

Key Clinical Message
We report a 4 years girl with congenital disorders of glycosylation (CDG) type Ib due to a novel homozygous mutation in MPI gene. She presented with diazoxide‐responsive hyperinsulinemic hypoglycemia. CDG should be considered in unexplained hypoglycemia particularly in consanguineous families. Diagnosis enables monitoring/prevention of disease comorbidities and early effective treatment.
Keywords: Glycation, hyperinsulinemia, hypoglycemia, mannose phosphate isomerase
Introduction
Congenital disorders of glycosylation (CDGs) are a genetically heterogeneous group of autosomal recessive disorders. They are caused by enzymatic defects in the synthesis and processing of asparagine (N)‐linked glycans or oligosaccharides on glycoproteins. These disorders are recognized by an abnormal isoelectric focusing profile of plasma transferrin 1. Most known cases belong to type I, which, has 12 defined subgroups (CDG‐Ia to CDG‐IL). CDG type 1b results from genetic mutations in the mannose phosphate isomerase gene on chromosome 15q24 that encodes mannose phosphate isomerase (MPI). The MPI gene contains eight exons and spans 5 kb 2.
Pelletier et al. (1986) first described the clinical features of CDG Ib. These features include secretory diarrhea with protein‐losing enteropathy, enterocolitis cystica superficialis, intestinal lymphangiectasia, hypoglycemia, coagulopathy, and congenital hepatic fibrosis 3. CDG Ib is clinically distinct from most other CDGs in two main features. Firstly, it lacks the central nervous system involvement and secondly, it can be effectively treated with oral mannose supplementation.
Niehues et al. reported an 11‐month‐old boy who presented with diarrhea and vomiting. Laboratory studies showed severe hypoproteinemia, anemia, decreased antithrombin III, and protein‐losing enteropathy.
Oral administration of mannose was found to be an effective therapy for CDG Ib as it corrected the clinical phenotype as well as the hypoglycosylation of serum glycoproteins 4. The authors identified a heterozygous mutation in the MPI gene resulting in an arg219‐to‐gln (R219Q) substitution inherited from the father. Subsequently, Schollen identified a a 1‐bp insertion in exon 3 of the MPI gene (116insC; 154550.0004) on the maternal allele in the same patient confirming compound heterozygosity and autosomal recessive inheritance 2. Subsequently, Schollen et al. identified seven other mutations in the MPI gene. Compound heterozygosity in the MPI gene was identified in another patient with a 304C‐T transition resulting in a ser102‐to‐leu (S102L) substitution, and a 413T‐C transition resulting in a met138‐to‐thr (M138T) substitution 5.
Hyperinsulinemic hypoglycemia has been described in several cases mostly in CDG Ib. In 1999, De Lonlay et al. reported a 3‐month‐old girl presenting with hypoglycemia. She had severe vomiting and diarrhea, congenital hepatic fibrosis, and coagulation factor deficiencies. Mannose therapy led to dramatic clinical improvement and normalization of several biochemical abnormalities 6. In the same year, another 2.5‐year‐old girl was reported with CDG Ib. She presented with severe persistent hypoglycemia and subsequently developed protein‐losing enteropathy, liver disease, and coagulopathy 7.
We report a 4 years old who presented with hyperinsulinemic hypoglycemia and was diagnosed with CDG Ib due to a novel mutation in the MPI gene.
Case Report
A 4 years old female of Emirati origin presented at the age of 8 months with hypoglycemia. Her hypoglycemia was severe and recurrent, and she was extensively investigated. She had multiple admissions for gastroenteritis and bronchiolitis during which she had hypoglycemia.
She was born at 36 weeks of gestation with a birthweight of 2450 kg. Her developmental milestones were normal. Parents were first‐degree cousins. She had seven healthy siblings. Mother had five first‐trimester abortions due to unknown cause. There was no family history of hypoglycemia or liver disease.
Clinical examination showed no dysmorphic features. She was normocephalic with weight of 14 kg (25th centile) and a height of 98 cm (25th centile). She had hair growth over her forehead and the side of the face. She had no edema. Abdominal examination showed a hepatomegaly of 5 cm below costal margin. Her neurological examination, particularly her muscle tone, was normal (Table 1).
Table 1.
