

Summary
T‐cell receptors (TCRs) can productively interact with many different peptides bound within the MHC binding groove. This property varies with the level of cross‐reactivity of TCRs; some TCRs are particularly hyper cross‐reactive while others exhibit greater specificity. To elucidate the mechanism behind these differences, we studied five TCRs in complex with the same class II MHC (1Ab)‐peptide (3K), that are known to exhibit different levels of cross‐reactivity. Although these complexes have similar binding affinities, the interface areas between the TCR and the peptide–MHC (pMHC) differ significantly. We investigated static and dynamic structural features of the TCR–pMHC complexes and of TCRs in a free state, as well as the relationship between binding affinity and interface area. It was found that the TCRs known to exhibit lower levels of cross‐reactivity bound to pMHC using an induced‐fitting mechanism, forming large and tight interfaces rich in specific hydrogen bonds. In contrast, TCRs known to exhibit high levels of cross‐reactivity used a more rigid binding mechanism where non‐specific π‐interactions involving the bulky Trp residue in CDR3β dominated. As entropy loss upon binding in these highly degenerate and rigid TCRs is smaller than that in less degenerate TCRs, they can better tolerate changes in residues distal from the major contacts with MHC‐bound peptide. Hence, our dynamics study revealed that differences in the peptide recognition mechanisms by TCRs appear to correlate with the levels of T‐cell cross‐reactivity.
Keywords: binding affinity–interface area relationship, CH‐π interactions, cross‐reactive T‐cell receptor recognition, fragment molecular orbital method, molecular dynamics simulation
Abbreviations
- CDR
complementarity determining regions
- FMO
fragment molecular orbital
- MD
molecular dynamics
- PDB
Protein Data Bank
- pMHC
peptide–MHC
- RMS
root‐mean‐square
- TCR
T‐cell receptor
Introduction
T‐cell receptors (TCRs) can cross‐reactively recognize different antigenic peptides presented by MHCs. The level of cross‐reactivity differs between TCRs; some possess hyper cross‐reactivity, whereas others exhibit low cross‐reactivity and somewhat greater specificity. The cross‐reactivity of a TCR may originate from a smaller number of existing TCRs compared with the number of potentially immunogenic antigenic peptides.1, 2 The number of existing TCRs is also much smaller than the theoretically possible number of TCRs based on combinatorial and junctional processes in TCR synthesis.3 The negative selection that occurs during TCR maturation, in which strongly self‐reactive TCRs are eliminated, is an important process to reduce a huge amount of TCR repertoires.4, 5 In this process, however, some self‐reactive TCRs with a weak binding affinity, which may induce autoimmune responses, survive.4, 6 Hence, the recognition mechanisms of peptide–MHC complexes (pMHCs) by TCRs must be diverse; it is challenging to comprehensively understand the various mechanisms.
Specificity in molecular recognition, i.e. one‐to‐one correspondence of two molecules, is an essential characteristic of biopolymers in a biological system. Several hypotheses have been proposed for the mechanism that generates this specificity; for example: lock‐and‐key, induced‐fit and conformational selection. On the other hand, cross‐reactivity of TCR, i.e. one‐to‐many correspondence, has provided new insights into specificity, and a number of mechanisms for this cross‐reactivity have been proposed.1, 3, 7, 8 Petrova et al. summarized cross‐reactivity in four conceptually distinct mechanisms with respect to their structures: (i) conformational plasticity of complementarity determining region (CDR) loops, i.e. so‐called induced fitting by TCR; (ii) altered TCR–pMHC docking geometry; (iii) flexibility of the peptide and MHC, i.e. induced fitting by pMHC; and (iv) structural degeneracy.3 Structural degeneracy has parallels with hydrophobic interactions, which are not necessarily strong and can easily slip.
Even if their interactions are degenerate,9, 10, 11, 12 TCR–pMHC complexes show some universality; i.e. TCR‐α and TCR‐β subunits interact with the α2‐ and α1‐helices of class I MHC, and the β and α subunits of class II MHC, respectively.13, 14, 15 Here, CDR3 is mainly responsible for the recognition of the peptide. In some cases, however, this universality does not hold. Moreover, even if it does, the interaction types formed at the interfaces differ among TCRs as shown in this study.
The recognition mechanism of TCR is similarly not fully understood from a thermodynamic perspective. It was initially hypothesized that, as the CDR loops of TCR are flexible in a free state but their flexibilities become suppressed by interactions with pMHCs upon binding, TCR binding to pMHC is entropically unfavourable. However, subsequent studies have shown that this is not always true.3, 8, 12 Furthermore, there are cases where the free energy changes upon binding are very similar, whereas the values of enthalpic and entropic contributions span a wide range, suggesting a compensatory mechanism between the entropic and enthalpic contributions, which is well known in the case of a ligand binding to an enzyme.3, 16
Dai et al. determined three crystal structures of TCR–pMHCs,17 in which three different TCRs – B3K506, 2W20 and YAe62 – recognize the same peptide, 3K, presented by class II MHC, 1Ab (Protein Data Bank (PDB) IDs: 3c5z, 3c6l and 3c60, respectively). These TCRs possess different levels of cross‐reactivity for the pMHC (1Ab‐3K); B3K506, 2W20 and YAe62 recognize 1Ab‐3K with low, moderate and high cross‐reactivity, respectively.18 Stadinski et al. later determined the structures of other TCRs – J809.B5 and 14.C6 – that specifically recognize 1Ab‐3K19, 20 (PDB IDs: 3rdt and 4p5t, respectively). Interestingly, the five TCR–pMHCs had similar binding affinities,21 although their interface areas differed significantly, as shown below.
A comparative study of the complexes is necessary to provide useful information regarding the following points: the difference between recognition mechanisms of TCRs exhibiting different levels of cross‐reactivity, and the relationship between interface area and binding affinity. For this purpose, we considered it necessary to study the structures of the TCR–pMHC complexes from not only static but also dynamic perspectives. In this study, we performed molecular dynamics (MD) simulations for both TCRs in a free state and TCR–pMHC complexes. On the basis of fluctuation patterns of CDRs in the five TCR–pMHC systems, which were obtained from the simulations, we identified two distinct mechanisms of peptide recognition. The TCRs exhibiting high levels of cross‐reactivity recognize 1Ab‐3K rigidly, leading to the formation of a small amount of interactions such as non‐specific CH‐π interactions, whereas those TCRs exhibiting low levels of cross‐reactivity recognize the pMHC flexibly by the induced‐fitting mechanism and form a large amount of specific hydrogen bond interactions. The fragment molecular orbital (FMO) method estimated non‐specific CH–π interactions quantitatively, which showed that the non‐specific interactions acquired sufficient binding energy. We concluded that the distinct entropic changes derived from the fluctuation patterns, which were different according to the levels of cross‐reactivity of TCRs, led to the non‐correlation between binding affinity and interface area. Hyper cross‐reactive TCRs have the potential to recognize self‐peptides as shown by Stadinski et al.,22 implying that our novel approach can be applied to the elucidation of the mechanism of cross‐reactive self‐peptide recognition by TCR. This approach may also help the design of novel peptides that control TCR recognition and T‐cell activation.
