

Abstract
The synthesis of shape‐persistent organic cage compounds is often based on the usage of multiple dynamic covalent bond formation (such as imines) of readily available precursors. By careful choice of the precursors geometry, the geometry and size of the resulting cage can be accurately designed and indeed a number of different geometries and sizes have been realized to date. Despite of this fact, little is known about the precursors conformational rigidity and steric preorganization of reacting functional groups on the outcome of the reaction. Herein, the influence of conformational rigidity in the precursors on the formation of a [4+4] imine cage with truncated tetrahedral geometry is discussed.
Keywords: covalent organic frameworks, gas sorption, imines, porous crystals, shape-persistent cages
Shape‐persistent covalent organic cage compounds and capsules are interesting target molecules, mainly because of their defined and precisely confined interior space, which can be used to host guest‐molecules of different sizes or for gas sorption.1 If the host–guest binding occurs selectively, the shape‐persistent organic cages can be used for example, for the molecular separation of (gas) mixtures either in the bulk,2 by embedding the cages into membranes,3 fill stationary columns with them for gas chromatographic purposes,2a, 4 or more sophisticated, to create porous liquids.5 Thin films of shape‐persistent organic cages have been deposited on quartz crystal microbalances for selective gravimetric sensing.6, 7 Organic cages have also been used for sensing in solution by selective fluorescence quenching in the presence of explosives.8
To efficiently synthesize shape‐persistent organic cages in high yields, dynamic covalent chemistry (DCC) turned out to be the synthetic method of choice.1a, 1b Cages of various sizes and geometries should be accessible by choosing the right precursors.9 And indeed a large number of geometries and topologies have been realized till date including prisms,10 tetrahedra,11 cubes,2c, 12 cuboctahedra,13 octahedra,14 tetrapods15 and others.16
However, the influence of the precursors degree of rigidity and pre‐orientation of functional groups to achieve the formation of shape‐persistent organic cages is still not much investigated and fully understood.17 We and others suggested that these precursors need to be well‐chosen and sometimes subtle changes leads to a complete failure of cage synthesis.18 For instance, Cooper et al. studied the effect of connecting aliphatic tethers of different lengths of diamines in the imine condensation with aromatic trialdehydes.19 Supported by theoretical calculations, an odd–even effect was predicted and confirmed in the product mixture, although the overall amount of non‐defined by‐products was still very high.19 A similar effect was investigated for cages stabilized by keto–enol tautomerization by the Banerjee group.20 Mukherjee et al. studied self‐sorting of a four component system based on precursors flexibility and demonstrated that only the cages are formed with at least one rigid component involved.21 Our group for instance investigated the degree of intrinsic flexibility of “linear” bis‐salicylaldehydes in the formation of [2+3] imine cages.10a In the case of the flexible precursor, the formation of defined [2+3] cages was significantly lower in yield.
Derivatives of 1,3,5‐triaminomethylbenzene 1 as well as 1,3,5‐triformylbenzene 2 have been used independently in the synthesis of imine‐based cages.11b, 22 Surprisingly, these two compounds have not been reported to be reacted together in imine condensation reactions.
Here, we present our initial studies on the formation of [4+4] imine cages with a truncated tetrahedral geometry by a twelve‐fold condensation of triamines 1 and trialdehydes 2; focusing on the influence of precursors rigidity and pre‐orientation of functional groups on cage formation (Scheme 1).
Scheme 1.
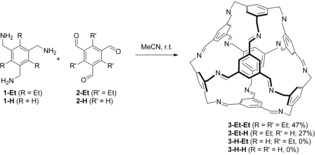
[4+4]‐condensation reactions of trimethylamines 1 and trialdehydes 2. R and R′ substituents were omitted for clarity in the cage structures.
