differentiation both among and within sites. In particular, Bayesian clustering revealed a significant genetic differentiation shaped by habitat heterogeneity. We also found high levels of epigenetic diversity but little epigenetic structure among populations within site, and random levels of DNA methylation across all populations. This result is consistent with the previous findings in salt marsh perennials (Foust et al., 2016), suggesting that genetic variation may be more strongly structured by environment than epigenetic variation. Although the scoring approaches are largely consistent when comparing the levels of diversity among populations, estimates of epigenetic diversity varied strongly (Schulz et al., 2013), and the deviation made by scoring approaches has observably affected our comparison of levels between genetic and epigenetic diversity. Consistent with previous studies of other species distributed across different habitats observing equal or higher epigenetic than genetic diversity (Foust et al., 2016; Herrera & Bazaga, 2010; Richards, Schrey, & Pigliucci, 2012; Schulz, Eckstein, & Durka, 2014), we found slightly higher levels of epigenetic than genetic diversity in V. negundo var. heterophylla. Although little differentiation between habitats within sites indicated strong gene flow, the AMOVA, Bayesian, and GLM analyses concluded congruently that genetic variation may play a more important role in habitat differentiation than epigenetic variation. The study of a floodplain herb emphasized the major role of environmentally induced epigenetic variation for adjustment to changing habitat conditions (Schulz et al., 2014), but our results indicated a very marginal role of epigenetic variation in the perennial shrub.
The main distinction between understory and open habitat is the availability of light, which may have strong effects on a series of other biotic and abiotic environmental factors. In fact, there are many other obvious environmental differences in arbor coverage, human disturbance, elevation, slope aspect, and climate, which we cannot control for in natural conditions across seven sites. Moreover, the understory and open habitats are dynamic and could rapidly transform. Although epigenetic variation is considered as a very potential component in the adaption to the changing environment, relationships among genetic and epigenetic variation and habitat may show different patterns in varying species. When running simple and partial Mantel tests, we obtained the same pattern as for Borrichia frutescens (Foust et al., 2016), failing to detect any relationship between epigenetic and habitat environment using crude binary data to describe complex habitats, as we had a larger number and range of sampling plots than the previous studies (Foust et al., 2016; Kim et al., 2016; Robertson, Schrey, Shayter, Moss, & Richards, 2017; Schulz et al., 2014). Limited studies have been conducted of epigenetic differentiation in natural populations across heterogeneous habitat conditions, and there is an urgent need to develop or replace the binary method to comprehensively characterize the complex habitat conditions. Here, we tried to introduce the functional traits for representing the environment.
The selected traits, including leaf water content, specific leaf area, level of resprout, and height‐diameter allometry, were plastic and could respond to the heterogeneous environment. For example, SLA, an indicator of ecophysiological characteristics with a strong phenotypic plasticity and substantial genetic effects (Scheepens, Frei, & Stöcklin, 2010), would increase in the understory habitat to optimize light harvesting (McIntyre & Strauss, 2014), as supported by our data. In addition, NR could be considered as a typical parameter of branching architecture for the shrub, which differs on the basis of browsing history, fire history, and other types of disturbance (Pérez‐Harguindeguy et al., 2013), especially access to light, water stress, and the human‐caused cutting for V. negundo var. heterophylla in our research region. We found phenotypic plasticity in response to controlled drought and shade treatment in our previous glasshouse experiments using V. negundo var. heterophylla (Du et al., 2010, 2012), and this field experiment also displayed adaptive plasticity in response to heterogeneous wild habitats. Nevertheless, assessing plant functional trait data separately may misrepresent the adaptive response to the complex habitat. Thus, we combined several typical fitness‐related phenotypes to quantify the divergence of adaptions.