Materials and methods
Calculation of interface area
To determine the interface areas between TCR, MHC and the peptide, the molecular surfaces (Connolly surfaces) for the TCR, MHC, peptide and pMHC were calculated separately using the program MSP.23 The output of the calculation is a set of vertices that are points of triangles covering the surface. We defined the sum of the areas of the triangles that satisfied the following condition as an interface area: at least two vertices of a triangle in one molecule are located within 3·0 Å of at least one vertex of a triangle in another molecule. As the interface areas differ between two molecules, the mean of the two values was defined as the interface area of the two molecules.24 We calculated the interface areas for the following three combinations: TCR and peptide, TCR and MHC, and TCR and pMHC.
Identification of interactions
We defined an interaction between two residues as when at least one atom in a residue was located within 4·5 Å of an atom in another residue. The interactions between a residue and a region (e.g. CDR1α) and those between a residue and a molecule (e.g. TCR) were defined in the same manner.
Similarly, atomic contact was defined as two atoms being located within 4·5 Å of each other.
The hydrogen bonds formed in TCR–pMHC complexes were calculated using the program HBPLUS (https://www.ebi.ac.uk/thornton-srv/software/HBPLUS/).25
Molecular dynamics simulations
Molecular dynamics simulations were performed for two kinds of systems – TCR–pMHC and free TCR – of the five complexes (PDB IDs: 3c5z, 4p5t, 3rdt, 3c6l and 3c60), which were crystal structures of TCR, B3K506, 14.C6, J809.B5, 2W20 and YAe62, respectively, in complex with peptide 3K presented by class II MHC 1Ab. As two asymmetric complexes were contained in the PDB data (except for the J809.B5 complex), we used the A, B, C and D chain data for TCR‐α, TCR‐β, MHC‐α and MHC‐β, respectively. The peptide bound to MHC was present in the D chain together with a linker polypeptide. Linker regions were removed in static analyses and simulations of TCR–pMHCs.
The gromacs package 4.6.7 (http://www.gromacs.org/Downloads)26 with AMBER99SB force field was used for MD simulation on the super computer UVs at RIKEN Quantitative Biology Centre and Osaka University. After energy minimization for ~ 3000 steps and equilibration for ~ 400 ps (200 ps of NVT MD followed by 200 ps of NPT MD), we performed MD production runs of 200 ns at 300 K in truncated octahedron boxes filled with TIP3P (transferable intermolecular potential with three‐points) water molecules. The MD product runs were terminated at 200 ns because the root‐mean‐square (RMS) deviations of the whole system were sufficiently stabilized.
Dynamic properties
The MD snapshots were taken every 5 ps from 20 to 200 ns from the production run. These were superimposed onto the snapshot at 200 ns (reference structure) in any of the simulations to remove translational and rotational motions of the systems. The fluctuation of a residue, i.e. RMS deviation of residue i represented by its Cα atom, F i, was obtained from N snapshots from 20 to 200 ns as follows:
| (1) |
where r i(k) and <r i> were position vectors of residue i in the kth snapshot structure and averaged over all snapshot structures, respectively.
The correlation coefficient of fluctuations between two residues i and j, C ij, was calculated as:
| (2) |
High positive and negative values of C ij indicate that the two residues move in the same and opposite directions, respectively, on average; on the other hand, a C ij value near zero indicates that the two residues move independently.
Clustering of residues in CDRs
To cluster the residues in the six CDRs, i.e. CDR1α–CDR3α and CDR1β–CDR3β, from a dynamic perspective, we estimated the similarity of residues i and j in motion by the RMS deviation of their correlation coefficients with the other residues m:
| (3) |
where M is the total number of residues in the six CDRs. The CDR residues were then clustered by the Ward.D2 clustering method of the hclust function in the program R (https://www.r-project.org/),27 using S ij as the distance between residues i and j.
FMO calculations
The FMO method28 was applied for the examination of π‐interactions formed by the aromatic residues, 94Phe and 95Trp in CDR3β, in 14.C6, J809.B5, 2W20 and YAe62 bound to 1Ab‐3K, where 2W20 contained only 95Trp. The FMO method calculates the interaction energies between ‘fragments,’ such as the energy between residues, or between a residue and a ligand, called inter‐fragment interaction energies. Moreover, the interaction energies are decomposed into four elements, electrostatic (ES), exchange‐repulsion (EX), charge‐transfer (CT) and dispersion (DI) interactions by applying pair interaction energy decomposition analysis.29
For the FMO calculation, we prepared the input coordinate files that included the structural information of the variable domain of TCR and pMHC in the crystal structures, by using MOE.30 The FMO calculation of the structure was performed at MP2/6‐31Gd level by ABINIT‐MP,31 which was installed on the K computer at RIKEN. The analysis of the results and the evaluation of CH‐π interactions were executed using biostation‐viewer in the MIZUHO/BioStation.31, 32
Results
Relationships between binding affinity and interface area in TCR–pMHC complexes
We analysed 31 structures of complexes between class II MHC–peptide and TCR in the PDB as of June 2016 (see Supplementary material, Fig. S1a for a structural summary of TCR–pMHC complexes). Of these structures, 15 had self‐peptides and another 16 had non‐self (i.e. foreign and synthesized) peptides. TCRs in these complexes exhibited different levels of cross‐reactivity.
It was considered that the interface area of a TCR with a pMHC is one of the factors determining their interaction modes. The calculated interface areas of the 31 TCR–pMHC complexes (see Materials and methods section) were very diverse (ranging from 750 to 1400 Å2), as shown in Fig. 1. The interface areas of a TCR with the binding partner MHC and that with the peptide were also diverse, and had high correlations with the interface areas of TCR with pMHC: the correlation coefficient for TCR–MHC interfaces was 0·96, and that for TCR–peptide interfaces was 0·84. These results indicate that TCR did not have a strong bias in favour of either MHC or peptide for its binding. In addition, we did not find any clear differences to distinguish the self‐peptide‐bound complexes (marked in Fig. 1) from non‐self‐peptide‐bound complexes with respect to the interface areas.
Figure 1.
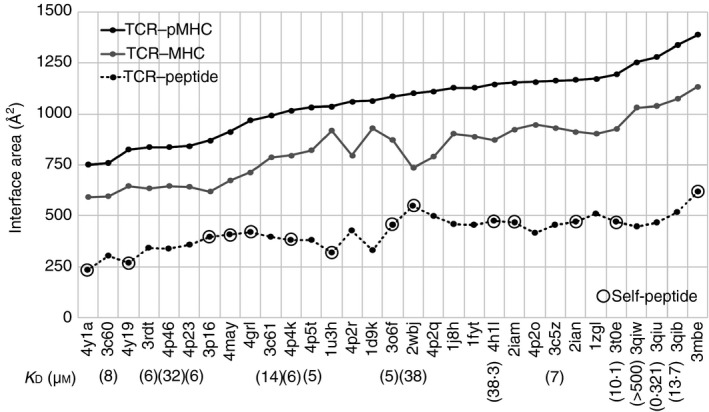
Interface areas in 31 class II MHC–peptide–T‐cell receptor (TCR) complexes. The black and grey solid lines and black dotted line indicate interface areas between TCR and peptide–MHC (pMHC), TCR and MHC, and TCR and peptide, respectively. Protein Data Bank (PDB) IDs of the complexes arranged in the order of interface size are shown on the horizontal axis. The binding affinity data are shown below the PDB IDs, if available in the primary citations. The complexes that according to the primary citations contain self‐peptides are marked with open black circles.
