

Summary
Zika virus (ZIKV) is responsible for a recent global epidemic that has been associated with congenital brain malformations in fetuses and with Guillain–Barré syndrome in adults. Within the last 2 years, a major effort has been made to develop murine models to study the mechanism of viral transmission, pathogenesis and the host immune response. Here, we discuss the findings from these models regarding the role that the innate and adaptive immune responses have in controlling ZIKV infection and pathogenesis. Additionally, we examine how innate and adaptive immune responses influence sexual and vertical transmission of ZIKV infection as well as how these responses can influence the ability of ZIKV to cross the placenta and to induce damage in the developing brain.
Keywords: innate receptors, neuroinflammation, reproductive Immunology, T cell, viral
Introduction
Zika virus (ZIKV) is a flavivirus, first discovered in Uganda in 1947 in a sentinel monkey, which probably contracted the virus via its primary vector, an Aedes mosquito. Historically, human cases were rare with symptoms including mild fever, rash, myalgia and conjunctivitis.1 However, recent outbreaks in French Polynesia in 2013/142 and in South America in 2015/163 showed an association between ZIKV infection and severe clinical outcomes including Guillain–Barré syndrome (GBS) in adults4 and microencephaly and congenital pathologies in fetuses and newborns.5 The congenital defects were associated with the ability of ZIKV to be transmitted vertically across the placental barrier.6 Additionally, ZIKV can be transmitted human‐ to‐ human sexually from males to females.7 Recent work has focused on the development of small‐animal models to better understand ZIKV pathogenesis and the role of the immune response elicited during infection in these processes.
Zika virus and the innate immune response
Developing useful murine models of ZIKV infection has been difficult. Early attempts required significant mouse adaptation through serial viral passaging in brain tissue to consistently observe disease.8 Even using ZIKV isolates from recent outbreaks demonstrated no obvious signs of disease and little to no detectable virus in tissues in wild‐type (WT) strains of mice (C57BL/6, BALB/c or CD‐1) following peripheral inoculation (Table 1, part a) suggesting that virus replication is effectively controlled in these animals.
Table 1.
Influence of innate and adaptive immune responses on Zika infection in mice
| Strain | Routea | Age | Virus strainsb | Dosec | Treatmentd | Clinical diseasee | Viraemia | Disseminationf:Brain | Spleen | Ovaries | Testes | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (a) Wild‐type | ||||||||||||
| C57BL/6 | s.c. | 7 weeks | FP2013 | 103 | – | None | n.d. | n.d. | 18 | |||
| C57BL/6 | s.c. | 5–6 weeks | UG1947 | 103 | – | None | 12 | |||||
| C57BL/6 | s.c. | 4–5 weeks | FP2013 | 102 | – | None | x(R) | x(R) | x(R) | 14 | ||
| C57BL/6 | s.c. | 5–6 weeks | FP2013, UG1947 | 102 | – | None | 14 | |||||
| C57BL/6 | i.v. | 5–7 weeks | CB2010 | 106 | None | 17 | ||||||
| C57BL/6 | i.v. | 5–6 weeks | FP2013, UG1947 | 102 | – | None | 14 | |||||
| C57BL/6 | i.p. | 5–6 weeks | SMGC‐1 | 106 | None | n.d. | n.d. | x(R) | 23 | |||
| C57BL/6 | i.p. | 6–8 weeks | PA2015 | 104 | – | None | n.d. | n.d. | n.d. | n.d. | 16 | |
| (b) Innate immune deficiencies | ||||||||||||
| A129 | i.p. | 11 weeks | CB2010 | 105 | – | None | x (PA) | 15 | ||||
| A129 | i.p. | 3 weeks | CB2010 | 105 | – | 6–7 dpi | x (PA) | 15 | ||||
| A129 | i.p. | 5 weeks | CB2010 | 105 | – | 8–9 dpi | x (PA) | 15 | ||||
| A129 | s.c. | 5–6 weeks | UG1962 | 106 | – | 4–6 dpi | x(R) | x(R) | x(R) | x(R) | x(R) | 72 |
| C57BL/6 | i.p. | 6–8 weeks | PA2015 | 104 | anti‐IFNAR1 | None | n.d. | x | n.d. | 16 | ||
| C57BL/6 | i.p. | 5 weeks | SN1984 | 106 | anti‐IFNAR1 | 7–12 dpi | x(R) | x(R) | x(R) | 20 | ||
| C57BL/6 | s.c. | 4–5 weeks | FP2013 | 102 | anti‐IFNAR1 | None | 3 dpi | 14 | ||||
| C57BL/6 | s.c. | 7 weeks | FP2013 | 103 | anti‐IFNAR1 | None | 3 dpi | x | 18 | |||
| C57BL/6 | s.c. | 5 weeks | SN1984 | 106 | anti‐IFNAR1 | 8–19 dpi (40%) | x(R) | x(R) | x(R) | 20 | ||
| Ifnar1 −/− | i.v. | 5–6 weeks | FP2013, UG1947 | 102 | – | 8–12 dpi | 14 | |||||
| Ifnar1 −/− | s.c. | 3–6 months | FP2013 | 103 | – | 7–18 dpi | x(R) | x(R) | 14 | |||
| Ifnar1 −/− | s.c. | 5–6 weeks | FP2013, UG1947 | 102 | – | 6–10 dpi | 14 | |||||
| Ifnar1 −/− | s.c. | 5–6 weeks | UG1947, SN1984 | 103 | – | 7–8 dpi | x(R) | x(R) | x(R) | x(R) | x(R) | 12 |
| Ifnar1 −/− HLA‐A*0101 | r.o. | 5 weeks | CB2010 | 104 | – | x(PA) | x(PA) | 25 | ||||
| Irf3 −/− Irf5 −/− Irf7 −/− | i.v. | 5–6 weeks | FP2013, UG1947 | 102 | – | 5–10 dpi | 14 | |||||
| Irf3 −/− Irf5 −/− Irf7 −/− | s.c. | 5–6 weeks | FP2013, UG1947 | 102 | – | 6–10 dpi | 14 | |||||
| Irf3 −/− | s.c. | 5–6 weeks | FP2013, UG1947 | 102 | – | None | 14 | |||||
| LysMCRE+IFN ARfl/fl | i.v. | 5–7 weeks | UG1947, CB2010 | 106 | – | 5–10 dpi | 17 | |||||
| Mavs −/− | i.p. | 6–8 weeks | PA2015 | 104 | – | None | n.d. | x(R) | n.d. | 16 | ||
| Mavs −/− | s.c. | 5–6 weeks | FP2013, UG1947 | 102 | – | None | 14 | |||||