Amazingly, we found 15 AFLP loci statistically correlated with habitat type, and the Bayesian clustering suggested a parallel genetic divergence that may result in microevolution from independent origins. This was also supported by MMRR showing potential effect of habitat environment on genetic differentiation. Although strong gene flow has homogenized the genetic differentiation across the genome, adaptive loci will remain due to similar pressure (Trucchi, Frajman, Haverkamp, Schönswetter, & Paun, 2017). Only several plastic functional traits were investigated, and very limited loci were detected by AFLP. Therefore, to test this hypothesis, it is necessary to find the adaptive phenotypes and adopt next‐generation sequencing methods. We did expect epigenetic differentiation between open and understory populations, but we only found one locus showing differentiation due to habitat type. Moreover, it remains a question whether this locus plays an independent role from genetic variation in adaptation to diverse habitats. It is possible either that the understory environment did not change any epigenetic variation between habitats, or that any existing epigenetic signature was too weak to be detected given the high epigenetic diversity between individuals among all populations.
The epigenetic mechanism may be restricted when natural plant populations endure some discrete human‐caused disturbance, such as heavy metal pollution (Kim et al., 2016), experimental disturbance (Herrera & Bazaga, 2016), and oil spills (Robertson et al., 2017). According to our field investigations and planting experiments (Du et al., 2012), Chinese chastetree tends to distribute in open habitats with plentiful light. Understory individuals might sometimes undergo aboveground dieback due to shade and drought, but individuals in open habitat were disturbed by human‐caused cutting in five (Fanggan, Liantaishan, Mengshan, Menglianggu, and Taishan) of seven study sites (see Figure 3a). Unlike the cyclic pattern of environmental factors such as light, rainfall, and salinity, plants may have not experienced these artificial stressors in evolutionary history. If plants do not have a plastic or regulatory response such as the epigenetic mechanism, genetic diversity is the vital resource of adaptive phenotypes and the genetic structure will be shaped deeply.
It is a challenge to search for epigenetic components independent from genetic variation in V. negundo var. heterophylla with such high genetic diversity. While many previous studies took advantage of low genetic diversity clonal or inbred species to minimize possibilities for genetic control (Richards et al., 2012; Schulz et al., 2014; Verhoeven, Jansen, van Dijk, & Biere, 2010), some recent studies attempted to use statistical approaches to uncover patterns of epigenetic variation that are not predictable from patterns of genetic variation (Foust et al., 2016; Gugger et al., 2016; Herrera, Medrano, & Bazaga, 2016). Simple and partial Mantel tests were proposed to combine the analyses of genetic and epigenetic variation and present the correlation between genetic or epigenetic variation and habitat while controlling for the correlation between genetic and epigenetic variation (Foust et al., 2016). A controlled planting experiment investigating epigenetic variation in response to warming failed to find significant correlations between epigenetic variation and phenotype or habitat using Mantel tests (Nicotra et al., 2015). In contrast, we found a weak but significant correlation between epigenetic variation and adaptive phenotype, with no significant correlation between genetic and adaptive phenotypes. The results indicated that epigenetic mechanisms may play a more important role in adaptive plasticity than genetic variation in some scenarios.
Finding direct casual links between epigenetic and variation and ecological phenotype is a key challenge in the study of epigenetic adaptation with nonmodel species (Richards et al., 2017). A glasshouse experiment with various epigenetic recombinant inbred lines of Arabidopsis thaliana concluded that heritable epigenetic variation can cause substantial variation in ecologically important plant traits (Zhang et al., 2013). To investigate the relationship between epigenetic variation and functional plant diversity in wild populations, genetic and epigenetic marker–trait association analyses for 20 functional traits in a perennial herb were conducted, showing that more MSAP markers than AFLP involved in significant association (Medrano et al., 2014). Due to the limited loci and phenotypes in our study, we did not perform a genotype–phenotype association analysis. Bayesian clustering and GLM revealed the genetic differentiation between habitats, but Mantel tests and MMRR indicated a significant correlation between epigenetic and phenotypic variation. This contradictory result suggests that the key stable phenotypes associated with habitat type were not considered in our study, as we only measured several plastic functional traits varying not only between habitats but also among all sites. However, our results added the indirect evidence that epigenetic variation can serve as an important source of intraspecific functional diversity in nature.