The binding affinity data were available at their primary citations for 15 of the 31 complexes, as shown below the relevant PDB IDs in Fig. 1. Binding affinities did not have any correlation with the interface areas, as mentioned previously.17 Hence, it was suggested that the interface size of TCR with pMHC, which was expected to be related to their tight and loose bindings, might not be the main factor to determine the binding affinity of TCR with pMHC.
Although the sizes of the interface between different TCRs and pMHCs are very diverse, as described above, we found that there are at least six specific MHC residues interacting with TCR in most of the complexes examined; three specific residues in MHC‐α, and three in MHC‐β as summarized in the Supplementary material (Table S1). The MHC residues involved in these ‘conserved interactions’ were located at the recognition helices for the peptide (see Supplementary material, Fig. S1b), and tended to interact with specific CDR loops in TCR (e.g. 55Glu or Asp in MHC‐α interacts with CDR3α; see Supplementary material, Table S1) in most cases. However, the counterpart residues and their positions in CDRs differ among the complexes because of the amino acid sequence diversity of the CDRs. On the other hand, about 70% of the atomic contacts between TCR and peptide on average were from contacts of CDR3α and CDR3β with the peptide in all complexes except for regulatory Treg complexes (4y19 and 4y1a) (see Supplementary material, Table S2). These data suggest that the six conserved TCR–MHC interactions, most of which were formed between CDR3α/CDR2β and MHC‐α and between CDR3β/CDR1α and MHC‐β, were the minimum requirement for TCR binding to pMHC, regardless of the diversity of interface areas between TCR and MHC. They also suggest that interface sizes and their tightness/looseness are determined by other mechanisms in TCR–pMHC interactions rather than the conserved TCR–MHC contacts. Elucidating the interaction mechanism may explain the relationship between interface size and binding affinity.
This implied that there are limitations to the use of the analysis of static structures in the understanding of interaction mechanisms, and that it is necessary to study their dynamic structures. This is because a crystal structure shows only one snapshot of a fluctuating protein, although such a structure has a high degree of probability of occurrence. In addition, the observation of other snapshots obtained from dynamic studies not only of the TCR–pMHC complex but also of TCR in a free state might facilitate our understanding of the mechanism of pMHC recognition by TCRs. As dynamics study required time‐consuming simulations, we focused on five of the 31 complexes, in which five different TCRs recognize the same pMHC, i.e. peptide 3K presented by class II MHC 1Ab (1Ab‐3K) (Table 1).17, 19, 20, 33
Table 1.
Summary of the characteristics of five T‐cell receptor–peptide–MHCs (TCR‐pMHCs); binding affinities and the levels of cross‐reactivity for 1Ab‐3K
| TCR | PDB ID | Binding affinity (μ m) | The levels of cross‐reactivity |
|---|---|---|---|
| B3K506 | 3c5z | 7 | Greatly specific |
| 14.C6 | 4p5t | 5 | Greatly specific |
| J809.B5 | 3rdt | 6 | Greatly specific |
| 2W20 | 3c6l | 14 | Moderately cross‐reactive |
| YAe62 | 3c60 | 8 | Highly cross‐reactive |
Changing TCR–pMHC interactions during MD simulations
As shown in Table 2, the five TCRs have different amino acid sequences and lengths of CDR3αs; from long to short, B3K506, 14.C6, J809.B5, YAe62 and 2W20. On the other hand, the amino acid sequences of CDR3βs in 14.C6, J809.B5 and YAe62, are identical and that in 2W20 is similar to these TCRs. It should be noted that the middle residues of the CDR3β loops are aromatic or non‐polar amino acids: 94Phe in 14.C6, J809.B5, and YAe62, 94Ala in 2W20, and 95Trp in all the four TCRs. In contrast, another TCR, B3K506, has polar amino acids (94Ser and 95Ser) in the same positions of CDR3β. In this study, we mainly focused on CDR3α and CDR3β in TCRs because the two CDR loops were involved in about 70% of the TCR–peptide interactions (last column in the Supplementary material, Table S2).
Table 2.
Summary of the characteristics of five T‐cell receptor–peptide–MHC (TCR‐pMHCs). Amino acid sequences and residue numbers with the chain ID of CDR3α and CDR3β
| TCR | CDR3α | CDR3β | ||
|---|---|---|---|---|
| Sequence | Residue number (length) | Sequence | Residue number | |
| B3K506 | ALVISNTNKVV | A:90–100 (11) | ASIDSSGNTLY | B:90–100 |
| 14.C6 | AASRDSGQKLV | A:91–101 (11) | ASGDFWGDTLY | B:90–100 |
| J809.B5 | AASKGADRLT | A:91–100 (10) | ASGDFWGDTLY | B:90–100 |
| 2W20 | AASDNRIF | A:90–100 (8) | ASGDAWGYEQY | B:90–100 |
| YAe62 | AANSGTYQR | A:90–100 (9) | ASGDFWGDTLY | B:90–100 |
The five TCR–pMHCs have different interface areas: from large to small, B3K506, 14.C6, 2W20, J809.B5 and YAe62 (Table 3). The size of the interface area does not correlate with the strength of the binding affinity: from high to low, 14.C6, J809.B5, B3K506, YAe62 and 2W20 (Table 1). The first four TCRs have very similar binding affinities. With respect to recognition of 1Ab‐3K, TCRs 2W20 and YAe62 are more cross‐reactive than B3K506, 14.C6 and J809.B5.
Table 3.
Summary of the characteristics of five T‐cell receptor–peptide–MHC (TCR‐pMHCs). Interface areas, change of fluctuations in CDR3α and CDR3β, and correlativity of the fluctuations
| TCR | Interface areas (Å2) (T‐pM/T‐M/T‐p)a | Change in fluctuations from the free to complex states | Correlativity of fluctuations between CDR3α and CDR3β | |||
|---|---|---|---|---|---|---|
| Crystal structures | MD (average) | CDR3α | CDR3β | Free state | Complex | |
| B3K506 | 1163/932/455 | 1230/998/456 | Decrease | Decrease | Non‐correlative | Correlative |
| 14.C6 | 1033/822/382 | 1126/903/385 | Decrease | Decrease | Non‐correlative | Correlative |
| J809.B5 | 836/635/343 | 1047/850/339 | Decrease | Decrease | Non‐correlative | Correlative |
| 2W20 | 992/787/396 | 923/716/329 | Not decrease | Not decrease | Non‐correlative | Correlative |
| YAe62 | 760/596/304 | 857/739/218 | Not decrease | Not decrease | Correlative | Non‐correlative |
T, TCR; pM, peptide‐MHC; M, MHC; p, peptide.