| Stat2 −/− | s.c. | 5–6 weeks | UG1947, SN1984 | 103 | – | 5–7 dpi | x(R) | x(R) | x(R) | x(R) | x(R) | 12 |
| Unc93b1 3D | i.p. | 6–8 weeks | PA2015 | 104 | – | None | n.d. | n.d. | n.d. | 16 | ||
| (c) Adaptive immune deficiencies | ||||||||||||
| C57BL/6 | i.p. | 6–8 weeks | PA2015 | 104 | anti‐CD4/CD8 | 7–14 dpi (<10% wl, recovered) | 16 | |||||
| Rag1 −/− | i.p. | 6–8 weeks | PA2015 | 104 | None | 16 | ||||||
| (d) Innate & adaptive immune deficiencies | ||||||||||||
| AG129 | i.d. | 3 weeks | CB2010 | 105 | – | 6 dpi | x(PA) | x | x | x | 15 | |
| AG129 | i.p. | 3 weeks | CB2010 | 105 | – | 6 dpi | x(PA) | 15 | ||||
| AG129 | s.c. | 8 weeks | FB2013 | 105 | – | 5–8 dpi | x(PA) | x | x | 13 | ||
| CD8 −/− | s.c. | 7 weeks | SN1984 | 105 | anti‐IFNAR1 | 12–18 dpi | x(PA) | x(PA) | x(PA) | 17 | ||
| Ifnar1 −/− | i.p. | 5–6 weeks | SMGC‐1 | 105 | a.t. of niave CD8+ T cells | 5–8 dpi | x(R) | 23 | ||||
| Ifnar1 −/− | i.p. | 5–6 weeks | SMGC‐1 | 105 | a.t. of ZIKV‐CD8+ T cells | None | n.d. | 23 | ||||
| Ifnar1 −/− HLA‐A*0101 | r.o. | 5 weeks | CB2010 | 104 | anti‐CD8 | x(PA) | x(PA) | 25 | ||||
| Ifnar1 −/− HLA‐A*0101 | r.o. | 5 weeks | CB2010 | 104 | ZIKV peptide | x(PA) | x(PA) | 25 | ||||
| Ifnar1 −/− HLA‐A*0101 | r.o. | 5 weeks | CB2010 | 104 | ZIKV peptide/anti‐CD8 | x(PA) | x(PA) | 25 | ||||
| Mavs −/− | i.p. | 6–8 weeks | PA2015 | 104 | anti‐CD4/CD8 | 7–14 dpi (>10% wl, recovered) | 16 | |||||
| Rag1 −/− | i.p. | 6–8 weeks | PA2015 | 104 | anti‐IFNAR1 | 9–17 dpi | x(PA) | x(R,H) | x(R) | x(R,H) | 16 | |
Route of inoculation: ai.d., intradermal; i.p., intraperitoneal; i.v., intravenous; r.o., retro‐orbital; s.c., subcutaenous.
bVirus strains were abbreviated as follows: Cambodia/2010 (FSS13025): CB2010; French Polynesia/2013 (H/PF/2013): FP2013; Uganda/1947 (MR‐766): UG1947; Paraiba_01/2015: PA2015; Senegal/1984: SN1984; Uganda/1962 (MP1751): UG1962; ZIKA‐SMGC‐1: SMGC‐1.
cPFU per mouse.
dTreatment of animals: a.t., adoptive transfer.
eReported clnical disease or weight loss (w.l.).
fx indicates detected. PA, plaque assay, R, RNA, H, Histology, n.d., not detected.
Studies analysing the type I interferon (IFN) responses in human and mouse cells identified a key difference between these species in their ability to control virus infection. In human cells, the virus NS5 protein antagonizes IFN signalling by promoting proteasomal degradation of the human signal transducer and activator of transcription 2 (STAT2) protein.9, 10 STAT2 is a transcription factor that mediates signalling through the IFN‐α/β receptor (IFNAR), leading to the production of IFN‐stimulated genes.11 Hence, ZIKV NS5 limits the type I IFN response during human infection. In contrast, ZIKV NS5 does not inhibit mouse STAT2, allowing for an efficient and effective type I IFN response that controls virus replication.9 Mice deficient in STAT2 are susceptible to ZIKV infection,12 as are mice deficient in the IFNAR1 gene (Ifnar1 −/−, A129). These mice develop a rapid wasting disease within 5–8 days and have high viral burdens in tissues13, 14, 15 (Table 1, part b). These findings show that the type I IFN response is essential for protection against ZIKV infection and explain, at least in part, disease susceptibility in humans.
Additional studies using knockout mice have further defined the role of type I IFN in regulating ZIKV pathogenesis. Complete deficiency in the IFN response, as observed with Ifnar1 −/−, A129 or Irf3 −/− Irf7 −/− Irf7 −/− mice, results in clinical disease by 5–10 days post infection with virus detection in multiple tissues including brain, spleen, ovaries and testes (Table 1, part b).14, 15, 16 This was also observed when IFNAR1 was specifically knocked out in myeloid cells, suggesting that these cells are critical for protection.17 The molecules necessary to mediate the type I IFN response to ZIKV infection are not as well defined, as deficiency in Mavs, the signalling molecule for RIG‐I‐like receptors resulted in only a short‐term viraemia,16 whereas deficiencies in Irf3 or the three‐dimensional mutation in Unc93b1, which results in deficient endosomal Toll‐like receptor responses, had no effect on ZIKV infection.14, 16 Hence, neither cytoplasmic RIG‐I‐like receptors nor endosomal Toll‐like receptors appear to be essential for protection against ZIKV, suggesting that other sensors of virus infection play a role in mediating the type I IFN response to ZIKV.
Partial disruption of the type I IFN response, to more effectively mimic the antagonism of the type I IFN response found in humans, has been achieved by treating mice with anti‐IFNAR antibodies. Treatment of WT mice with a single, large bolus of anti‐IFNAR1 blocking antibody (MAR1‐53A) does not result in clinical signs such as wasting disease, but can result in detectable virus in peripheral tissues.14, 18, 19 Furthermore, the concentration of blocking antibody used can influence tissue‐specific viral load14 and result in disease if repeatedly administered.20 In the future, it may be possible to use this approach as a model of ZIKV‐associated GBS because ZIKV infection of Ifnar1 −/− mice can result in peripheral neuron infection and apoptosis21, 22 (Figure 1c).