Clinical characteristics of the patient reported
| Patient characteristics | |
| Current age | 4 years |
| Age at presentation | 8 months |
| Main presenting feature | Hyperinsulinemic Hypoglycemia |
| Family history | Emirati, first‐degree cousins |
| No history of hypoglycemia or liver disease | |
| Clinical features | Normal development |
| Normal growth | |
| Hepatomegaly | |
| Biochemical investigations | High liver transaminases |
| Low albumin and total protein | |
| Hypoglycemia | |
| Positive urinary ketones | |
| High insulin level | |
| Positive glucagon challenge | |
| Imaging | Hepatomegaly with a liver nodule |
| Genetic test result | Novel homozygous mutation (p.Ala288Val) at the MPI gene |
| Both parents are carriers | |
| Treatment | Diazoxide‐responsive |
Investigations
Investigations showed microcytic anemia with a hemoglobin level of 8.2 g, hematocrit of 0.26. Her liver transaminases were elevated with ALT of 61(NR below 31) and AST of 87 (NR below 40). Her total protein and albumin were both low at 44 g/L and 30 g/L, respectively, and her coagulation screen was normal. Alpha‐fetoprotein was normal.
Investigations during hypoglycemia showed positive urine ketones, normal lactate, ammonia, free fatty acid, serum amino acid, urine organic acid, total acyl, and free carnitine. Her growth hormone and cortisol level were both normal. At a glucose level of 42 mg, her insulin level was detectable at 8.54 μIU/mL, and her c‐peptide was 1.85 ng/mL (NR 0.8–3.1). Glucagon challenge test after fasting resulted in an increase of glucose from 2.3 mmol (42 mg) to 5.3 mmol (96 mg).
Liver MRI showed hepatomegaly with 1.7 cm sized low density subcapsular nodular lesion.
Genetic testing
Whole exome sequencing showed a novel homozygous mutation (p.Ala288Val) at the MPI gene. Both parents were found to be carriers of the mutant gene. Based on the clinical presentation and the genetic testing results, she was diagnosed with congenital disorder of glycosylation type 1b (OMIM_#602579).
Treatment and progress
During infancy, she had multiple hypoglycemic episodes with documented low glucose level of 1.9 mmol (34 mg). She was put on complex carbohydrate at night time and advised to avoid prolonged fast. She continued to have episodes of hypoglycemia particularly during febrile illness of gastroenteritis and bronchiolitis. At the age of 2 years, the diagnosis of hyperinsulinemic hypoglycemia due to MPI gene defect was confirmed, and she was started on diazoxide at a dose range of 4–7 mg/kg/day in three divided doses. Her typical daily glucose profile showed a median glucose of 3.8 mmol (69 mg) with an interquartile range of 2.8 mmol (51 mg) – 4.0 mmol (72 mg) (Fig. 1). On the above dose, she developed marked hirsutism. Reducing diazoxide to below 4 mg/kg/day resulted in recurrence of hypoglycemia attacks (Fig. 2). On follow‐up, she continued to grow along the 25th centile and had no features of malabsorption. Her liver function profile continued to show 1–2 folds elevation of liver transaminases with borderline low albumin and total protein. Her coagulation screen remained normal. Repeated liver MRI showed no change in size of the nodular lesion previously detected.
Figure 1.
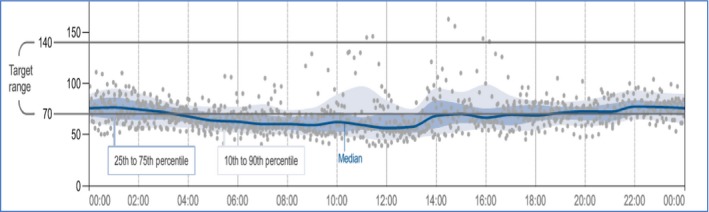
Typical daily glucose profile as shown by ambulatory glucose monitoring.
Figure 2.
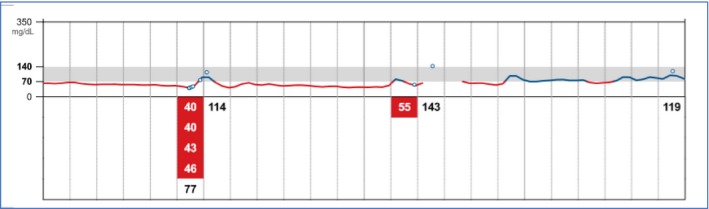
Glucose trace showing hypoglycemia down to 40 mg (2.2 mmol) on reducing the diazoxide dose.