We performed MD simulations of the five TCR–pMHC complexes and TCRs in a free state to obtain the dynamic structural properties of TCR–pMHC interactions (see Materials and methods section). TRC regions interacting with pMHC in the crystal structures and in the final snapshots of TCR–pMHC simulations (at 200 ns) are shown in Fig. 2. The interface areas in the crystal structures and in the 10 snapshots at every 20 ns from the MD trajectories are summarized in Table 3 and in the Supplementary material (Table S3); Table 3 shows only the average of interface areas over the 10 MD snapshots. The interface sizes increased during the simulations in all complexes, except for 2W20. Particularly in J809.B5 and YAe62, the TCR–MHC interacting areas increased significantly; the interfaces in the crystal structures localized to the interactions of CDRβ loops with pMHC spread to the interactions of CDRα with pMHC (Fig. 2c,e). These changes were observed in the atomic contacts of TCR‐α and TCR‐β with pMHC during the simulation, as shown in the Supplementary material (Table S4); the former atomic contacts (particularly Tα‐M) increased, as the latter ones (Tβ‐M) decreased.
Figure 2.

T‐cell receptor (TCR) regions interacting with peptide–MHC (pMHC). (a–e) MHC and peptide‐binding footprints on TCR surface in B3K506, 14.C6, J809.B5, 2W20 and YAe62, respectively, in the crystal structure (upper figures) and the MD snapshot at 200 ns (lower figures). From left to right, the binding footprints of peptide, MHC and pMHC, respectively, are shown in purple. CDR regions are defined based on the IMGT 3D structure‐DB 39 and Dunbrack definition,40 as summarized in Table 2 for CDR3 loops, and shown in Figure 3. CDR1α–3α and CDR1β–3β are coloured cyan, orange, green, blue, pink and yellow, respectively. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 4.
Summary of the characteristics of five TCR‐pMHCs. Atomic contacts
| TCR | T‐pM/T‐M/T‐pa | Tα‐M/Tβ‐Ma | Tα‐p/Tβ‐pa | |||
|---|---|---|---|---|---|---|
| Crystal structures | MD (average) | Crystal structures | MD (average) | Crystal structures | MD (average) | |
| B3K506 | 288/200/88 | 374/284/90 | 92/108 | 149/134 | 50/38 | 56/34 |
| 14.C6 | 328/232/96 | 274/194/81 | 50/182 | 68/125 | 49/47 | 46/35 |
| J809.B5 | 279/202/77 | 286/217/69 | 11/191 | 55/162 | 26/51 | 31/38 |
| 2W20 | 301/200/101 | 248/170/78 | 39/161 | 83/87 | 46/55 | 45/33 |
| YAe62 | 274/227/47 | 238/196/41 | 41/186 | 58/138 | 8/39 | 5/36 |
T, TCR; pM, peptide‐MHC; M, MHC; p, peptide; Tα, α subunit of TCR; Tβ, β subunit of TCR.
We indicated above that there were six conserved TCR–MHC interactions formed in the crystal structures. In fact, all of the six interactions were observed in B3K506, 14.C6 and 2W20, but one and two interactions were missing in J809.B5 and YAe62, respectively (see Supplementary material, Fig. S2a,b). During the simulations, all the interactions formed in the crystal structures were maintained, except for two residues in MHC‐β of 2W20, i.e. 92Glu and 103Thr, which were lost. The missing interactions in J809.B5 (77Thr in MHC‐β) and in YAe62 (92Glu in MHC‐β) in the crystal structures were formed and maintained during the simulations. Another missing interaction in YAe62 (56Asp in MHC‐α) was formed, but only temporarily maintained.
As its name implies, peptide 3K contains three Lys residues, which can be recognized by TCRs. All Lys residues in the five TCR–pMHCs interact with the TCRs in the crystal structures. During the simulations, all the Lys residues formed the interactions most of the time, except for one Lys residue in 2W20, i.e. 10Lys (see Supplementary material, Fig. S2c).
These results suggested that B3K506 and 14.C6 formed stable and balanced interfaces with 1Ab‐3K. Such an interface was formed during the simulation in J809.B5. Although the interface area on YAe62 with 1Ab spread widely, that area with 3K decreased. On the other hand, the interface size on 2W20 decreased, differently from the others, and the conserved and important pMHC interactions were incomplete during the simulation. The MD snapshot structures at 200 ns shown in the Supplementary material (Fig. S3d,e) for 2W20 and YAe62, respectively, illustrate larger deviations of the peptides and MHCs during the simulation compared with B3K506, 14.C6 and J809.B5 (see Supplementary material, Fig. S3a–c, respectively). These results imply tight bindings of TCR and pMHC in B3K506, 14.C6 and J809.B5 and loose ones in 2W20 and YAe62.
Correlative fluctuations of TCR residues in the free and complex states
We then examined fluctuations of TCR residues during the simulations. On the basis of differences between the dynamic characters of TCRs in the free and complex states, we considered the recognition mechanisms of 1Ab‐3K by these TCRs.
Figure 3 shows RMS fluctuations of the residues in the six CDR loops of TCR in the free and complex states during simulations (see Materials and methods section). In B3K506, 14.C6 and J809.B5, although the residues in the turn region of the CDR loops largely fluctuated in the free state, their fluctuations were considerably reduced by the formation of the complex, particularly in CDR1 and CDR3. On the other hand, in 2W20 and YAe62, the changes in fluctuations caused by the formation of the complex were small, compared with the above three TCRs. It was considered that the much larger reduction in the fluctuations of CDR residues in B3K506, 14.C6 and J809.B5 than in 2W20 and YAe62 was an important implication for the difference in recognition mechanisms of 1Ab‐3K.
Figure 3.