According to the illustration of genetic and epigenetic isolation‐by‐distance scenarios along with hypothesized causes (Herrera et al., 2016), our data revealed the moderate recent epigenetic variation between generations in natural populations of V. negundo var. heterophylla. Consistent with the results from a perennial herb (Herrera, Medrano, & Bazaga, 2017), geographic distance explained genetic differentiation better than epigenetic differentiation, and epigenetic variation contributed to the divergence in functional traits in the perennial shrub. It is necessary to conduct transplantation or common garden experiments with offspring from populations in different habitats to distinguish the contribution of induced and inherited epigenetic variation to adaptive phenotypes. As the MSAP marker only provides limited anonymous loci that are difficult to link to functional genomic elements or phenotype, a reduced representation bisulfite sequencing approach based on next‐generation sequencing methods is the next level of epigenetic study in natural populations (Gugger et al., 2016; Platt et al., 2015; Robertson & Richards, 2015; Trucchi et al., 2016).
5. CONCLUSION
Our study used functional traits, AFLP, and MSAP to analyze phenotypic, genetic, and epigenetic variation in natural populations of V. negundo var. heterophylla from an extensive variety of habitat environments. Two scoring approaches for MSAP marker data were conducted, and the results were consistent in most cases. The analysis demonstrated significant habitat‐related adaptation in phenotypic and genetic differentiation, suggesting an evident process of natural selection. However, we did not find a direct correlation between functional phenotypic and genetic variation, and Mantel tests and MMRR revealed a significant relationship between epigenetic and genetic or phenotypic variation. This result ultimately implied a potential intermediary role of epigenetic mechanisms in the adaption to heterogeneous habitats.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTION
LL, WG, and ND conceived the idea and designed the study. LL, CP, and ND contributed to the field survey. CP and LL performed the molecular laboratory work. LL, XG, and WG analyzed and interpreted the data. LL drafted the manuscript and all authors participated in manuscript modifications and gave final approval for publication.
ACKNOWLEDGMENTS
We want to thank Cui Wenqiang, Liu Shuna, Chen Bin, and Ma Haoran for their assistance with fieldwork. We are also grateful to Dr. Cai Yunfei for his guidance in the molecular biology experiments. Financial support was obtained from the National Science Foundation of China (Nos. 31770361;31470402) and the Ministry of Science and Technology of China (No. 2015FY110300).
Lele L, Ning D, Cuiping P, Xiao G, Weihua G. Genetic and epigenetic variations associated with adaptation to heterogeneous habitat conditions in a deciduous shrub. Ecol Evol. 2018;8:2594–2606. https://doi.org/10.1002/ece3.3868
REFERENCES
- Abràmoff, M. D. , Magalhães, P. J. , & Ram, S. J. (2004). Image processing with ImageJ. Biophotonics International, 11(7), 36–42. [Google Scholar]
- Bonnet, E. , & Van de Peer, Y. (2002). zt: A software tool for simple and partial Mantel tests. Journal of Statistical Software, 7(10), 1–12. [Google Scholar]
- Bossdorf, O. , Richards, C. L. , & Pigliucci, M. (2008). Epigenetics for ecologists. Ecology Letters, 11(2), 106–115. [DOI] [PubMed] [Google Scholar]