Figure 1.
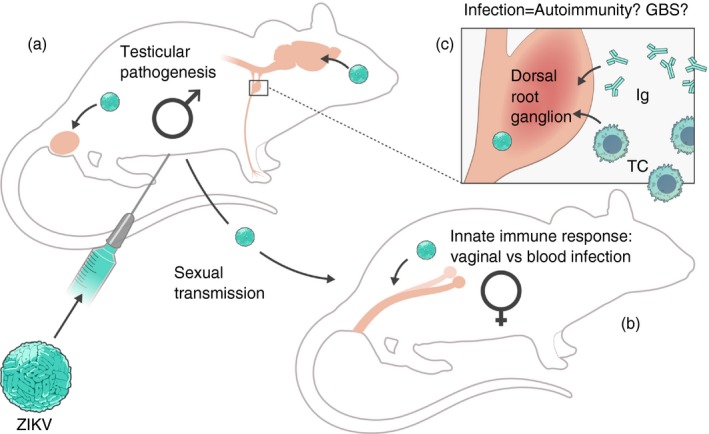
Immune responses to Zika virus (ZIKV) are critical to prevent pathogenesis and transmission but may also contribute to autoimmunity. (a) Models of ZIKV infection in mice have demonstrated that elements of the innate immune response are critical for preventing infection of the testes in males. Experiments with mice deficient in innate immune signalling have shown testicular pathology and prolonged infection of cells in the testes, including sperm, which allow for animal‐to‐animal sexual transmission of ZIKV. These models are reminiscent of findings in humans. (b) Additionally, sexual transmission or intravaginal inoculation of mice demonstrates that the female reproductive tract is permissive to ZIKV infection. Furthermore, these modes of transmission in mice deficient in innate immune signalling can result in ZIKV‐associated disease and vertical transmission to fetuses. (c) In mice with innate immune signalling deficiencies, ZIKV can infect peripheral nerves within the dorsal root ganglia. Such an infection could induce an autoimmune response reminiscent of Guillain–Barré syndrome, which in humans is associated with ZIKV infection. TC, T cell; Ig, immunoglobulin.
Adaptive immune response to ZIKV
Although the innate immune response is clearly necessary for controlling viral replication and preventing disease, the adaptive T‐cell and B‐cell responses also contribute to protection. Several studies, including work from our laboratory, have demonstrated a short‐lived, but strong T‐cell response around 7 days post infection in ZIKV‐infected WT mice.16, 23, 24 CD4+ T cells proliferate rapidly and show a classical T helper type 1 antigen‐experienced cytokine profile, expressing IFN‐γ, tumour necrosis factor‐α (TNF‐α) and interleukin‐2 (IL‐2). Concomitantly, CD8+ T cells are proliferating, are activated and express cytotoxic markers, suggesting a virus‐specific cytotoxic T‐cell response. However, depletion of T cells using anti‐CD4 and anti‐CD8 antibodies or deficiency of T cells in Rag1 −/− mice did not result in clinical disease following ZIKV infection16 (Table 1, part c). Hence, a functional innate response appears to be sufficient to control ZIKV infection in mice, even in the absence of functional T cells.
Combined influence of innate and adaptive immune responses
Combined deficiencies in components of the innate and adaptive immune response have shown that both arms of the immune system influence ZIKV pathogenesis (Table 1, part d). AG129 mice, which lack both IFN type I (α/β) and type II (γ), develop disease in an age‐dependent manner with younger mice being more susceptible.13, 15 These mice have similar infection kinetics to A129 mice but with exaggerated disease signs. Depletion or deficiency of T cells in IFN‐antagonized mice results in high ZIKV titres and associated disease, demonstrating that the adaptive immune response is critical to controlling infection, when the type I IFN response is suboptimal.16, 17 Furthermore, adoptive transfer of ZIKV‐specific CD8 T cells also prevents disease in Ifnar1 −/− mice, suggesting that a vaccination strategy could be effective at preventing disease, even in the absence of strong IFN responses17, 23, 25 (Table 1, part d).
In addition to the cellular adaptive response, there is some evidence from mouse models implicating the humoral response in preventing ZIKV‐associated disease. Although B cells are not activated during ZIKV infection in WT animals,16 a strong neutralizing antibody response is correlated with recovery from ZIKV infection in highly susceptible Ifnar −/− mice.26 Additionally, monoclonal antibodies as well as antibodies derived from convalescent patient serum can inhibit disease in ZIKV‐susceptible mice.27, 28, 29, 30 These antibodies target the envelope (E) glycoprotein, which is required for flavivirus entry into the cell.31 Multiple candidate ZIKV vaccine platforms are currently being developed to prevent human disease.32, 33, 34 So far, all effective candidates have demonstrated induction of a potent sterilizing neutralizing antibody titre and a robust T‐cell response, further demonstrating the importance of the adoptive immune response to controlling ZIKV infection.
Influence of immune responses on sexual transmission
Although ZIKV is primarily transmitted to humans by the bite of a mosquito, human‐to‐human transmission can be observed sexually, from infected males to females,7 and vertically, from a pregnant woman to her fetus.1, 35, 36 Mouse models have shown that both innate and adaptive immune responses influence both sexual and vertical transmission. For example, Ifnar1 −/− male mice or anti‐IFNAR1 treatment of WT male mice develop infection in the testes, which is associated with apoptotic cells following ZIKV infection (Fig. 1a, Table 2, part a).18, 37, 38 Similar findings were observed in BALB/c mice treated with dexamethasone.39 These findings, in association with reports of long‐lived infectious virus in human male testes,40, 41, 42 suggest that ZIKV infection could impact male reproductive health (Fig. 1a). Additionally, several studies have shown sexual transmission of ZIKV from infected immune‐compromised males to naive immune‐compromised females (Fig. 1b).26, 43 Direct animal‐to‐animal sexual transmission results in infection of reproductive organs in the female (Fig. 1b).26 Enhanced infection of female reproductive tissues by intravaginal inoculation has also been shown in several studies in mice with suppressed type I IFN responses (Irf3 −/− Irf7 −/−, Ifnar1 −/− and LysMCre+ Ifnar fl/fl mice).44, 45 In contrast, WT mice or mice without severe IFN suppression (Mx1, Rag2 −/−) more readily controlled vaginal infection. Hence, sexual transmission of ZIKV requires an impairment of the type I IFN response in mice and infection of reproductive tissue in both males and females is enhanced in this context.
Table 2.