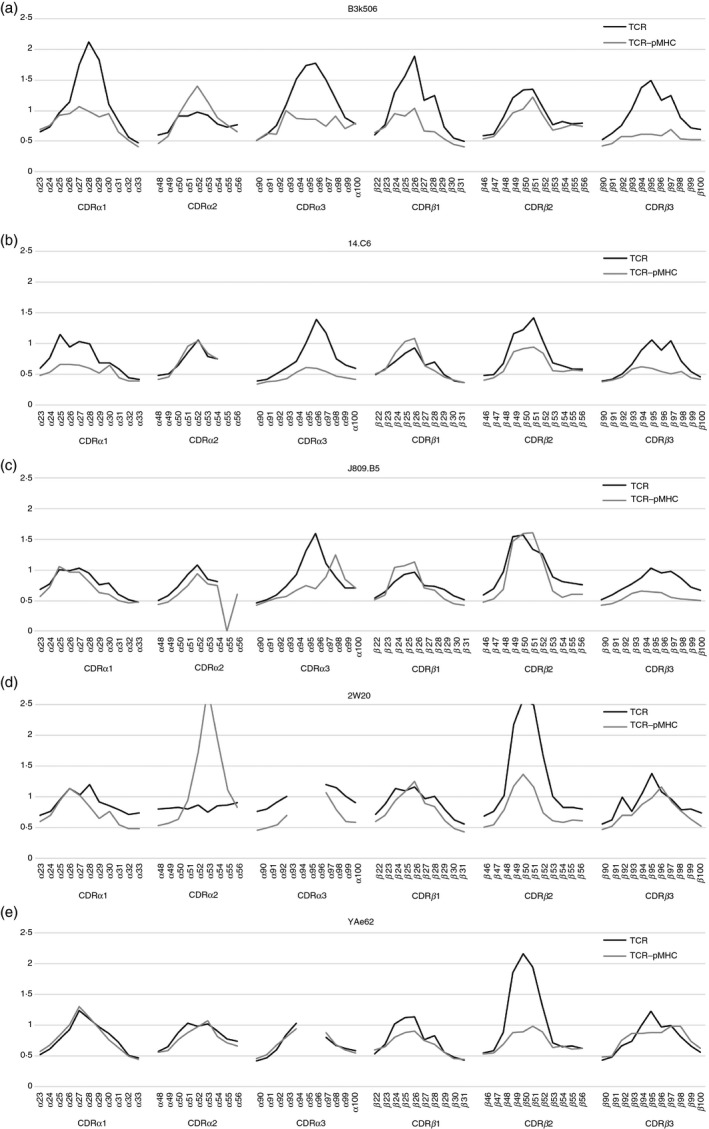
Fluctuations of T‐cell receptor (TCR) residues in the free and complex states. (a–e) The root‐mean‐square (RMS) fluctuations of the CDR residues in B3K506, 14.C6, J809.B5, 2W20 and YAe62, respectively, are shown. The fluctuations were calculated applying Eq. (1) to the molecular dynamics (MD) snapshots from 20 to 200 ns. The black and grey lines indicate the fluctuations in the free TCR and complex states, respectively.
The correlation between residue fluctuations in TCR was calculated from MD trajectories of TCR in the free and complex states. Then, the CDR residues were clustered based on their correlation coefficient values C ij in Eq. (2) in Materials and methods (see clustering dendrograms in the Supplementary material, Fig. S4, where more positively correlated residues are grouped into the same cluster). In the free state, the residues in the turn regions of CDR3α and CDR3β belonged to the distant clusters in the dendrograms, except for YAe62. On the other hand, in the complex state, they belong to the same or neighbouring clusters in all TCR–pMHCs. These results indicated that the residues in the turn regions of the CDR3α and CDR3β fluctuated independently before binding to 1Ab‐3K, but they fluctuated in a positively correlated manner after forming the complexes in B3K506, 14.C6, J809.B5 and 2W20. In contrast, the CDR3α and CDR3β in YAe62 fluctuated correlatively in both the free and complex states.
We examined the interactions between CDR3α and CDR3β to confirm whether the inter‐CDR loop interactions, particularly between CDR3α and CDR3β, are responsible for their correlative fluctuations. In the free state of 14.C6, J809.B5, 2W20 and YAe62, the bulky aromatic residue 95Trp in CDR3β was located close to CDR3α during the simulations (see Supplementary material, Table S5). In 2W20 and YAe62, the main‐chain atoms of 95Trp were involved in 65 and 72% of inter‐CDR loop contacts, respectively, differently from 14.C6 and J809.B5 (14 and 2%, respectively). Although these inter‐CDR loop contacts may affect the correlative fluctuations between CDR3α and CDR3β in YAe62, these contacts had less influence on correlative fluctuations in 2W20, probably because of the shortness of the CDR3α loop.
Specific and non‐specific interaction modes
CDR3α and CDR3β mainly contribute to the recognition of pMHC, as described above. Different types of amino acid residues in the turn regions of CDR3α and CDR3β loops are probably key factors to determine the interaction modes, such as a hydrogen bond, which is a typical specific and stable polar residue interaction, or a π‐interaction related to aromatic residues. The difference in interaction modes may affect the antigen recognition dynamics.
First, we examined hydrogen bond interactions observed in crystal structures and MD snapshots, and categorized them into three types: hydrogen bonds formed in the crystal structure and maintained during the simulation (maintained hydrogen bond), hydrogen bonds found in the crystal structure but lost during the simulation (lost hydrogen bond), and, finally, hydrogen bonds not observed in the crystal structure but newly formed and maintained during the simulation (added hydrogen bond) (see Supplementary material, Table S6 footnote for more detail). TCRs that recognize 1Ab‐3K with greater specificity, such as B3K506, 14.C6 and J809.B5, formed over 10 hydrogen bonds with the peptide and MHC and maintained them during the simulations (involving both maintained and added hydrogen bonds). On the other hand, TCRs that cross‐reactively recognize pMHCs, such as 2W20 and YAe62, formed six and seven hydrogen bonds, respectively. In all complexes, CDR3α and CDR3β were involved in the formation of many hydrogen bonds, except for YAe62. In addition, all TCRs formed at least one hydrogen bond with the important Lys residue in the 3K peptide, mostly by CDR3α and/or CDR3β.
Although the residues in the middle of CDR3α, 95Asn and 96Thr, were involved in hydrogen bond formation in B3K506, the residues in the off‐middle of CDR3α, 94Arg and 99Lys in 14.C6 and 94Lys, 97Asp and 98Arg in J809.B5, also took part in establishing hydrogen bonds. The residues in the middle of CDR3α, 95Asp and 96Ser in 14.C6 and 96Ala in J809.B5, interacted with a bulky residue, 95Trp, in CDR3β during the simulations in TCR–pMHC complex state (see Supplementary material, Table S5b,c). On the other hand, in B3K506, there is no Trp residue in CDR3β, and 97Asn in CDR3α and 97Asn CDR3β in the off‐middle of CDR3s formed inter‐CDR loop contacts instead (see Supplementary material, Table S5a). These observations suggest that, although the CDR3α loops in B3K506, 14.C6 and J809.B5 are similar in length (11, 11 and 10 residues, respectively), the amino acid positions to form specific interactions depend on the sequence of the interacting partner, CDR3β. Interestingly, 92Asn, 93Ser and 97Thr in CDR3α of YAe62 did not form any hydrogen bonds with the pMHC. The strong inter‐CDR loop interactions with 95Trp and 96Gly in CDR3β may be dominant over hydrogen bonds.
Next, we examined non‐specific π‐interactions, particularly CH‐π interactions, of aromatic residues, 94Phe and 95Trp, in CDR3β of 14.C6, J809.B5, 2W20 (94Ala instead of 94Phe) and YAe62 (Table 2), which were considered to play an important role in the binding of TCR with pMHC. Although π‐interactions are weak and can gain only a small amount of energy (~1 kcal/mol), they can be formed non‐specifically with many atoms simultaneously. Therefore, we examined π‐interactions established by aromatic residues in CDR3β using the FMO method, which is one of the methods to calculate molecular orbitals of macro‐molecules (see Materials and methods for more detail).28 In this study, we were particularly interested in charge‐transfer (CT) and dispersion (DI) interaction energies among the four elements of interacting energies between TCR and pMHC because these energies corresponded to π‐interactions.