We started our investigation with the triethyl‐substituted triamine 1‐Et 23 and trisaldehyde 2‐H.24 1‐Et is known to be thermodynamically most stable in its conformation with alternating substituents, thus “fixing” all methylamines on one side of the benzene ring.25 It turned out that the cage is formed within three days only in the absence of catalytic traces of acid (TFA) in acetonitrile at room temperature, suggesting that the cage is rather a kinetic than a thermodynamic product (see also discussion below). A substantial amount of the as‐synthesized product mixture is not soluble in common organic solvents. Therefore, purification by extraction with DCM was possible to isolate [4+4] imine cage 3‐Et‐H in 27 % yield. The cage 3‐Et‐H was characterized by NMR spectroscopy, MALDI MS (m/z=1430.817 [M+H]+) and single‐crystal diffraction analysis (see below). By DOSY experiments in C6D6 (T=298 K) a diffusion coefficient of D=4.17×10−10 m2s−1 was measured, corresponding to a solvodynamic radius of r s=0.87 nm.
Next, we were interested, if the preorganization of the three amines by the adjacent ethyl groups is essential or not. By MM2 calculations (see Supporting Information), the rotational barrier around the Csp2−Csp3 bond of a methylamine substituent is with +227.7 kcal mol−1 much higher for 1‐Et than for 1‐H (+3.5 kcal mol−1), concluding that the functional groups in 1‐H are rotating relative fast under reaction conditions and thus are not preoriented. The most stable conformation of the trialdehyde 2‐H is the C 3h‐symmetric one with all formyl groups found in the molecular plane. The rotational barrier from the C 3h‐ to the C s‐symmetric conformation is calculated to be +42.4 kcal mol−1. Therefore, triamine 1‐H was reacted with 2‐H. No formation of the corresponding cage 3‐H‐H could be observed, although the insoluble product showed the typical imine stretching band at ṽ=1639 cm−1 and no aldehyde band at 1688 cm−1 anymore, revealing that an imine polymer with a typical morphology of globular units sticking together (see also SE micrograph image in Figure 1) has been formed instead. In 2‐Et, the compound contains three ethyl groups between the formalydehyde functions. Here, the thermodynamically most stable conformation is the C 1 symmetric one, although the C 3v‐symmetric one is only about 2 kcal mol−1 less stable. However, due to the steric strain of the ethyl moieties, the aldehyde groups are forced a bit out of plane (dihedral angle: ≈40°), but still the C=O bonds are not orthogonally oriented to the π‐plane. Counterintuitively, the rotational barrier is with +34.5 kcal mol−1 even smaller than found in the trisaldehyde 2‐H. In consequence, it is not surprising that in the combination 1‐H and 2‐Et, again an insoluble imine polymer is formed rather than cage 3‐Et‐H. In contrast, the combination 1‐Et and 2‐Et again gives the corresponding cage 3‐Et‐Et in 46 % isolated yield, thus suggesting that the functional groups need to be highly pre‐oriented at least in one of the reactants.26
Figure 1.

Secondary electron micrograph images of the H‐H‐Polymer (top) and the 3‐Et‐H cage (bottom) after activation with ethane and gas sorption measurement.
The outer diameter of 3‐Et‐Et is roughly the same as for 3‐Et‐H (see discussion on X‐ray structures below), but due to the absence of the second set of ethyl chains, the pore windows are more open. Most interestingly, this subtle structural change makes no significant difference to the molecular diffusion measured by DOSY‐NMR in C6D6 at 298 K (D=4.07×10−10 m2s−1, r S=0.89 nm).
As mentioned above, the cages were formed only in the absence of acid, suggesting that the product is kinetic rather than thermodynamic. Mixing both cages with each other or with the other aldehyde under reaction conditions gave no scrambled products (see Supporting Information), which would have been expected in case of a thermodynamically controlled reaction. As soon as acid is added to the reaction mixture, cages degrade to finally give with increasing acid content the insoluble imine polymer as the thermodynamic product.