Models of Zika virus (ZIKV) transmission and influence of infection on central nervous system development
| (a) Sexual transmission | ||||||||||||||
| Routea | Strain | ageb | Virus strainsc | Dose | Treatment | Clinical diseased | Viraemia | Disseminatione | Spleen | Ovaries | Vagina | Testes | Placenta | References |
| i.p. to males | AG129 x AG129 or CD‐1 | 13–18 weeks | PR2015 | 103 | – | 13–28 dpi (transmitter) 9–21 dpi (recipient) | x(R) | x(R) | 43 | |||||
| i.p. to males | Rag1 −/− (m) x Ifnar1 −/− (f) | 6–8 weeks | PA2015 | 104 | anti‐IFNAR1 | 16–17 dpi (transmitter) 7–12 dpi (recipient) | x (R/H) | x (R) | x(R) | x (R/H) | x (R/H) | 26 | ||
| i.vag. | C57BL/6 | 7–22 weeks | CB2010 | 104 | Depo‐Provera | – | n.d. | n.d. | x(PA) | 44 | ||||
| i.vag. | Mx1 | 7–22 weeks | CB2010 | 104 | Depo‐Provera | – | n.d. | 44 | ||||||
| i.vag. | Rag2 −/− | 7–22 weeks | CB2010 | 104 | Depo‐Provera | – | n.d. | 44 | ||||||
| i.vag. | Tlr7 −/− Mavs −/− Mx1 | 7–22 wks | CB2010 | 104 | Depo‐Provera | – | n.d. | 44 | ||||||
| i.vag. | Irf3 −/− Irf7 −/− | 7–22 weeks | CB2010 | 104 | Depo‐Provera | weight loss (<10%) | x(R) | x(R) | 44 | |||||
| i.vag. | Ifnar1 −/− | 7–22 weeks | CB2010 | 104 | Depo‐Provera | 8–9 dpi | x(R) | x(R) | x(R) | x(R) | x(R) | 44 | ||
| i.vag. | AG129, LysMCre+IFNARfl/fl | 8–12 weeks | CB2010 | 105 | progest. PMSG | 13–22 dpi | x(R) | x(H) | x(H) | x(R/H) | 45 | |||
| (b) Vertical transmission | ||||||||||||||
| Routea | Strain | Age infected/analysed | Virus strains | Dose | Treatment | Clinical disease/pathology | Placenta | Disseminatione: fet. CNS | fet. body | fet. lymph | mat. blood | mat. spleen | mat. brain | References |
| dam i.p. | SJL | E13‐15/birth | PA2015 | 1012 | – | FGR, cortical thinning, ocular malformations | x(R) | x(R) | 56 | |||||
| dam i.p. pup i.vent | C57BL/6 | E13·5/E17·5‐P1 | CH2016 | 102 | – | cortical thinning, < VZ/SVZ thickness, <ventricular volume and surface area | x(R) | x(R/H) | x(R) | 73 | ||||
| dam i.p. | Rag1 −/− | E7/E17‐E19 or P0‐P2 | PA2015 | 104 | anti‐IFNAR1 | – | x (R/H) | x (R/H) | x (R/H) | x (R/H) | x (R/H) | x (R/H) | 26 | |
| i.vag. | C57BL/6NCrl | E4·5 or E8·5/E18·5 | CB2010 | 104 | – | modest FGR | x(R) | x(H) | 44 | |||||
| i.vag. | Irf3 −/− Irf7 −/− | E4·5 or E8·5/E18·5 | CB2010 | 104 | – | FGR | x(R) | x(R) | x(R) | 44 | ||||
| i.vag. | Ifnar1 −/− x C57BL/6NCrl | E4·5 or E8·5/E18·5 | CB2010 | 104 | – | resorption, FGR | x(R) | x(R) | x(R) | 44 | ||||
| dam i.v. | FVB/NJ, C57BL/6J | E5 or E7·5‐9·5 or 12·5/various to E18·5 | BZ2016 | 105 | – | dysraphia, hydrocephalus, FGR | x | x | x (W) | x | n.d. | 57 | ||
| dam s.c. | Ifnar1 −/− (dam) x C57BL/6 (sire) | E6·5 or 7·5/E13·5 or 15·5 | FP2013 | 103 | FGR, fetal CNS apoptosis | x(FA) | x(FA) | x(FA) | x(FA) | x(FA) | 19 | |||
| dam s.c. | C57BL/6 | E6·5 or 7·5/E13·5 or 15·5 | FP2013 | 103 | anti‐IFNAR1 | Modest FGR | x(FA) | x(FA) | x(FA) | x(FA) | x(FA) | 19 | ||
| (c) Developmental pathogenesis | ||||||||||||||
| dam i.p. | SJL | E13‐15/birth | PA2015 | 1012 | – | FGR, cortical thinning, ocular malformations | x(R) | x(R) | 56 | |||||
| i.c. | BALBc | E13·5/E18·5 or P1/clinical | VZ2016 | 101 | – | cortical thinning (E13·5) 4‐25 dpi (P1) | x (PA, H) | 74 | ||||||
| s.c. | Swiss | P3/clinical | PA2015 | 106 | – | 9–18 dpi | x(H) | 59 | ||||||
| i.c., i.p., s.c. | Swiss | P1 or 2 wks/clinical | BZ2015 | 10% BS | – | 6 dpi (i.c.), 12 dpi (s.c.), gliosis, infiltration, neuronal death, WM damage | x(H) | 75 | ||||||
| i.c. | C57BL/6 | P7 or P21/4dpi | UG1974 | 107 | – | decreased brain volume, gliosis, < NPC proliferation, neuronal death | x(H) | 76 | ||||||
| (c) Developmental pathogenesis | ||||||||||||||
| Routea | Strain | Age infected/analysed | Virus strains | Dose | Treatment | Clinical disease/pathology | Placenta | Disseminationc: fet. CNS | fet. body | fet. lymph | mat. blood | mat. spleen | mat. brain | References |
| i.c. | CD‐1 | E13·5/P3‐5 | CH2016 | 105 | – | cortical thinning, >cell death, >progenitor cell cycle exit | x(H) | 77 | ||||||
| i.c. | C57BL/6 | E14·5/P3‐5 | MX2016 | 106 | – | FGR, <brain size, cortical thinning, neuronal death, <NPC proliferation, gliosis, blood–brain barrier leakage | x(H) | 78 | ||||||
aRoute of transmission: i.c., intracerebral; i.p., intraperitoneal; i. vag, intravaginal; i.vent, intraventricular; s.c. subcutaneous.
bAge at infection, E: embryonic day; P: post birth day.
cVirus strains: BZ2015: Brazil/2015 (HS‐2015‐BA‐01); CB2010: Cambodia/2010 (FSS13025); CH2016: China 2016; FP2013: French Polynesia/2013 (H/PF/2013); MX2016: Mexico/2016(MEX1‐44); PA2015: Paraiba_01/2015; PR2015: Puerto Rico/2015 (PRVABC58); UG1947: Uganda/1947 (MR‐766); VZ2016: Venezula/2016.
dBS, brain suspension; FGR, fetal growth restriction; NPC, neural progenitor cell; VZ/SVZ, ventricular zone/subventricular zone; WM, white matter.
eFA, focus forming assay; H, Histology; n.d., not detected; PA, plaque assay; R, RNA; w, Western blot.