The interaction energies between TCR and pMHC residues were calculated for crystal structures and MD snapshot structures at 100 and 200 ns in the above four TCR–pMHC complexes using the FMO method. The biostation‐viewer program31, 32 identified CH‐π interactions of 94Phe and 95Trp in CDR3β with Lys, Gln and Val residues in the pMHC, based on interaction energies and geometries of the residues (see Supplementary material, Table S7). Figure S5 (in the Supplementary material) illustrates the CH‐π interactions of 94Phe and 95Tpr in CDR3β with 7Lys in the peptide and 62Gln and 66Val in MHC‐α in YAe62. As mentioned above, a CH‐π interaction is weak; however, CH‐π interactions have an advantage in their ability to form interactions among various residues and in various conformations of the same residue. In fact, even if the counterpart residues (Lys, Gln and Val in Fig. S5, see Supplementary material) changed their conformations, it is likely that the aromatic ring formed CH‐π interactions with the same atoms or other counterpart atoms neighbouring them. Hence, CH‐π interactions added up to a significant amount.
Discussion
In this study, we focused on the five TCR–pMHC complexes, where TCRs with different levels of cross‐reactivity are bound to the same pMHC. We were also interested in why they had similar binding affinities, whereas the interface areas between TCR and pMHC differ significantly. Comparative analyses based on the dynamic and static structural features revealed the differences in the recognition mechanisms and interaction modes among the five TCR–pMHCs, and so provided clues to understanding these features.
The above results show that the collaborations between CDR3α and CDR3β, which were characterized by their amino acid sequences, mainly contribute to the cross‐reactivity (or specificity) of TCRs and interaction modes between TCR and pMHC. The CDR3α loops differ in their lengths, content of polar and charged residues, and positions. During the MD simulation of the complex between J809.B5 and 1Ab‐3K, the interface area localized in a crystal structure spread widely. In contrast, the interface area of the complex between 2W20 and pMHC decreased. As a result, the order of the interface sizes in the MD simulation structures changed from that in the crystal structures: B3K506 (1163→1230 Å2), 14.C6 (1033→1126 Å2), J809.B5 (836→1047 Å2), 2W20 (992→923 Å2), and YAe62 (760→857 Å2) (Table 3 and see Supplementary material, Table S3). Interestingly, the first three and last two TCRs exhibit low and high levels of cross‐reactivity, respectively (Table 1).
Examination of the fluctuations of the TCRs during the MD simulations in free and complex states showed that the complex formation led to considerable decrease in the fluctuations of CDR3α and CDR3β only in the three TCRs exhibiting a specificity for 1Ab‐3K greater than the others. This result indicated that the greatly specific TCRs formed a tight interface with 1Ab‐3K, whereas the cross‐reactive TCRs formed a loose interface with the pMHC. Examination of the correlative motions of the CDR residues showed that many of the residues in CDR3α and CDR3β loops fluctuated in a correlative manner in the complex state. In contrast, CDR3α and CDR3β fluctuate independently in the free state, except for YAe62, where CDR3α and CDR3β fluctuate correlatively even in the free state, similarly to that in the complexed state. These observations indicate that the CDR3α and CDR3β in YAe62 approached 1Ab‐3K rigidly, and formed a loose complex with the pMHC. The larger deviations of pMHC in the MD snapshot structures at 200 ns shown in Fig. S3 (see Supplementary material) for 2W20 and YAe62 also support this inference.
Examination of the interactions showed that over 10 hydrogen bonds were formed during the simulations in the complexes of B3K506, 14.C6 and J809.B5 with 1Ab‐3K; this finding was different from that of 2W20 and YAe62. On the other hand, 14.C6, J809.B5, 2W20 and YAe62 formed several CH‐π interactions between the aromatic residues in CDR3β and the residues in pMHC. These observations indicated that the greatly specific TCRs, B3K506, 14.C6 and J809.B5, recognize 1Ab‐3K principally by forming specific hydrogen bonds, whereas the moderate and hyper cross‐reactive TCRs, 2W20 and YAe62, respectively, recognize the pMHC by predominantly forming non‐specific CH‐π interactions.
The above observations led to the following scenario of TCR binding with pMHC. In B3K506, 14.C6 and J809.B5, the TCRs exhibiting low levels of cross‐reactivity and somewhat specific for 1Ab‐3K, the large and non‐correlative fluctuations of the CDR3α and CDR3β facilitated the formation of many interactions with specific counterpart pMHC atoms, and thereby the tight complex with the pMHC, by changing its conformation if necessary. This can be referred to as ‘induced‐fit’ binding. To compensate for the large entropy loss following the binding with pMHC, the formation of tight complexes with numerous specific interactions is required. Hence, it is possible for the long and polar/charged residue‐rich CDR3α loops that can form hydrogen bonds in B3K506, 14.C6 and J809.B5 to gain enough enthalpy.
In contrast, in YAe62, hyper cross‐reactive TCR, the moderate and mutually correlated fluctuations of the CDR3α and CDR3β in the free state led to a rigid approach of the TCR to the pMHC, resulting in the formation of a smaller number of interactions and the loose complex with pMHC. This may be referred to as the ‘lock‐and‐key’ model. However, it should be noted that the complex between YAe62 and 1Ab‐3K is formed loosely, in contrast to the popular perception that the lock‐and‐key is tightly adjusted. In this case, many specific interactions are not required because the entropy loss owing to the binding of the TCR with the pMHC is much smaller than in the cases of the greatly specific TCRs. The non‐specific interactions such as CH‐π and π‐π interactions and hydrophobic interactions of CDR3β are sufficient for this requirement. Moreover, the ‘loose’ interaction may be synonymous with ‘structural degeneracy’ described in a previous report.3
The above‐mentioned recognition mechanisms may answer the following question, which was our major incentive for this study: Why do interfaces with significantly different features have similar binding affinities?
As summarized in the Supplementary material (Table S4), the aromatic residue 95Trp in CDR3β dominated the interactions with not only the peptide but also MHC in 14.C6, J809.B5, 2W20 and YAe62. The bulky 95Trp was involved in the inter‐CDR loop contacts with the middle residues of the CDR3α, as well as in the hydrogen bond formations and CH‐π interactions. In YAe62, the formation of inter‐CDR loop contacts by 95Trp in CDR3β governed the dynamics of the 1Ab‐3K recognition by TCR in the free state, following which the aromatic residue was involved in about 58 and 26% of the TCR‐peptide and TCR–pMHC contacts, respectively, in the complex state during the simulation. The aromatic residue functioned as a hub of the dense network of TCR–pMHC interactions at least in YAe62 (see Supplementary material, Fig. S6), which seems to be a ‘hotspot’ in the sense that the aromatic residue contributed significantly to binding affinity.
Such a situation was found in other TCR–pMHC complexes. TCR Hy.1B11, which originates from a patient with multiple sclerosis and recognizes the self‐antigen myelin basic protein, has the aromatic residue 95Phe in CDR3α. The structures of the TCR in complex with three different self‐peptides, MBP, PMM and UL15, presented by class II MHC DQ1 have already been solved (PDB ID: 3pl6, 4grl and 4may, respectively34, 35). The residue 95Phe in CDR3α covers 62, 55 and 76% of the atomic contacts with the peptides, and 27, 22 and 31% of all atomic contacts, respectively. It indicates that this aromatic residue is a hotspot for the interactions between TCR and pMHC and contributes significantly to the binding affinity. Several ‘hotspot’ peptide motifs that can be cross‐reactively recognized by a TCR have been identified previously.36, 37, 38 In this study, however, we focused on the CDR residues that determined the cross‐reactivity (or specificity) of the TCRs.