For both cage compounds, suitable single crystals for X‐ray diffraction were grown (Figure 2). For 3‐Et‐Et two polymorphic structures (α and β) have been found from different crystallization conditions. Polymorph α (trigonal space‐group R3) was crystallized from CHCl3. Here, the cage is T d‐symmetric with all ethyl chains oriented to the outside of the cage. The alternating substitution pattern at the former triamine is preserved and all substituents at the other aromatic ring are syn‐oriented and nearly orthogonal to the aromatic π‐plane, with the imine protons pointing inside the cavity. In polymorph β (space‐group P41212; from toluene), the alternating substitution pattern at the former triamine unit is also preserved, but in two of the other aromatic rings, one ethyl chain each is oriented inwards (up‐up‐down conformation), which reflects the smaller rotational barrier of the substituents in this subunit (see discussion above). Crystals (monoclinic space‐group P21/n) of 3‐Et‐H were grown from DCM. Again, the alternating substitution pattern at the triamine is preserved. Since the triimine unit does not inhibit ethyl chains, the imine units are coplanar to the aromatic ring, which is reasonable due to conjugation. By this subtle difference, the cage 3‐Et‐H is somewhat larger in size (1.2 nm inner diameter) than 3‐Et‐Et (1.1 nm).
Figure 2.
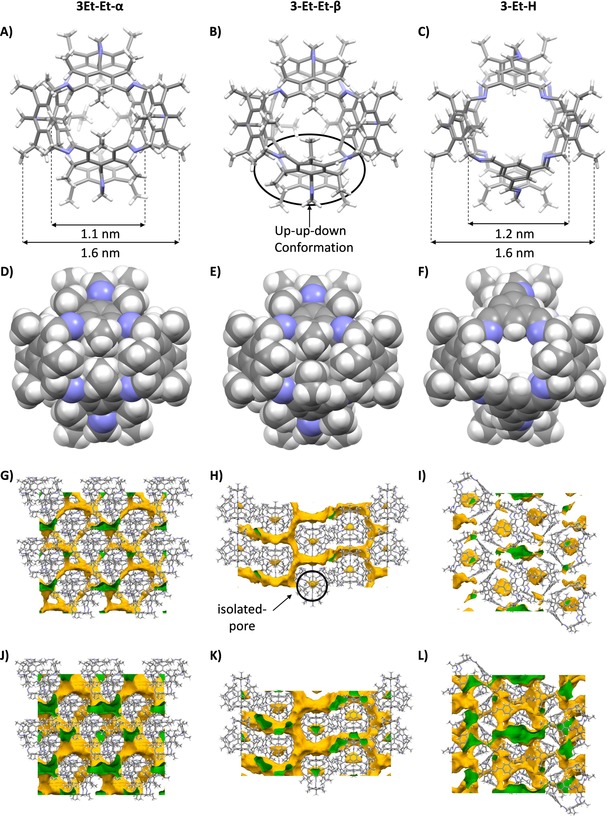
Single‐crystal structure analysis. Left column: 3‐Et‐Et‐α, central column: 3‐Et‐Et‐β, right column: 3‐Et‐H. A–C) capped stick models. D–F) space filling models. G–I) solvent accessible pores calculated for a probe with radius 1.82 Å. J–L) solvent accessible pores calculated for a probe with radius 1.2 Å.
By comparing space‐filling models of 3‐Et‐Et and 3‐Et‐H it becomes clear that in contrast to 3‐Et‐H the cage windows of 3‐Et‐Et are blocked by the additional alkyl chains. Analyzing the pore structure of in silico desolvated crystals of 3‐Et‐Et by their solvent accessible surfaces reveal that for both polymorphs α and β the cage void is isolated for probes with radii 1.82 Å as well as 1.20 Å. For both polymorphs three‐dimensional connected extrinsic pores filled with non‐resolved solvate molecules surrounding the cages can be found already for probes with radius 1.82 Å. Because these solvate‐filled pores are almost entirely surrounding the cages, the solvate molecules are probably largely contributing to the stability of the crystals and in both structures, interactions between adjacent cages are limited to a few point‐contacts of ethyl chains by weak dispersion interactions, thus expecting that by desolvation the structural arrangement will collapse.