Immune‐mediated mechanisms of ZIKV transmission across the placental barrier
Vertical transmission of ZIKV from infected dam to the fetus has also been successfully modelled in mice (Fig. 2, Table 2, part b). This includes infection of immune‐competent and immune‐compromised mice, by intraperitoneal, subcutaneous or intravaginal routes at either early (E4–5–E7) or late (E13–15) stages of fetal development. Virus has been detected in the fetal central nervous system (CNS) by RNA, histological and focus‐forming assay with pathology demonstration of fetal growth restriction, as well as cortical thinning and apoptosis of neurons in the fetal brain. The timing of infection during pregnancy may greatly influence this response as younger embryos/placentas in infected mice are more likely to develop fetal insufficiency rather than develop microcephaly.46
Figure 2.
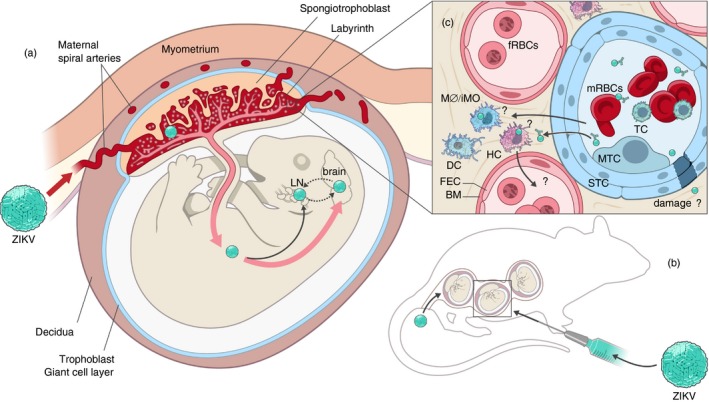
Zika virus (ZIKV) vertical transmission has been demonstrated in murine models, but further study is required to determine the specific mechanism of transmission. (a) In both mice and humans, the developing fetus and placenta are separated from the maternal myometrium by the decidua and the trophoblast giant cell layer. The placenta serves as the primary interface between the fetal and maternal blood where nutrient exchange occurs. In several mouse vertical transmission models, ZIKV has been shown to heavily infect the placenta. If infection occurs early in development, fetuses typically are reabsorbed or are not viable at birth. Infection later in development can result in vertical transmission and infection of the fetal brain and lymphatic tissue. (b) Vertical transmission in mice can occur if virus is inoculated intravaginally or through a peripheral route and is not necessary transmitted to all fetuses in a litter. Further experimentation with these models is required to determine the rate and timing of transmission. (c) ZIKV must cross the placental barrier, which is formed by syncytiotrophoblasts (STC), in order to be vertically transmitted. Multiple crossing mechanisms are possible. These include damage to STC cells by maternal or fetal immune cells, antibody‐dependent viral transcytosis, infection of maternal immune cells that cross the STC and or infection of HCs that cross back into fetal blood. FEC, fetal endothelial cells, BM, basement membrane, HC, Hoffbauer cells, DC, dendritic cells, MO/iMO, monocyte/macrophage, fRBCs, fetal red blood cells, MTC, mononuclear trophoblast cell, STC, syncytiotrophoblast cells.
These models allow investigators to address an important question in terms of the ZIKV pathogenesis, how does ZIKV cross the placental barrier? This barrier is established with in several days of conception47 and is necessary for maintaining the pregnancy. A critical function of the placenta is to connect the maternal and fetal blood (Fig. 2a,c). In both humans and mice, this critical interface is maintained and controlled by syncytiotrophoblasts,47, 48 suggesting that the mouse may be a useful model to study transmission. ZIKV may enter the fetal blood through various mechanisms involving both the maternal and fetal immune systems. Maternal immune cells may become infected and be transported across the syncytiotrophoblast layer into close proximity to the fetal blood supply (Fig. 2c). Alternatively, syncytiotrophoblasts may be damaged by maternal immune cells through direct or indirect cytotoxic mechanisms leading to breakdown of the placental barrier (Fig. 2c). It is also possible that ZIKV may be trafficked across the placental barrier via immunoglobulin‐mediated transcytosis, as has been suggested for other placenta‐invasive viruses (Fig. 2c).49 This mechanism could be facilitated by dengue‐specific cross‐reactive antibody, which has been shown to enhance ZIKV infection.50
On the fetal side of the barrier, resident macrophage Hofbauer cells are known to be activated by ZIKV infection.51 These cells may cross the fetal endothelium and become infected before returning to the fetal blood or may damage the fetal endothelium in response to infection (Fig. 2c). Although any of these mechanisms may contribute to ZIKV crossing the placenta, caution must be applied when drawing conclusions from murine models. Mice are haemotrichorial in that three layers of syncytiotrophoblasts separate maternal from fetal blood whereas humans are haemochorial with a single layer of syncytiotrophoblasts. Hence, findings from murine models should be verified in other haemochorial systems such as guinea pigs.52
Effect of fetal CNS inflammation on brain development and pathogenesis
Important questions remain regarding ZIKV infection and vertical transmission such as how the virus mediates damage to the developing brain and the role of the immune response in mediating this damage. Direct intravaginal inoculation of the dam or intracerebral injection of ZIKV into fetuses at late stages of embryonic development or early points post birth have shown clear damage to the developing brain, including cortical thinning, decreased brain size, decreased neuroprogenitor cell numbers and gliosis (Fig. 2, Table 2). However, the role of the immune response in mediating this damage is only starting to be examined. Exposure to virus in utero can elicit a strong local immune response within the fetus, which is mediated by pro‐inflammatory cytokines such as TNF, IL‐1β, IFN‐β and IFN‐γ.53 Such exposure can cause both behavioural and CNS structural pathology in offspring, which is correlated with changes in developmental gene expression within the CNS.54 Experiments using poly(I:C) injected into pregnant mice to mimic viral infection have demonstrated that the cytokine profile induced in the fetal CNS is dependent on the gestational age when exposed to the stimulus.55 Fetuses exposed at earlier embryonic time‐points express higher levels of pro‐inflammatory cytokines in their brain, which correlates with increased CNS pathology and worsened behavioural outcomes. Hence, immune responses generated in the developing fetal brain caused by early exposure to ZIKV may account for the more severe phenotypic outcomes seen in these models of ZIKV vertical transmission, such as fetal abortion/resorption, (Table 2, part b).19, 44, 56, 57 In contrast, CD8 T cells may play a role in clearing virus from the brain as they are the predominant infiltrate in a model of ZIKV infection in neonatal immunocompetent mice.58