In the modestly cross‐reactive TCR, 2W20, the fluctuations of the CDR3α and CDR3β did not decrease by complex formation, as is the case with YAe62. This finding suggested that 2W20 formed loose interactions with 1Ab‐3K; in other words, it could gain a small amount of enthalpy. In contrast, the CDR3α and CDR3β did not show correlative fluctuations clearly in the free‐state TCR, differently from YAe62. This is probably because the CDR3α loop is too short to fluctuate correlatively with CDR3β. These observations might suggest that the entropy loss in the complex formation of 2W20 was larger than that of YAe62. The findings may explain why the binding affinity in 2W20 for 1Ab‐3K is slightly smaller than that of the other TCR–pMHC complexes.
Dai et al. have commented on the aromatic residues in YAe6217. They considered that hydrophobic interaction of the aromatic residues, which were non‐specific, was responsible for the cross‐reactivity of YAe62. They introduced the double mutations, Ser94Phe and Ser95Trp, into CDR3β of B3K506 and showed that these mutations led to the cross‐reactive recognition of a self‐peptide.22
Incidentally, we have not mentioned hydrophobic interactions so far. One of the reasons is that it is hard to theoretically estimate hydrophobic interactions; another is that the concept of hydrophobic interactions is still controversial. Most researchers may implicitly include the van der Waals interactions between non‐polar side chains, which form a hydrophobic core in the protein, in the hydrophobic interaction, besides the transfer of free energy from water to a non‐aqueous environment. In fact, we estimated the van der Waals interactions by counting the atomic contacts. It should be emphasized, however, that the FMO calculations described above highlighted the importance of aromatic residues in CDRs. The results suggested that the interaction between TCR and pMHC of YAe62 could not be sufficiently explained by only non‐specific interactions such as hydrophobic and van der Waals interactions, and that the aromatic residues significantly contributed to the binding affinity in the relatively small interface area, as shown by the FMO calculations. Since the interactions involving the aromatic residues are less specific than those involving hydrogen bonds, they presumably make the high level of cross‐reactivity of YAe62 (and 2W20) possible.
Disclosures
The authors declare no financial and commercial conflicts of interest.
Author contributions
YT designed the research study and analysed the data. YN YT and HT performed the MD simulations. HT performed the FMO calculations. YT wrote the manuscript. HW and HT revised the manuscript and supervised all of the research.
Supporting information
Figure. S1. Structural features of a T‐cell receptor–peptide–MHC (TCR–pMHC) complex and the positions of conserved TCR–MHC interactions on MHC.
Figure. S2. Interactions between T‐cell receptor (TCR) and peptide–MHC (pMHC) in crystal structures and molecular dynamics (MD) snapshots.
Figure. S3. Structural features of crystal structures and molecular dynamics (MD) snapshot structures at 200 ns.
Figure. S4. Hierarchical clustering dendrograms of CDR residues of T‐cell receptors in the free and complex states.
Figure. S5. CH‐π interactions of 94Phe and 95Trp in CDR3β of YAe62.
Figure. S6. Scattered (B3k506 in a) and concentrated (YAe62 in b) interaction networks, both in complex with 1Ab‐3K.
Table S1. Conserved T‐cell receptor–MHC (TCR‐MHC) interactions.
Table S2. The number of T‐cell receptor (TCR) atoms in contact with the peptide and classification of the TCR atoms based on the CDRs they belong to for 31 complexes.
Table S3. Interface areas in crystal structures and molecular dynamics (MD) snapshots
Table S4. Number of atomic contacts in the crystal structure and molecular dynamics (MD) snapshots
Table S5. The numbers of inter‐CDR loop atomic contacts in the molecular dynamics (MD) snapshots.
Table S6. Maintained, added, and lost hydrogen bonds between T‐cell receptor (TCR) and peptide/MHC during the molecular dynamics (MD) simulations
Table S7. CH‐π interactions and the dispersion (DI) and charge‐transfer (CT) interaction energies in CDR3β
Acknowledgements
Part of this research was carried out as activities of the FMO drug design consortium (FMODD), by using computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project (project ID: hp150160 and hp160103 for HT). We thank Y. Eguchi for advice on analysing the FMO data. This work was partially supported by JSPS Grant‐in‐Aid for Scientific Research (C) (Grant No. 16K00407 for HW and No. 25330349 for HT).
Contributor Information
Yuko Tsuchiya, Email: tsuchiya@protein.osaka-u.ac.jp.
Hiromichi Tsurui, Email: tsurui@juntendo.ac.jp.
References
- 1. Yin Y, Mariuzza RA. The multiple mechanisms of T cell receptor cross‐reactivity. Immunity 2009; 31:849–51. [DOI] [PubMed] [Google Scholar]
- 2. Mason D. A very high level of crossreactivity is an essential feature of the T‐cell receptor. Immunol Today 1998; 19:395–404. [DOI] [PubMed] [Google Scholar]
- 3. Petrova G, Ferrante A, Gorski J. Cross‐reactivity of T cells and its role in the immune system. Crit Rev Immunol 2012; 32:349–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C et al How the T cell repertoire becomes peptide and MHC specific. Cell 2005; 122:247–60. [DOI] [PubMed] [Google Scholar]
- 5. Stone JD, Harris DT, Kranz DM. TCR affinity for p/MHC formed by tumor antigens that are self‐proteins: impact on efficacy and toxicity. Curr Opin Immunol 2015; 33:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wucherpfennig KW, Call MJ, Deng L, Mariuzza R. Structural alterations in peptide‐MHC recognition by self‐reactive T cell receptors. Curr Opin Immunol 2009; 21:590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L et al Structural basis of plasticity in T cell receptor recognition of a self peptide‐MHC antigen. Science 1998; 279:1166–72. [DOI] [PubMed] [Google Scholar]
- 8. Colf LA, Bankovich AJ, Hanick NA, Bowerman NA, Jones LL, Kranz DM et al How a single T cell receptor recognizes both self and foreign MHC. Cell 2007; 129:135–46. [DOI] [PubMed] [Google Scholar]
- 9. Bello M, Correa‐Basurto J. Energetic and flexibility properties captured by long molecular dynamics simulations of a membrane‐embedded pMHCII‐TCR complex. Mol BioSyst 2016; 12:1350–66. [DOI] [PubMed] [Google Scholar]
- 10. Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB et al Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell 2003; 12:1367–78. [DOI] [PubMed] [Google Scholar]
- 11. Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Two‐step binding mechanism for T‐cell receptor recognition of peptide MHC. Nature 2002; 418:552–6. [DOI] [PubMed] [Google Scholar]
- 12. Armstrong KM, Baker BM. A comprehensive calorimetric investigation of an entropically driven T cell receptor‐peptide/major histocompatibility complex interaction. Biophys J 2007; 93:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beringer DX, Kleijwegt FS, Wiede F, van der Slik AR, Loh KL, Petersen J et al T cell receptor reversed polarity recognition of a self‐antigen major histocompatibility complex. Nat Immunol 2015; 16:1153–61. [DOI] [PubMed] [Google Scholar]