This is different for 3‐Et‐H. Here, more narrow contacts with π‐surfaces involved can be found. The compound seems to be non‐porous for example for dinitrogen, if an average kinetic diameter of 3.64 Å is taken into account. Because Jelfs et al. recently reported that the pore system of cage crystals are not as static as derived from crystal structures,27 applying a minimum diameter of 3.10 Å for dinitrogen on the static crystal structure is more reasonable.28 The solvent accessible surface area for probes with a radius <1.6 Å show a three‐dimensional pore system, the cage voids included. By this assumption, pores of 3‐Et‐H should be accessible after desolvation.
Therefore, both cage compounds have been investigated by gas sorption. Thermal treatment (120 °C for 3 h) under vacuum led not to a reasonable activation of the pores. In each case, the apparent specific surface area (Brunauer–Emmett–Teller model) never exceeded 11 m2 g−1 (3‐Et‐Et) or 27 m2 g−1 (3‐Et‐H) as measured by nitrogen sorption at 77 K. Because the pore‐windows of both cages are narrow, we exchanged solvent first with n‐pentane at room temperature,29 sonicated the immersion for 15 min, before exchanging with liquid ethane at 157 K for 10 min and activated at 40 °C in vacuum. Comparison of PXRD patterns show in both cases that the materials activated with ethane are much more crystalline than in case of thermal activation and the sample of cage 3‐Et‐H show very distinct sharp peaks (see Supporting Information). Additionally, the crystalline character is also confirmed by secondary electron micrographs (see Figure 1). The activated sample of cage compound 3‐Et‐Et adsorbed 58 cm3 g−1 N2 at 77 K. The isotherm can best be described as a mixture of type I (microporous) and type II (non‐ or macroporous).30 By NL‐DFT, a broad distribution of pores is calculated. The determined specific surface area (BET) is with 71 m2 g−1 larger in comparison to the thermally activated material. The compound adsorbs 0.34 wt % H2 (at 77 K and 1 bar), as well as 8.26 wt % CO2 and 1.14 wt % CH4 (both at 273 K, 1 bar). For cage 3‐Et‐H, the gas sorption behavior changed significantly, when activated by this method. The compound adsorbs at 77 K 139 cm3 g−1 N2 and the isotherms are almost of an ideal type I (Figure 3). The NLDFT pore size distribution show two sharp peaks at 7.0 and 8.2 Å, which is in about the same order as the pore diameter determined by SCXRD (minus two times the van‐der‐Waals radius of carbon). The specific BET surface area is with 443 m2 g−1 much larger than accessible by thermal activation and comparable to smaller cages for example, from Cooper et al.,11b but significantly lower than for larger cages.13, 31 Furthermore, the material adsorbs 0.93 wt % H2 (at 77 K and 1 bar), 13.8 wt % CO2 and 2.17 wt % CH4 (both at 273 K, 1 bar). All derived polymer‐materials have also been investigated by nitrogen sorption at 77 K, turning out to be of low porosity with specific surface areas below 26 m2 g−1.
Figure 3.
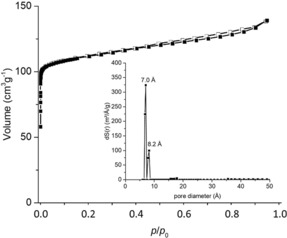
Nitrogen sorption at 77 K for 3‐Et‐H cage activated with ethane. Filled symbols: adsorption, open symbols: desorption. Inset: NL‐DFT pore size distribution.
In conclusion, it was shown that the rotational freedom of Csp2−Csp3 bonds at least in one of the precursors needs to be highly restricted for the formation of [4+4] imine cages with truncated tetrahedral geometry. The geometrical pre‐orientation of reacting groups is crucial, because the cage formation is kinetically controlled. The thermodynamic products are less structurally ordered polymers, which are formed as soon as an acid is present. Both cages were investigated by gas sorption. Whereas thermal activation led to more amorphous and less porous materials, mild activation by exchange with first n‐pentane and then liquid ethane gave significantly higher BET surface areas up to 443 m2 g−1 for cage 3‐Et‐H and a high degree of crystallinity was maintained.