Fetal mice exposed to ZIKV later in development generally present with neurodevelopmental pathologies such as cortical thinning, neuronal death, reduced neural progenitor proliferation, white matter damage and gliosis (Table 2, part c). One study found that neonatal mice injected intracerebrally with ZIKV have increased levels of Tnf, Il6, Il1b, Nos2 and Ccxcl1 mRNA in their brains at the peak of viral replication, which are associated with increased incidence of seizure.59 Hence, virus‐associated neuroinflammation may contribute to these neuropathologies. Similar behavioural abnormalities are observed when neonatal mice are directly injected intracerebrally with TNF‐α, indicating impaired CNS development.60 Furthermore, IL‐6 when applied to neural progenitor cells in vitro decreases their differentiation into mature neurons and increases programmed cell death, suggesting that pro‐inflammatory cytokines may be detrimental to neurodevelopment.61, 62 Interleukin‐1β and TNF have similar effects when applied to rat embryonic primary cortical neurons.63 In this way, virus‐associated pro‐inflammatory cytokines produced in the CNS during development could, in part, account for the reduced brain volumes and impaired cortical patterning observed in ZIKV‐associated cases of microcephaly.64
In related studies, intracerebral injection of TNF and IL‐1β into neonatal rat brain results in increased astrogliosis and microgliosis,65 suggesting that these cytokines could contribute to the reactive gliosis observed in models of ZIKV infection in developing brains (Table 2, part c). Likewise, these cytokines, regardless of whether they are directly injected or are induced by hypoxic injury in the neonatal brain, have been shown to induce oligodendrocyte death and hypomyelination of axons.65, 66 Hence, pro‐inflammatory cytokines may also contribute to the white‐matter injury and hypomyelination associated with ZIKV infection in the developing brain (Table 2, part c).67, 68 Furthermore, microglia have been associated with synaptic pruning during development,69 which is a necessary process for cellular patterning and function. Stimulation of developmentally immature microglia with poly(I:C) can shift these cells toward a more mature, reactive phenotype.70 Hence, ZIKV infection may predispose the developing brain to abnormal synaptic pruning, as is the case with Fragile‐X syndrome.71 Collectively these data suggest that activation of glial responses during CNS development will probably have a negative outcome during fetal development.
Conclusions and future directions
The ability to use a large variety of knockout mice and antibody treatments have provided the tools to gain a basic understanding of the key innate and adaptive immune responses that are essential for controlling ZIKV infection and preventing both pathogenesis and transmission. As one of the most harrowing outcomes of ZIKV infection is microcephaly, the use of vertical transmission models and CNS developmental models to dissect the mechanisms by which ZIKV induces damage to the developing CNS is essential. Therapeutic studies have indicated a potential for improving CNS developmental outcomes during fetal viral infection. For example, administration of anti‐TNF antibodies reduced the incidence of seizures in neonatal mice injected intracerebrally with ZIKV.59 Hence, measures taken to minimize the pro‐inflammatory response in the CNS of fetuses with ZIKV infection may improve developmental outcomes. However, further studies are needed to determine the efficacy of any such intervention and should ensure that the innate immune viral clearance mechanisms within the CNS cells are not impaired.
Disclosures
The authors have no competing interests with the publication of this manuscript.
Acknowledgements
Thanks to Ryan Kissinger for his excellent illustrative work on the figures in this review. CWW and KEP are supported by the NIAID Division of Intramural Research.
This work was supported by the National Institute of Allergy and Infectious Diseases Division of Intramural Research.
References
- 1. Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika Virus. N Engl J Med 2016; 374:1552–63. [DOI] [PubMed] [Google Scholar]
- 2. Cao‐Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP et al Zika virus, French Polynesia, South Pacific, 2013. Emerg Infect Dis 2014; 20:1085–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campos GS, Bandeira AC, Sardi SI. Zika virus outbreak, Bahia, Brazil. Emerg Infect Dis 2015; 21:1885–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao‐Lormeau VM, Blake A, Mons S, Lastere S, Roche C, Vanhomwegen J et al Guillain–Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case–control study. Lancet 2016; 387:1531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brasil P, Pereira JP Jr, Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M et al Zika virus infection in pregnant women in Rio de Janeiro. N Engl J Med 2016; 375:2321–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Driggers RW, Ho CY, Korhonen EM, Kuivanen S, Jaaskelainen AJ, Smura T et al Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 2016; 374:2142–51. [DOI] [PubMed] [Google Scholar]
- 7. D'Ortenzio E, Matheron S, Yazdanpanah Y, de Lamballerie X, Hubert B, Piorkowski G et al Evidence of sexual transmission of Zika virus. N Engl J Med 2016; 374:2195–8. [DOI] [PubMed] [Google Scholar]
- 8. Dick GW. Zika virus. II. Pathogenicity and physical properties. Trans R Soc Trop Med Hyg 1952; 46:521–34. [DOI] [PubMed] [Google Scholar]
- 9. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M et al Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 2016; 19:882–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W et al Zika virus inhibits type‐I interferon production and downstream signaling. EMBO Rep 2016; 17:1766–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blaszczyk K, Nowicka H, Kostyrko K, Antonczyk A, Wesoly J, Bluyssen HA. The unique role of STAT2 in constitutive and IFN‐induced transcription and antiviral responses. Cytokine Growth Factor Rev 2016; 29:71–81. [DOI] [PubMed] [Google Scholar]