- 14. Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol 2009; 10:143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gras S, Burrows SR, Turner SJ, Sewell AK, McCluskey J, Rossjohn J. A structural voyage toward an understanding of the MHC‐I‐restricted immune response: lessons learned and much to be learned. Immunol Rev 2012; 250:61–81. [DOI] [PubMed] [Google Scholar]
- 16. Armstrong KM, Insaidoo FK, Baker BM. Thermodynamics of T‐cell receptor‐peptide/MHC interactions: progress and opportunities. J Mol Recognit 2008; 21:275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dai S, Huseby ES, Rubtsova K, Scott‐Browne J, Crawford F, Macdonald WA et al Crossreactive T Cells spotlight the germline rules for αβ T cell‐receptor interactions with MHC molecules. Immunity 2008; 28:324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yin L, Huseby E, Scott‐Browne J, Rubtsova K, Pinilla C, Crawford F et al A single T cell receptor bound to major histocompatibility complex class I and class II glycoproteins reveals switchable TCR conformers. Immunity 2011; 35:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stadinski BD, Trenh P, Smith RL, Bautista B, Huseby PG, Li G et al A role for differential variable gene pairing in creating T cell receptors specific for unique major histocompatibility ligands. Immunity 2011; 35:694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stadinski BD, Trenh P, Duke B, Huseby PG, Li G, Stern LJ et al Effect of CDR3 sequences and distal V gene residues in regulating TCR‐MHC contacts and ligand specificity. J Immunol 2014; 192:6071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huseby ES, Crawford F, White J, Marrack P, Kappler JW. Interface‐disrupting amino acids establish specificity between T cell receptors and complexes of major histocompatibility complex and peptide. Nat Immunol 2006; 7:1191–9. [DOI] [PubMed] [Google Scholar]
- 22. Stadinski BD, Shekhar K, Gomez‐Tourino I, Jung J, Sasaki K, Sewell AK et al Hydrophobic CDR3 residues promote the development of self‐reactive T cells. Nat Immunol 2016; 17:946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Connolly ML. Solvent‐accessible surfaces of proteins and nucleic acids. Science 1983; 221:709–13. [DOI] [PubMed] [Google Scholar]
- 24. Tsuchiya Y, Kinoshita K, Nakamura H. Analyses of homo‐oligomer interfaces of proteins from the complementarity of molecular surface, electrostatic potential and hydrophobicity. Protein Eng Des Sel 2006; 19:421–9. [DOI] [PubMed] [Google Scholar]
- 25. McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol 1994; 238:777–93. [DOI] [PubMed] [Google Scholar]
- 26. Páll S, Abraham MJ, Kutzner C, Hess B, Lindahl E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. Volume 8759 of the series Lecture Notes in Computer Science 2015: 3–27.
- 27. R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2015. [Google Scholar]
- 28. Nakano T, Kaminuma T, Sato T, Fukuzawa K, Akiyama Y, Uebayasi M et al Fragment molecular orbital method: use of approximate electrostatic potential. Chem Phys Lett 2002; 351:475–80. [Google Scholar]
- 29. Kitaura K, Morokuma K. A new energy decomposition scheme for molecular interactions within the Hartree–Fock approximation. Int J Quant Chem 1976; 10:325–40. [Google Scholar]
- 30. Chemical Computing Group Inc . Molecular Operating Environment (MOE). 2017.
- 31. Nakano T, Mochizuki Y. ABINIT‐MP 5.0 and BioStation Viewer 13.01 (MIZUHO Rev.). Research and Development of Innovative Simulation Software 2011.
- 32. Umezawa Y, Nishio M. CH/pi interactions as demonstrated in the crystal structure of guanine‐nucleotide binding proteins, Src homology‐2 domains and human growth hormone in complex with their specific ligands. Bioorg Med Chem 1998; 6:493–504. [DOI] [PubMed] [Google Scholar]
- 33. Liu X, Dai S, Crawford F, Fruge R, Marrack P, Kappler J. Alternate interactions define the binding of peptides to the MHC molecule IA(b). Proc Natl Acad Sci U S A 2002; 99:8820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sethi DK, Schubert DA, Anders AK, Heroux A, Bonsor DA, Thomas CP et al A highly tilted binding mode by a self‐reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med 2011; 208:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sethi DK, Gordo S, Schubert DA, Wucherpfennig KW. Crossreactivity of a human autoimmune TCR is dominated by a single TCR loop. Nat Commun 2013; 4:2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bulek AM, Cole DK, Skowera A, Dolton G, Gras S, Madura F et al Structural basis for the killing of human beta cells by CD8+ T cells in type 1 diabetes. Nat Immunol 2012; 13:283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J et al Deconstructing the peptide‐MHC specificity of T cell recognition. Cell 2014; 157:1073–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cole DK, Bulek AM, Dolton G, Schauenberg AJ, Szomolay B, Rittase W et al Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross‐reactivity. J Clin Invest 2016; 126:2191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ehrenmann F, Lefranc MP. IMGT/3Dstructure‐DB: querying the IMGT database for 3D structures in immunology and immunoinformatics (IG or antibodies, TR, MH, RPI, and FPIA). Cold Spring Harb Protoc 2011; 2011:750–61. [DOI] [PubMed] [Google Scholar]
- 40. North B, Lehmann A, Dunbrack RL Jr. A new clustering of antibody CDR loop conformations. J Mol Biol 2011; 406:228–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure. S1. Structural features of a T‐cell receptor–peptide–MHC (TCR–pMHC) complex and the positions of conserved TCR–MHC interactions on MHC.
Figure. S2. Interactions between T‐cell receptor (TCR) and peptide–MHC (pMHC) in crystal structures and molecular dynamics (MD) snapshots.
Figure. S3. Structural features of crystal structures and molecular dynamics (MD) snapshot structures at 200 ns.
Figure. S4. Hierarchical clustering dendrograms of CDR residues of T‐cell receptors in the free and complex states.
Figure. S5. CH‐π interactions of 94Phe and 95Trp in CDR3β of YAe62.
Figure. S6. Scattered (B3k506 in a) and concentrated (YAe62 in b) interaction networks, both in complex with 1Ab‐3K.
Table S1. Conserved T‐cell receptor–MHC (TCR‐MHC) interactions.
Table S2. The number of T‐cell receptor (TCR) atoms in contact with the peptide and classification of the TCR atoms based on the CDRs they belong to for 31 complexes.
Table S3. Interface areas in crystal structures and molecular dynamics (MD) snapshots
Table S4. Number of atomic contacts in the crystal structure and molecular dynamics (MD) snapshots
Table S5. The numbers of inter‐CDR loop atomic contacts in the molecular dynamics (MD) snapshots.
Table S6. Maintained, added, and lost hydrogen bonds between T‐cell receptor (TCR) and peptide/MHC during the molecular dynamics (MD) simulations
Table S7. CH‐π interactions and the dispersion (DI) and charge‐transfer (CT) interaction energies in CDR3β