- Bradburd, G. S. , Ralph, P. L. , & Coop, G. M. (2013). Disentangling the effects of geographic and ecological isolation on genetic differentiation. Evolution, 67(11), 3258–3273. https://doi.org/10.1111/evo.12193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräutigam, K. , Vining, K. J. , Lafon‐Placette, C. , Fossdal, C. G. , Mirouze, M. , Marcos, J. G. , … Cervera, M. T. (2013). Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecology and Evolution, 3(2), 399–415. https://doi.org/10.1002/ece3.461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douhovnikoff, V. , & Hazelton, E. L. G. (2014). Clonal growth: Invasion or stability? A comparative study of clonal architecture and diversity in native and introduced lineages of Phragmites australis (Poaceae). American Journal of Botany, 101(9), 1577–1584. https://doi.org/10.3732/ajb.1400177 [DOI] [PubMed] [Google Scholar]
- Doyle, J. J. , & Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin, 19, 11–15. [Google Scholar]
- Du, N. , Guo, W. , Zhang, X. , & Wang, R. (2010). Morphological and physiological responses of Vitex negundo L. var. heterophylla (Franch.) Rehd. to drought stress. Acta Physiologiae Plantarum, 32(5), 839–848. https://doi.org/10.1007/s11738-010-0468-z [Google Scholar]
- Du, N. , Wang, R. , Liu, J. , Zhang, X. , Tan, X. , Wang, W. , Chen, H. , Guo, W. (2012). Morphological response of Vitex negundo var. heterophylla and Ziziphus jujuba var. spinosa to the combined impact of drought and shade. Agroforestry Systems, 87(2), 403–416. [Google Scholar]
- Earl, D. A. , & vonHoldt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4(2), 359–361. https://doi.org/10.1007/s12686-011-9548-7 [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14(8), 2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2007). Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Molecular Ecology Notes, 7(4), 574–578. https://doi.org/10.1111/j.1471-8286.2007.01758.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust, C. M. , Preite, V. , Schrey, A. W. , Alvarez, M. , Robertson, M. H. , Verhoeven, K. J. F. , & Richards, C. L. (2016). Genetic and epigenetic differences associated with environmental gradients in replicate populations of two salt marsh perennials. Molecular Ecology, 25(8), 1639–1652. https://doi.org/10.1111/mec.13522 [DOI] [PubMed] [Google Scholar]
- Gugger, P. F. , Fitz‐Gibbon, S. , PellEgrini, M. , & Sork, V. L. (2016). Species‐wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Molecular Ecology, 25(8), 1665–1680. https://doi.org/10.1111/mec.13563 [DOI] [PubMed] [Google Scholar]
- Guillot, G. , & Rousset, F. (2013). Dismantling the Mantel tests. Methods in Ecology and Evolution, 4(4), 336–344. https://doi.org/10.1111/2041-210x.12018 [Google Scholar]
- Herrera, C. M. , & Bazaga, P. (2008). Population‐genomic approach reveals adaptive floral divergence in discrete populations of a hawk moth‐pollinated violet. Molecular Ecology, 17(24), 5378–5390. https://doi.org/10.1111/j.1365-294X.2008.04004.x [DOI] [PubMed] [Google Scholar]
- Herrera, C. M. , & Bazaga, P. (2010). Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytologist, 187(3), 867–876. https://doi.org/10.1111/j.1469-8137.2010.03298.x [DOI] [PubMed] [Google Scholar]
- Herrera, C. M. , & Bazaga, P. (2013). Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and nonprickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. Botanical Journal of the Linnean Society, 171(3), 441–452. https://doi.org/10.1111/boj.12007 [Google Scholar]
- Herrera, C. M. , & Bazaga, P. (2016). Genetic and epigenetic divergence between disturbed and undisturbed subpopulations of a Mediterranean shrub: A 20‐year field experiment. Ecology and Evolution, 6(11), 3832–3847. https://doi.org/10.1002/ece3.2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera, C. M. , Medrano, M. , & Bazaga, P. (2016). Comparative spatial genetics and epigenetics of plant populations: Heuristic value and a proof of concept. Molecular Ecology, 25(8), 1653–1664. https://doi.org/10.1111/mec.13576 [DOI] [PubMed] [Google Scholar]