Discussion
Patients with MPI‐CDG have a syndromal presentation with a hepatointestinal phenotype of chronic diarrhea (protein‐losing enteropathy), coagulation defects, and liver disease, without the presence of dysmorphic features and with normal development. De Lonlay et al. reported the clinical, biologic, and molecular analysis of 26 patients with CDG I, including two CDG Ib 8. The two patients with CDG Ib had severe liver disease, protein‐losing enteropathy, and hyperinsulinemic hypoglycemia and no neurologic involvement. The latter was the case in our patient; however, she did not have features of coagulation defect. Her liver disease was not pronounced, and her transaminase elevation was mild. In CDG type Ib, liver fibrosis might appear in childhood or in young adulthood when liver failure may occur. Successful liver transplant has been reported in this condition 9. In one reported patient, severe hypoglycemia was the initial feature of presentation, but liver disease with coagulopathy subsequently developed highlighting the need for follow‐up of liver function 7.
Different molecular defects have been reported in patients with CDG Ib. Compound heterozygosity of arg219‐to‐gln (R219Q) and a 1‐bp insertion in exon 3 was confirmed in an 11 months old who was diagnosed with CDG Ib 2, 4. Another compound heterozygosity in the MPI gene was identified in a patient who had a 304C‐T transition resulting in a ser102‐to‐leu (S102L) substitution, and a 413T‐C transition with a met138‐to‐thr (M138T) substitution 5. Homozygous mutation in the MPI was first reported by Vuillaumier‐Barrot who identified the first patient with homozygous mutation in exon 7 of the MPI gene; R295H (G884A) 10. Our patient had a homozygous mutation (p.Ala288Val) with both parents confirmed to be carriers. This mutation has not been reported before. The clinical phenotype of our patient is different from the other patient reported with the homozygous mutation who was a French boy born to nonconsanguinous parents. Unlike our patient, he presented with diarrhea related to protein‐losing enteropathy with edema, hypoalbuminemia, hyponatremia. His coagulation profile was deranged with a decreased coagulation factor XI and an increased prothrombin time.
Hyperinsulinemic hypoglycemia has been described in several cases in CDG but mostly in CDG Ib. Severe and persistent hypoglycemia has also been reported in patients with CDG Ib. Hypoglycemia could be the presenting clinical feature which can be followed by protein‐losing enteropathy, liver disease, and coagulopathy 7. Treatment of CDG Ib has been proven to be effective. In 1999, De Lonlay et al. reported a 3‐month‐old girl presenting with hypoglycemia. She was successfully treated with mannose which led to normalization of several biochemical abnormalities including hypoglycemia and liver enzyme derangement 6. In 2008, the first case of CDG Ib was reported in Spain. The patient presented at 6 months with hypoglycemia, failure to thrive, and elevated liver transaminases. He subsequently developed an enteropathy with subtotal villous atrophy on biopsy. His genetic defect was confirmed to be due to a mutation in MPI gene at R219Q and R56fs. His clinical and biochemical parameters normalized after treatment with mannose 1 11. Our patient responded to diazoxide treatment, and her liver disease was mild. Accordingly, mannose treatment was not introduced.
At the molecular level, response to diazoxide led to the hypothesis that there is element of hypoglycosylation at the sulfonylurea receptor‐1. The response to diazoxide indicates the presence of some functional ATP‐sensitive K+ channels on the cell surface 12.
In addition to Hyperinsulinemic hypoglycemia in CDG Ib, it was also reported in CDG‐Ia and CDG‐Ic. In a patient with CDG Id due to homozygous point mutation (512G>A) in the hALG3 gene, the patient had severe hyperinsulinemic hypoglycemia and died at 19 days of age. She had profound hypotonia and facial dysmorphism. Autopsy revealed islet cell hyperplasia with increased β‐cell mass 13. CGD Ib is a rare disease. However, it might be underdiagnosed as it has been reported in asymptomatic adults misdiagnosed with a more common disease. Helander et al. detected a homozygous missense mutation in the MPI gene (p.R219Q) in a 32‐year‐old woman who had a highly elevated serum level of carbohydrate‐deficient transferrin, a biomarker for excessive alcohol consumption, during a routine health check 14.
In summary, our patient is the first to be reported with CDG Ib from the region. Her mutation is novel. Her phenotype is unique with mild liver involvement, normal coagulation, and diazoxide‐responsive hyperinsulinemic hypoglycemia. We recommend that CDG Ib should be considered in the differential diagnosis of patients with unexplained hypoglycemia particularly in consanguineous families. Early diagnosis allows introduction of effective therapy and enables genetic counseling.
Consent
Informed consent was obtained from the patient's mother to report the child's case.
Authorship
AD: is the named physician of the patient. She investigated the patient and followed her up. Dr Deeb: finalized and submitted the manuscript. AA: revised the literature and wrote the initial draft of the manuscript.