- 12. Tripathi S, Balasubramaniam VR, Brown JA, Mena I, Grant A, Bardina SV et al A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog 2017; 13:e1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aliota MT, Caine EA, Walker EC, Larkin KE, Camacho E, Osorio JE. Characterization of lethal Zika virus infection in AG129 mice. PLoS Negl Trop Dis 2016; 10:e0004682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ et al A mouse model of Zika virus pathogenesis. Cell Host Microbe 2016; 19:720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ et al Characterization of a novel murine model to study Zika virus. Am J Trop Med Hyg 2016; 94:1362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Winkler CW, Myers LM, Woods TA, Messer RJ, Carmody AB, McNally KL et al Adaptive immune responses to Zika virus are important for controlling virus infection and preventing infection in brain and testes. J Immunol 2017; 198:3526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elong Ngono A, Vizcarra EA, Tang WW, Sheets N, Joo Y, Kim K et al Mapping and role of the CD8+ T cell response during primary Zika virus infection in mice. Cell Host Microbe 2017; 21:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Govero J, Esakky P, Scheaffer SM, Fernandez E, Drury A, Platt DJ et al Zika virus infection damages the testes in mice. Nature 2016; 540:438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH et al Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 2016; 165:1081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith DR, Hollidge B, Daye S, Zeng X, Blancett C, Kuszpit K et al Neuropathogenesis of Zika virus in a highly susceptible immunocompetent mouse model after antibody blockade of type I interferon. PLoS Negl Trop Dis 2017; 11:e0005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swartwout BK, Zlotnick MG, Saver AE, McKenna CM, Bertke AS. zika virus persistently and productively infects primary adult sensory neurons in vitro . Pathogens 2017; 6:pii: E49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oh Y, Zhang F, Wang Y, Lee EM, Choi IY, Lim H et al Zika virus directly infects peripheral neurons and induces cell death. Nat Neurosci 2017; 20:1209–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang H, Li S, Zhang Y, Han X, Jia B, Liu H et al CD8+ T cell immune response in immunocompetent mice during Zika virus infection. J Virol 2017; 91:Pii: e00900‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pardy RD, Rajah MM, Condotta SA, Taylor NG, Sagan SM, Richer MJ. Analysis of the T cell response to Zika virus and identification of a novel CD8+ T cell epitope in immunocompetent mice. PLoS Pathog 2017; 13:e1006184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wen J, Tang WW, Sheets N, Ellison J, Sette A, Kim K et al Identification of Zika virus epitopes reveals immunodominant and protective roles for dengue virus cross‐reactive CD8+ T cells. Nat Microbiol 2017; 2:17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winkler CW, Woods TA, Rosenke R, Scott DP, Best SM, Peterson KE. Sexual and vertical transmission of Zika virus in anti‐interferon receptor‐treated Rag1‐deficient mice. Sci Rep 2017; 7:7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ et al Structural basis of Zika virus‐specific antibody protection. Cell 2016; 166:1016–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sapparapu G, Fernandez E, Kose N, Bin C, Fox JM, Bombardi RG et al Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 2016; 540:443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dai L, Song J, Lu X, Deng YQ, Musyoki AM, Cheng H et al Structures of the Zika virus envelope protein and its complex with a Flavivirus broadly protective antibody. Cell Host Microbe 2016; 19:696–704. [DOI] [PubMed] [Google Scholar]
- 30. Wang J, Bardelli M, Espinosa DA, Pedotti M, Ng TS, Bianchi S et al A human bi‐specific antibody against Zika virus with high therapeutic potential. Cell 2017; 171:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dowd KA, Pierson TC. Antibody‐mediated neutralization of flaviviruses: a reductionist view. Virology 2011; 411:306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du L, Zhou Y, Jiang S. The latest advancements in Zika virus vaccine development. Expert Rev Vaccines 2017; 16:951–4. [DOI] [PubMed] [Google Scholar]
- 33. Shan C, Muruato AE, Jagger BW, Richner J, Nunes BTD, Medeiros DBA et al A single‐dose live‐attenuated vaccine prevents Zika virus pregnancy transmission and testis damage. Nat Commun 2017; 8:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shan C, Muruato AE, Nunes BTD, Luo H, Xie X, Medeiros DBA et al A live‐attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat Med 2017; 23:763–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects – reviewing the evidence for causality. N Engl J Med 2016; 374:1981–7. [DOI] [PubMed] [Google Scholar]
- 36. van der Eijk AA, van Genderen PJ, Verdijk RM, Reusken CB, Mogling R, van Kampen JJ et al Miscarriage associated with Zika virus infection. N Engl J Med 2016; 375:1002–4. [DOI] [PubMed] [Google Scholar]
- 37. Ma W, Li S, Ma S, Jia L, Zhang F, Zhang Y et al Zika virus causes testis damage and leads to male infertility in mice. Cell 2016; 167:e10. [DOI] [PubMed] [Google Scholar]
- 38. Uraki R, Hwang J, Jurado KA, Householder S, Yockey LJ, Hastings AK et al Zika virus causes testicular atrophy. Sci Adv 2017; 3:e1602899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chan JF, Zhang AJ, Chan CC, Yip CC, Mak WW, Zhu H et al Zika virus infection in dexamethasone‐immunosuppressed mice demonstrating disseminated infection with multi‐organ involvement including orchitis effectively treated by recombinant type I interferons. EBioMedicine 2016; 14:112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mansuy JM, Dutertre M, Mengelle C, Fourcade C, Marchou B, Delobel P et al Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect Dis 2016; 16:405. [DOI] [PubMed] [Google Scholar]
- 41. Mansuy JM, Suberbielle E, Chapuy‐Regaud S, Mengelle C, Bujan L, Marchou B et al Zika virus in semen and spermatozoa. Lancet Infect Dis 2016; 16:1106–7. [DOI] [PubMed] [Google Scholar]
- 42. de Laval F, Matheus S, Labrousse T, Enfissi A, Rousset D, Briolant S. Kinetics of Zika viral load in semen. N Engl J Med 2017; 377:697–9. [DOI] [PubMed] [Google Scholar]
- 43. Duggal NK, Ritter JM, Pestorius SE, Zaki SR, Davis BS, Chang GJ et al Frequent Zika virus sexual transmission and prolonged viral RNA shedding in an immunodeficient mouse model. Cell Rep 2017; 18:1751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yockey LJ, Varela L, Rakib T, Khoury‐Hanold W, Fink SL, Stutz B et al Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 2016; 166:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang WW, Young MP, Mamidi A, Regla‐Nava JA, Kim K, Shresta S. A mouse model of Zika virus sexual transmission and vaginal viral replication. Cell Rep 2016; 17:3091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jagger BW, Miner JJ, Cao B, Arora N, Smith AM, Kovacs A et al Gestational stage and IFN‐λ signaling regulate ZIKV infection in utero . Cell Host Microbe 2017; 22:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet 2001; 2:538–48. [DOI] [PubMed] [Google Scholar]