- Herrera, C. M. , Medrano, M. , & Bazaga, P. (2017). Comparative epigenetic and genetic spatial structure of the perennial herb Helleborus foetidus: Isolation by environment, isolation by distance, and functional trait divergence. American Journal of Botany, 104(8), 1195–1204. https://doi.org/10.3732/ajb.1700162 [DOI] [PubMed] [Google Scholar]
- Hu, P. , Li, D. , Wang, K. , Wang, H. , Wang, Z. , Li, Z. , & Hua, H. (2015). New phenolic compounds from Vitex negundo var. heterophylla and their antioxidant and NO inhibitory activities. Journal of Functional Foods, 19, 174–181. https://doi.org/10.1016/j.jff.2015.09.016 [Google Scholar]
- Jablonka, E. , & Raz, G. (2009). Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. The Quarterly Review of Biology, 84(2), 131–176. https://doi.org/10.1086/598822 [DOI] [PubMed] [Google Scholar]
- Kilvitis, H. J. , Alvarez, M. , Foust, C. M. , Schrey, A. W. , Robertson, M. , & Richards, C. L. (2014). Ecological Epigenetics In Landry C. R., & Aubin‐Horth N. (Eds.), Ecological genomics (pp. 191–210). Dordrecht, the Netherlands: Springer; https://doi.org/10.1007/978-94-007-7347-9 [DOI] [PubMed] [Google Scholar]
- Kim, N.‐S. , Im, M.‐J. , & Nkongolo, K. (2016). Determination of DNA methylation associated with Acer rubrum (red maple) adaptation to metals: Analysis of global DNA modifications and methylation‐sensitive amplified polymorphism. Ecology and Evolution, 6(16), 5749–5760. https://doi.org/10.1002/ece3.2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, J. A. , & Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics, 11(3), 204–220. https://doi.org/10.1038/nrg2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre, P. , Fortin, M. J. , & Borcard, D. (2015). Should the Mantel test be used in spatial analysis? Methods in Ecology and Evolution, 6(11), 1239–1247. https://doi.org/10.1111/2041-210X.12425 [Google Scholar]
- Li, S. , Yang, B. , & Wu, D. (2008). Community succession analysis of naturally colonized plants on coal gob piles in Shanxi mining areas, China. Water, Air, and Soil Pollution, 193(1–4), 211–228. https://doi.org/10.1007/s11270-008-9684-1 [Google Scholar]
- McIntyre, P. J. , & Strauss, S. Y. (2014). Phenotypic and transgenerational plasticity promote local adaptation to sun and shade environments. Evolutionary Ecology, 28(2), 229–246. https://doi.org/10.1007/s10682-013-9670-y [Google Scholar]
- McLean, E. H. , Prober, S. M. , Stock, W. D. , Steane, D. A. , Potts, B. M. , Vaillancourt, R. E. , & Byrne, M. (2014). Plasticity of functional traits varies clinally along a rainfall gradient in Eucalyptus tricarpa. Plant, Cell & Environment, 37(6), 1440–1451. https://doi.org/10.1111/pce.12251 [DOI] [PubMed] [Google Scholar]
- Medrano, M. , Herrera, C. M. , & Bazaga, P. (2014). Epigenetic variation predicts regional and local intraspecific functional diversity in a perennial herb. Molecular Ecology, 23(20), 4926–4938. https://doi.org/10.1111/mec.12911 [DOI] [PubMed] [Google Scholar]
- Nicotra, A. B. , Atkin, O. K. , Bonser, S. P. , Davidson, A. M. , Finnegan, E. J. , Mathesius, U. , … van Kleunen, M. (2010). Plant phenotypic plasticity in a changing climate. Trends in Plant Science, 15(12), 684–692. https://doi.org/10.1016/j.tplants.2010.09.008 [DOI] [PubMed] [Google Scholar]