Conflict of Interest
Authors have no conflict of interest to declare.
References
- 1. Leroy, J. G. 2006. Congenital disorders of N‐glycosylation including diseases associated with O‐ as well as N‐glycosylation defects. Pediatr. Res. 60:643–656. [DOI] [PubMed] [Google Scholar]
- 2. Schollen, E. , Dorland L., De Koning T. J., Van Diggelen O. P., Huijmans J. G. M., Marquardt T., et al. 2000. Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG‐Ib). Hum. Mutat. 16:247–252. [DOI] [PubMed] [Google Scholar]
- 3. Pelletier, V. A. , Galéano N., Brochu P., Morin C. L., Weber A. M., and Roy C. C.. 1986. Secretory diarrhea with protein‐losing enteropathy, enterocolitis cystica superficialis, intestinal lymphangiectasia, and congenital hepatic fibrosis: a new syndrome. J. Pediatr. 108:61–65. [DOI] [PubMed] [Google Scholar]
- 4. Niehues, R. , Hasilik M., Alton G., Körner C., Schiebe‐Sukumar M., Koch H. G., et al. 1998. Carbohydrate‐deficient glycoprotein syndrome type Ib: phosphomannose isomerase deficiency and mannose therapy. J. Clin. Invest. 101:1414–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jaeken, J. , Matthijs G., Saudubray J. M., Dionisi‐Vici C., Bertini E., de Lonlay P., et al. 1998. Phosphomannose isomerase deficiency: a carbohydrate‐deficient glycoprotein syndrome with hepatic‐intestinal presentation. (Letter). Am. J. Hum. Genet. 62:1535–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Lonlay, P. , Cuer M., Vuillaumier‐Barrot S., Beaune G., Castelnau P., Kretz M., et al. 1999. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: a new manifestation of carbohydrate‐deficient glycoprotein syndrome treatable with mannose. J. Pediatr. 135:379–383. [DOI] [PubMed] [Google Scholar]
- 7. Babovic‐Vuksanovic, D. , Patterson M. C., Schwenk W. F., O'Brien J. F., Vockley J., Freeze H. H., et al. 1999. Severe hypoglycemia as a presenting symptom of carbohydrate‐deficient glycoprotein syndrome. J. Pediatr. 135:775–781. [DOI] [PubMed] [Google Scholar]
- 8. de Lonlay, P. , Seta N., Barrot S., Chabrol B., Drouin V., Gabriel B. M., et al. 2001. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J. Med. Genet. 38:14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janssen, M. C. , de Kleine R. H., van den Berg A. P., Heijdra Y., van Scherpenzeel M., Lefeber D. J., et al. 2014. Successful liver transplantation and long‐term follow‐up in a patient with MPI‐CDG. Pediatrics 134:e279–e283. [DOI] [PubMed] [Google Scholar]
- 10. Vuillaumier‐Barrot, S. , Le Bizec C., De Lonlay P., Barnier A., Mitchell G., Pelletier V., et al. 2002. Protein losing enteropathy‐hepatic fibrosis syndrome in Saguenay‐Lac St‐Jean, Quebec is a congenital disorder of glycosylation type Ib. J. Med. Genet. 39:849–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martín, H. E. , Vega P. A., Pérez G. B., Ecay C. M., Leal P. F., Manzanares L. M. J., et al. 2008. Congenital disorder of glycosylation type 1b. Experience with mannose treatment. An. Pediatr. (Barc.) 69:358–365. [DOI] [PubMed] [Google Scholar]
- 12. Böhles, H. , Sewell A. C., Gebhardt B., Reinecke‐Lüthge A., Klöppel G., and Marquardt T.. 2001. Hyperinsulinaemic hypoglycaemia‐leading symptom in a patient with congenital disorder of glycosylation Ia (phosphomannomutase deficiency). J. Inherit. Metab. Dis. 24:858–862. [DOI] [PubMed] [Google Scholar]
- 13. Sun, L. , Eklund E. A., Chung W. K., Wang C., Cohen J., and Freeze H. H.. 2005. Congenital disorder of glycosylation presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J. Clin. Endocrinol. Metab. 90:4371–4375. [DOI] [PubMed] [Google Scholar]
- 14. Helander, A. , Jaeken J., Matthijs G., and Eggertsen G.. 2014. Asymptomatic phosphomannose isomerase deficiency (MPI‐CDG) initially mistaken for excessive alcohol consumption. Clin. Chem. Acta. 20:15–18. [DOI] [PubMed] [Google Scholar]