- 48. Coyne CB, Lazear HM. Zika virus – reigniting the TORCH. Nat Rev Microbiol 2016; 14:707–15. [DOI] [PubMed] [Google Scholar]
- 49. Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor‐mediated transcytosis. Am J Pathol 2006; 168:1210–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba‐Spaeth G, Duangchinda T et al Dengue virus sero‐cross‐reactivity drives antibody‐dependent enhancement of infection with Zika virus. Nat Immunol 2016; 17:1102–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwartz DA. Viral infection, proliferation, and hyperplasia of Hofbauer cells and absence of inflammation characterize the placental pathology of fetuses with congenital Zika virus infection. Arch Gynecol Obstet 2017; 295:1361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kumar M, Krause KK, Azouz F, Nakano E, Nerurkar VR. A guinea pig model of Zika virus infection. Virol J 2017; 14:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rostovsky I, Davis C. Induction of an embryonic mouse innate immune response following inoculation in utero with minute virus of mice. J Virol 2015; 89:2182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kneeland RE, Fatemi SH. Viral infection, inflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2013; 42:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I et al The time of prenatal immune challenge determines the specificity of inflammation‐mediated brain and behavioral pathology. J Neurosci 2006; 26:4752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP et al The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016; 534:267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xavier‐Neto J, Carvalho M, Pascoalino BD, Cardoso AC, Costa AM, Pereira AH et al Hydrocephalus and arthrogryposis in an immunocompetent mouse model of ZIKA teratogeny: a developmental study. PLoS Negl Trop Dis 2017; 11:e0005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Manangeeswaran M, Ireland DD, Verthelyi D. Zika (PRVABC59) infection is associated with T cell infiltration and neurodegeneration in CNS of immunocompetent neonatal C57BL/6 mice. PLoS Pathog 2016; 12:e1006004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Souza INO, Frost PS, Franca JV, Barbosa J, Neris RLS, Freitas L et al . Neonatal Zika virus infection causes epileptic seizures, viral persistence and long‐term behavioral impairment. bioRxiv 2017:195172. [Google Scholar]
- 60. Babri S, Doosti MH, Salari AA. Tumor necrosis factor‐α during neonatal brain development affects anxiety‐ and depression‐related behaviors in adult male and female mice. Behav Brain Res 2014; 261:305–14. [DOI] [PubMed] [Google Scholar]
- 61. Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003; 302:1760–5. [DOI] [PubMed] [Google Scholar]
- 62. Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. Microglia‐derived interleukin‐6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur J Neurosci 2007; 25:649–58. [DOI] [PubMed] [Google Scholar]
- 63. Gilmore JH, Fredrik Jarskog L, Vadlamudi S, Lauder JM. Prenatal infection and risk for schizophrenia: IL‐1β, IL‐6, and TNFα inhibit cortical neuron dendrite development. Neuropsychopharmacology 2004; 29:1221–9. [DOI] [PubMed] [Google Scholar]
- 64. Mlakar J, Korva M, Tul N, Popovic M, Poljsak‐Prijatelj M, Mraz J et al Zika virus associated with microcephaly. N Engl J Med 2016; 374:951–8. [DOI] [PubMed] [Google Scholar]
- 65. Cai Z, Lin S, Pang Y, Rhodes PG. Brain injury induced by intracerebral injection of interleukin‐1β and tumor necrosis factor‐α in the neonatal rat. Pediatr Res 2004; 56:377–84. [DOI] [PubMed] [Google Scholar]
- 66. Deng Y, Xie D, Fang M, Zhu G, Chen C, Zeng H et al Astrocyte‐derived proinflammatory cytokines induce hypomyelination in the periventricular white matter in the hypoxic neonatal brain. PLoS ONE 2014; 9:e87420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Soares de Oliveira‐Szejnfeld P, Levine D, Melo AS, Amorim MM, Batista AG, Chimelli L et al Congenital brain abnormalities and Zika virus: what the radiologist can expect to see prenatally and postnatally. Radiology 2016; 281:203–18. [DOI] [PubMed] [Google Scholar]
- 68. Hazin AN, Poretti A, Di Cavalcanti Souza Cruz D, Tenorio M, van der Linden A, Pena LJ et al Computed tomographic findings in microcephaly associated with Zika virus. N Engl J Med 2016; 374:2193–5. [DOI] [PubMed] [Google Scholar]
- 69. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al Synaptic pruning by microglia is necessary for normal brain development. Science 2011; 333:1456–8. [DOI] [PubMed] [Google Scholar]
- 70. Matcovitch‐Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S et al Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016; 353:aad8670. [DOI] [PubMed] [Google Scholar]
- 71. Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile‐X mental retardation syndrome. Cereb Cortex 2000; 10:1038–44. [DOI] [PubMed] [Google Scholar]
- 72. Dowall SD, Graham VA, Rayner E, Atkinson B, Hall G, Watson RJ et al A susceptible mouse model for Zika virus infection. PLoS Negl Trop Dis 2016; 10:e0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wu KY, Zuo GL, Li XF, Ye Q, Deng YQ, Huang XY et al Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 2016; 26:645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yuan L, Huang XY, Liu ZY, Zhang F, Zhu XL, Yu JY et al A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017; 358:933–6. [DOI] [PubMed] [Google Scholar]
- 75. Fernandes NC, Nogueira JS, Ressio RA, Cirqueira CS, Kimura LM, Fernandes KR et al Experimental Zika virus infection induces spinal cord injury and encephalitis in newborn Swiss mice. Exp Toxicol Pathol 2017; 69:63–71. [DOI] [PubMed] [Google Scholar]
- 76. Huang WC, Abraham R, Shim BS, Choe H, Page DT. Zika virus infection during the period of maximal brain growth causes microcephaly and corticospinal neuron apoptosis in wild type mice. Sci Rep 2016; 6:34793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X et al Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 2016; 19:120–6. [DOI] [PubMed] [Google Scholar]
- 78. Shao Q, Herrlinger S, Zhu YN, Yang M, Goodfellow F, Stice SL et al The African Zika virus MR‐766 is more virulent and causes more severe brain damage than current Asian lineage and Dengue virus. Development 2017; 144:4114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]