Experimental Section
For all experimental details, see Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank A. Widera (RG Himmel, Ruprecht‐Karls‐Universität Heidelberg) for recording PXRD data. Funding of this project (CaTs N DOCs, grant no.725765) by the ERC is highly acknowledged.
J. C. Lauer, W.-S. Zhang, F. Rominger, R. R. Schröder, M. Mastalerz, Chem. Eur. J. 2018, 24, 1816.
References
- 1.
- 1a. Zhang G., Mastalerz M., Chem. Soc. Rev. 2014, 43, 1934–1947; [DOI] [PubMed] [Google Scholar]
- 1b. Mastalerz M., Angew. Chem. Int. Ed. 2010, 49, 5042–5053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5164–5175; [Google Scholar]
- 1c. Rue N. M., Sun J., Warmuth R., Isr. J. Chem. 2011, 51, 743–768; [Google Scholar]
- 1d. Evans J. D., Sumby C. J., Doonan C. J., Chem. Lett. 2015, 44, 582–588; [Google Scholar]
- 1e. Hasell T., Cooper A. I., Nat. Rev. Mater. 2016, 1, 16053. [Google Scholar]
- 2.
- 2a. Chen L., Reiss P. S., Chong S. Y., Holden D., Jelfs K. E., Hasell T., Little M. A., Kewley A., Briggs M. E., Stephenson A., Thomas K. M., Armstrong J. A., Bell J., Busto J., Noel R., Liu J., Strachan D. M., Thallapally P. K., Cooper A. I., Nat. Mater. 2014, 13, 954–960; [DOI] [PubMed] [Google Scholar]
- 2b. Hasell T., Miklitz M., Stephenson A., Little M. A., Chong S. Y., Clowes R., Chen L., Holden D., Tribello G. A., Jelfs K. E., Cooper A. I., J. Am. Chem. Soc. 2016, 138, 1653–1659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Elbert S. M., Rominger F., Mastalerz M., Chem. Eur. J. 2014, 20, 16707–16720. [DOI] [PubMed] [Google Scholar]
- 3. Song Q., Jiang S., Hasell T., Liu M., Sun S., Cheetham A. K., Sivaniah E., Cooper A. I., Adv. Mater. 2016, 28, 2629–2637. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Kewley A., Stephenson A., Chen L., Briggs M. E., Hasell T., Cooper A. I., Chem. Mater. 2015, 27, 3207–3210; [Google Scholar]
- 4b. Zhang J.-H., Xie S.-M., Wang B.-J., He P.-G., Yuan L.-M., J. Chromatogr. A 2015, 1426, 174–182. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Giri N., Del Pópolo M. G., Melaugh G., Greenaway R. L., Rätzke K., Koschine T., Pison L., Gomes M. F. C., Cooper A. I., James S. L., Nature 2015, 527, 216; [DOI] [PubMed] [Google Scholar]
- 5b. Mastalerz M., Nature 2015, 527, 174. [DOI] [PubMed] [Google Scholar]
- 6. Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Adv. Mater. 2012, 24, 6049–6052. [DOI] [PubMed] [Google Scholar]
- 7. Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Chem. Commun. 2013, 49, 8398–8400. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Acharyya K., Mukherjee P. S., Chem. Eur. J. 2015, 21, 6823–6831; [DOI] [PubMed] [Google Scholar]
- 8b. Acharyya K., Mukherjee P. S., Chem. Commun. 2014, 50, 15788–15791. [DOI] [PubMed] [Google Scholar]
- 9. Santolini V., Miklitz M., Berardo E., Jelfs K. E., Nanoscale 2017, 9, 5280–5298. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Schneider M. W., Oppel I. M., Mastalerz M., Chem. Eur. J. 2012, 18, 4156–4160; [DOI] [PubMed] [Google Scholar]
- 10b. Dhara A., Beuerle F., Chem. Eur. J. 2015, 21, 17391–17396. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Skowronek P., Gawronski J., Org. Lett. 2008, 10, 4755–4758; [DOI] [PubMed] [Google Scholar]
- 11b. Tozawa T., Jones J. T. A., Swamy S. I., Jiang S., Adams D. J., Shakespeare S., Clowes R., Bradshaw D., Hasell T., Chong S. Y., Tang C., Thompson S., Parker J., Trewin A., Bacsa J., Slawin A. M. Z., Steiner A., Cooper A. I., Nat. Mater. 2009, 8, 973–978. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Hong S., Rohman M. R., Jia J., Kim Y., Moon D., Kim Y., Ko Y. H., Lee E., Kim K., Angew. Chem. Int. Ed. 2015, 54, 13241–13244; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13439–13442; [Google Scholar]
- 12b. Klotzbach S., Scherpf T., Beuerle F., Chem. Commun. 2014, 50, 12454–12457. [DOI] [PubMed] [Google Scholar]
- 13. Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 1516–1520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1542–1546. [Google Scholar]
- 14.