- Nicotra, A. B. , Segal, D. L. , Hoyle, G. L. , Schrey, A. W. , Verhoeven, K. J. F. , & Richards, C. L. (2015). Adaptive plasticity and epigenetic variation in response to warming in an Alpine plant. Ecology and Evolution, 5(3), 634–647. https://doi.org/10.1002/ece3.1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall, R. O. D. , & Smouse, P. E. (2006). GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6(1), 288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Harguindeguy, N. , Díaz, S. , Garnier, E. , Lavorel, S. , Poorter, H. , Jaureguiberry, P. , … Cornelissen, J.H.C. (2013). New handbook for standardised measurement of plant functional traits worldwide. Australian Journal of Botany, 61(3), 167–234. https://doi.org/10.1071/BT12225 [Google Scholar]
- Pigliucci, M. (2001). Phenotypic plasticity: Beyond nature and nurture. Baltimore: JHU Press. [Google Scholar]
- Platt, A. , Gugger, P. F. , Pellegrini, M. , & Sork, V. L. (2015). Genome‐wide signature of local adaptation linked to variable CpG methylation in oak populations. Molecular Ecology, 24(15), 3823–3830. https://doi.org/10.1111/mec.13230 [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, E. J. (2011). Natural epigenetic variation in plant species: A view from the field. Current Opinion in Plant Biology, 14(2), 204–209. https://doi.org/10.1016/j.pbi.2011.03.009 [DOI] [PubMed] [Google Scholar]
- Richards, C. L. , Alonso, C. , Becker, C. , Bossdorf, O. , Bucher, E. , Colome‐Tatche, M. , … Verhoeven, K.J.F. (2017). Ecological plant epigenetics: Evidence from model and non‐model species, and the way forward. Ecology Letters, 20(12), 1576–1590. [DOI] [PubMed] [Google Scholar]
- Richards, C. L. , Schrey, A. W. , & Pigliucci, M. (2012). Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecology Letters, 15(9), 1016–1025. https://doi.org/10.1111/j.1461-0248.2012.01824.x [DOI] [PubMed] [Google Scholar]
- Robertson, M. , & Richards, C. (2015). Opportunities and challenges of next‐generation sequencing applications in ecological epigenetics. Molecular Ecology, 24(15), 3799–3801. https://doi.org/10.1111/mec.13277 [DOI] [PubMed] [Google Scholar]
- Robertson, M. , Schrey, A. , Shayter, A. , Moss, C. J. , & Richards, C. (2017). Genetic and epigenetic variation in Spartina alterniflora following the Deepwater Horizon oil spill. Evolutionary Applications, 10(8), 792–801. https://doi.org/10.1111/eva.12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda, F. , Ambrose, L. , Walter, G. M. , Liu, H. L. , Schaul, A. , Lowe, A. , … Ortiz‐Barrientos, D. (2013). Genomic evidence for the parallel evolution of coastal forms in the Senecio lautus complex. Molecular Ecology, 22(11), 2941–2952. https://doi.org/10.1111/mec.12311 [DOI] [PubMed] [Google Scholar]
- Scheepens, J. F. , Frei, E. S. , & Stöcklin, J. (2010). Genotypic and environmental variation in specific leaf area in a widespread Alpine plant after transplantation to different altitudes. Oecologia, 164(1), 141–150. https://doi.org/10.1007/s00442-010-1650-0 [DOI] [PubMed] [Google Scholar]
- Schrey, A. W. , Alvarez, M. , Foust, C. M. , Kilvitis, H. J. , Lee, J. D. , Liebl, A. L. , … Robertson, M. (2013). Ecological epigenetics: Beyond MS‐AFLP. Integrative and Comparative Biology, 53(2), 340–350. https://doi.org/10.1093/icb/ict012 [DOI] [PubMed] [Google Scholar]
- Schulz, B. , Eckstein, R. L. , & Durka, W. (2013). Scoring and analysis of methylation‐sensitive amplification polymorphisms for epigenetic population studies. Molecular Ecology Resources, 13(4), 642–653. https://doi.org/10.1111/1755-0998.12100 [DOI] [PubMed] [Google Scholar]
- Schulz, B. , Eckstein, R. L. , & Durka, W. (2014). Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Molecular Ecology, 23(14), 3523–3537. https://doi.org/10.1111/mec.12835 [DOI] [PubMed] [Google Scholar]
- Slotkin, R. K. , & Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nature Reviews Genetics, 8(4), 272–285. https://doi.org/10.1038/nrg2072 [DOI] [PubMed] [Google Scholar]
- Su, H. , Qu, L.‐J. , He, K. , Zhang, Z. , Wang, J. , Chen, Z. , & Gu, H. (2003). The Great Wall of China: A physical barrier to gene flow? Heredity, 90(3), 212–219. https://doi.org/10.1038/sj.hdy.6800237 [DOI] [PubMed] [Google Scholar]
- Trucchi, E. , Frajman, B. , Haverkamp, T. H. A. , Schönswetter, P. , & Paun, O. (2017). Genomic analyses suggest parallel ecological divergence in Heliosperma pusillum (Caryophyllaceae). New Phytologist, 216(1), 267–278. https://doi.org/10.1111/nph.14722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trucchi, E. , Mazzarella, A. B. , Gilfillan, G. D. , Lorenzo, M. T. , Schönswetter, P. , & Paun, O. (2016). BsRADseq: Screening DNA methylation in natural populations of non‐model species. Molecular Ecology, 25(8), 1697–1713. https://doi.org/10.1111/mec.13550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven, K. J. F. , Jansen, J. J. , van Dijk, P. J. , & Biere, A. (2010). Stress‐induced DNA methylation changes and their heritability in asexual dandelions. New Phytologist, 185(4), 1108–1118. https://doi.org/10.1111/j.1469-8137.2009.03121.x [DOI] [PubMed] [Google Scholar]
- Verhoeven, K. J. F. , vonHoldt, B. M. , & Sork, V. L. (2016). Epigenetics in ecology and evolution: What we know and what we need to know. Molecular Ecology, 25(8), 1631–1638. https://doi.org/10.1111/mec.13617 [DOI] [PubMed] [Google Scholar]
- Vos, P. , Hogers, R. , Bleeker, M. , Reijans, M. , van de Lee, T. , Hornes, M. , … Zabeau, M. (1995). AFLP: A new technique for DNA fingerprinting. Nucleic Acids Research, 23(21), 4407–4414. https://doi.org/10.1093/nar/23.21.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, I. J. (2013). Examining the full effects of landscape heterogeneity on spatial genetic variation: A multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution, 67(12), 3403–3411. https://doi.org/10.1111/evo.12134 [DOI] [PubMed] [Google Scholar]
- Warton, D. I. , Duursma, R. A. , Falster, D. S. , & Taskinen, S. (2012). smatr 3– an R package for estimation and inference about allometric lines. Methods in Ecology and Evolution, 3(2), 257–259. https://doi.org/10.1111/j.2041-210X.2011.00153.x [Google Scholar]
- Williams, G. C. (2008). Adaptation and natural selection: A critique of some current evolutionary thought. Princeton: Princeton University Press. [Google Scholar]
- Wilschut, R. A. , Oplaat, C. , Snoek, L. B. , Kirschner, J. , & Verhoeven, K. J. F. (2016). Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Molecular Ecology, 25(8), 1759–1768. https://doi.org/10.1111/mec.13502 [DOI] [PubMed] [Google Scholar]
- Xie, H. J. , Li, H. , Liu, D. , Dai, W. M. , He, J. Y. , Lin, S. , … Qiang, S. (2015). ICE1 demethylation drives the range expansion of a plant invader through cold tolerance divergence. Molecular Ecology, 24(4), 835–850. https://doi.org/10.1111/mec.13067 [DOI] [PubMed] [Google Scholar]
- Zhang, Y.‐Y. , Fischer, M. , Colot, V. , & Bossdorf, O. (2013). Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytologist, 197(1), 314–322. https://doi.org/10.1111/nph.12010 [DOI] [PubMed] [Google Scholar]
- Zhang, Z.‐Y. , Zheng, X.‐M. , & Ge, S. (2007). Population genetic structure of Vitex negundo (Verbenaceae) in three‐gorge area of the Yangtze River: The riverine barrier to seed dispersal in plants. Biochemical Systematics and Ecology, 35(8), 506–516. https://doi.org/10.1016/j.bse.2007.01.014 [Google Scholar]