- 14a. Mastalerz M., Chem. Commun. 2008, 4756–4758; [DOI] [PubMed] [Google Scholar]
- 14b. Sun J., Warmuth R., Chem. Commun. 2011, 47, 9351–9353; [DOI] [PubMed] [Google Scholar]
- 14c. Liu X., Liu Y., Li G., Warmuth R., Angew. Chem. Int. Ed. 2006, 45, 901–904; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 915–918. [Google Scholar]
- 15. Briggs M. E., Jelfs K. E., Chong S. Y., Lester C., Schmidtmann M., Adams D. J., Cooper A. I., Cryst. Growth Des. 2013, 13, 4993–5000. [Google Scholar]
- 16.
- 16a. Skowronek P., Warzajtis B., Rychlewska U., Gawronski J., Chem. Commun. 2013, 49, 2524–2526; [DOI] [PubMed] [Google Scholar]
- 16b. Liu Y., Liu X., Warmuth R., Chem. Eur. J. 2007, 13, 8953–8959. [DOI] [PubMed] [Google Scholar]
- 17. Klotzbach S., Beuerle F., Angew. Chem. Int. Ed. 2015, 54, 10356–10360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10497–10502. [Google Scholar]
- 18.
- 18a. Mastalerz M., Chem. Eur. J. 2012, 18, 10082–10091; [DOI] [PubMed] [Google Scholar]
- 18b. Briggs M. E., Cooper A. I., Chem. Mater. 2017, 29, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jelfs K. E., Eden E. G. B., Culshaw J. L., Shakespeare S., Pyzer-Knapp E. O., Thompson H. P. G., Bacsa J., Day G. M., Adams D. J., Cooper A. I., J. Am. Chem. Soc. 2013, 135, 9307–9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bera S., Basu A., Tothadi S., Garai B., Banerjee S., Vanka K., Banerjee R., Angew. Chem. Int. Ed. 2017, 56, 2123–2126; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2155–2158. [Google Scholar]
- 21. Acharyya K., Mukherjee S., Mukherjee P. S., J. Am. Chem. Soc. 2013, 135, 554–557. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Francesconi O., Ienco A., Moneti G., Nativi C., Roelens S., Angew. Chem. Int. Ed. 2006, 45, 6693–6696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6845–6848; [Google Scholar]
- 22b. Içli B., Christinat N., Tönnemann J., Schüttler C., Scopelliti R., Severin K., J. Am. Chem. Soc. 2009, 131, 3154–3155; [DOI] [PubMed] [Google Scholar]
- 22c. Mateus P., Delgado R., Brandão P., Félix V., J. Org. Chem. 2009, 74, 8638–8646; [DOI] [PubMed] [Google Scholar]
- 22d. Granzhan A., Schouwey C., Riis-Johannessen T., Scopelliti R., Severin K., J. Am. Chem. Soc. 2011, 133, 7106–7115; [DOI] [PubMed] [Google Scholar]
- 22e. Guillet G. L., Sloane F. T., Ermert D. M., Calkins M. W., Peprah M. K., Knowles E. S., Cizmar E., Abboud K. A., Meisel M. W., Murray L. J., Chem. Commun. 2013, 49, 6635–6637; [DOI] [PubMed] [Google Scholar]
- 22f. Mateus P., Delgado R., Brandão P., Félix V., Chem. Eur. J. 2011, 17, 7020–7031; [DOI] [PubMed] [Google Scholar]
- 22g. Chen H.-Y., Gou M., Wang J.-B., Chem. Commun. 2017, 53, 3524–3526; [DOI] [PubMed] [Google Scholar]
- 22h. Arunachalam M., Ravikumar I., Ghosh P., J. Org. Chem. 2008, 73, 9144–9147; [DOI] [PubMed] [Google Scholar]
- 22i. De Rycke N., Marrot J., Couty F., David O. R. P., Tetrahedron Lett. 2010, 51, 6521–6525; [Google Scholar]
- 22j. Bojdys M. J., Briggs M. E., Jones J. T. A., Adams D. J., Chong S. Y., Schmidtmann M., Cooper A. I., J. Am. Chem. Soc. 2011, 133, 16566–16571; [DOI] [PubMed] [Google Scholar]
- 22k. Giri N., Davidson C. E., Melaugh G., Del Popolo M. G., Jones J. T. A., Hasell T., Cooper A. I., Horton P. N., Hursthouse M. B., James S. L., Chem. Sci. 2012, 3, 2153–2157. [Google Scholar]
- 23. Vacca A., Nativi C., Cacciarini M., Pergoli R., Roelens S., J. Am. Chem. Soc. 2004, 126, 16456–16465. [DOI] [PubMed] [Google Scholar]
- 24. Zhang J.-H., Xie S.-M., Chen L., Wang B.-J., He P.-G., Yuan L.-M., Anal. Chem. 2015, 87, 7817–7824. [DOI] [PubMed] [Google Scholar]
- 25. Wang X., Hof F., Beilstein J. Org. Chem. 2012, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.It is worth mentioning, that such a restriction of rotational freedom seems to play a minor role in recent systems based on flexible TREN (here the rotational barrier is about +3 kcal mol−1), where cages have been reported to form quantitatively. Unfortunately no details on reaction conditions have been given: Jiao T., Chen L., Yang D., Li X., Wu G., Zeng P., Zhou A., Yin Q., Pan Y., Wu B., Hong X., Kong X., Lynch V. M., Sessler J. L., Li H., Angew. Chem. Int. Ed. 2017, 56, 8226–8230; [Google Scholar]; Angew. Chem. 2017, 129, 8338–8342. [Google Scholar]
- 27. Holden D., Jelfs K. E., Cooper A. I., Trewin A., Willock D. J., J. Phys. Chem. C 2012, 116, 16639–16651. [Google Scholar]
- 28. Holden D., Jelfs K. E., Trewin A., Willock D. J., Haranczyk M., Cooper A. I., J. Phys. Chem. C 2014, 118, 12734–12743. [Google Scholar]
- 29. Mastalerz M., Oppel I. M., Angew. Chem. Int. Ed. 2012, 51, 5252–5255; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5345–5348. [Google Scholar]
- 30. Thommes M., Kaneko K., Neimark Alexander V., Olivier James P., Rodriguez-Reinoso F., Rouquerol J., Sing Kenneth S. W., Pure Appl. Chem. 2015, 87, 1051. [Google Scholar]
- 31.
- 31a. Schneider M. W., Oppel I. M., Ott H., Lechner L. G., Hauswald H.-J. S., Stoll R., Mastalerz M., Chem. Eur. J. 2012, 18, 836–847; [DOI] [PubMed] [Google Scholar]
- 31b. Mastalerz M., Schneider M. W., Oppel I. M., Presly O., Angew. Chem. Int. Ed. 2011, 50, 1046–1051; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1078–1083. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary