

Summary
Genes encoding antigen‐presenting molecules within the human major histocompatibility complex (MHC) account for the highest component of genetic risk for many neurological diseases, such as multiple sclerosis, neuromyelitis optica, Parkinson's disease, Alzheimer's disease, schizophrenia, myasthenia gravis and amyotrophic lateral sclerosis. Myriad genetic, immunological and environmental factors may contribute to an individual's susceptibility to neurological disease. Here, we review and discuss the decades long research on the influence of genetic variation at the MHC locus and the role of immunogenetic killer cell immunoglobulin‐like receptor (KIR) loci in neurological diseases, including multiple sclerosis, neuromyelitis optica, Parkinson's disease, Alzheimer's disease, schizophrenia, myasthenia gravis and amyotrophic lateral sclerosis. The findings of immunogenetic association studies are consistent with a polygenic model of inheritance in the heterogeneous and multifactorial nature of complex traits in various neurological diseases. Future investigation is highly recommended to evaluate both coding and non‐coding variation in immunogenetic loci using high‐throughput high‐resolution next‐generation sequencing technologies in diverse ethnic groups to fully appreciate their role in neurological diseases.
Keywords: immunogenetics, neurological disease
Introduction
Rare or common neurological diseases are associated with an impaired central nervous system (CNS) and/or peripheral nervous system (PNS). Neurological disease impacts approximately 1 billion individuals around the world, comprising individuals of all age groups and races, in diverse geographical locations and with diverse socio‐economic status.1 Together, neurological diseases represent 7·1% of the total global burden of disease, evaluated in disability‐adjusted life years, across all causes and ages.2
Despite significant progress in the management of many neurological diseases, we are still lacking complete and coherent models of pathogenesis, and as a result the repertoire of available therapies is imperfect. Recent advances in genomic sciences have set in place the foundation for understanding and decoding the rules of inheritable risk for chronic neurological diseases, which may translate into improved prognosis of outcomes and new therapeutic options. The genetic signals associated with susceptibility to the majority of neurological diseases remains inadequately explained due to the heterogeneous and multifactorial nature of these complex traits.3 Myriad genetic, immunological and environmental factors may contribute to an individual's susceptibility to neurological disease. However, the clear implication of genetic factors in the causation of many neurological diseases has been gleaned from heritability studies in families, twins and adopted individuals.3 Familial studies have reported increased incidence of several neurological diseases in offspring or siblings of affected individuals, twin studies have reported higher disease concordance in monozygotic than in dizygotic twins and studies in adopted individuals have suggested a high disease concordance in monozygotic twins raised in diverse environments.
Genes encoding antigen‐presenting molecules within the human major histocompatibility complex (MHC) account for the highest component of the genetic risk for many neurological diseases. Risk or protection for a variety of neurological diseases, including multiple sclerosis, neuromyelitis optica, Parkinson's disease, Alzheimer's disease, schizophrenia, myasthenia gravis and amyotrophic lateral sclerosis, has been mapped to this region (Fig. 1). However, the precise mechanisms underpinning these effects in these neurological diseases remain elusive. Here, we aim to review and discuss the decades long research on the influence of genetic variation at immunogenetic loci in neurological disease. We focus primarily on the well‐studied loci of the MHC, and later turn our attention to the killer immunoglobulin‐like receptor (KIR) complex, whose protein products functionally interact with some loci of the MHC.
Figure 1.
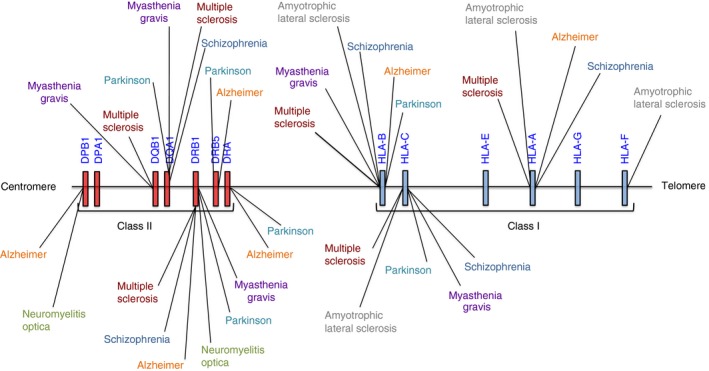
Genomic discovery in neurological disease mapped to the extended MHC region on chromosome 6.
The human major histocompatibility complex region and neurological disease
The balance of innate and adaptive immunity is now appreciated as an important component in the determination of neurological disease outcome. The molecules encoded by the major histocompatibility complex (MHC) regions regulate the innate and adaptive arms of human immune response through antigen presentation, inflammation regulation and the complement system and the impact of this region in various immune‐mediated conditions, including neurological diseases, has long been recognized.4, 5, 6, 7, 8, 9 The human MHC gene family maps to chromosome 6. With a size of nearly 5 Mbp it encodes approximately 165 protein‐coding genes, many of them immune‐related,10 and comprises approximately 0·13% of the human genome.9 After the first discovery of the mouse MHC in 193611 the human equivalent, the human leucocyte antigen (HLA), was subsequently mapped and extensively studied for both gene content and allelic variation. Thus, the HLA region became the most investigated region in vertebrate genomes. This region is considered the densest region of the human genome and with the effort of the MHC sequencing consortium the complete sequence and gene map of this region was first generated in 1999.12
The HLA region is characterized by an extreme level of polymorphism and extensive patterns of linkage disequilibrium (LD), which varies among populations. The genes of this region are divided into five subregions: (i) the extended class I, (ii) class I, (iii) class III, (iv) class II and (v) extended class II regions.9 The extended MHC region comprises greater than 400 annotated genes and pseudogenes.10 The HLA class I region consists of three classical loci, HLA‐A, HLA‐B and HLA‐C, along with three non‐classical loci: HLA‐G, HLA‐E and HLA‐F. The non‐classical HLA class I molecules are characterized by a more limited degree of polymorphism compared to the their classical counterparts.9 HLA class I molecules are expressed on all nucleated cells and their main function is presentation of non‐self antigens originated from intracellular sources to cytotoxic (CD8+) T‐cells for killing of the antigen‐presenting cells (APCs).13 Similarly, the class II region of HLA comprises three classical loci, HLA‐DP, HLA‐DQ and HLA‐DR, along with two non‐classical loci, HLA‐DO and HLA‐DM.9 The genes of classical HLA class II loci are expressed on the surface of professional APCs, which generally present antigens of extracellular origin,14 such as those derived from food (metabolites) or bacteria for the presentation to helper (CD4+) T‐cells. The HLA class III region consists of inflammatory regulatory genes, such as complement (C2, C4, CFB), cytokine genes (e.g. TNF, LTA, LTB) and other genes with non‐immune or unknown functions.9
The CNS is considered an immune privileged site, and it was long considered that typical neurons did not express HLA class I. However, this notion was rejected following the detection of HLA class I mRNA and/or protein expression in various neuronal populations, comprising motor nuclei, substantia nigra pars compacta,15, 16 dorsal root ganglia neurons,17 dopaminergic nigral cells,18 developing and adult hippocampal pyramidal cells,19, 20 sensory neurons of the vomeronasal organ,21, 22 brainstem,15, 18 and spinal,15, 23 motor neurons and cortical pyramidal cells.16, 20 More recently, a direct link has been established for HLA class I in functional and structural synapse pruning in the CNS.24, 25 Further, the capacity of microglia, the brain's resident macrophage, to present antigen through the class II MHC to T‐cells permits these typically quiescent cells to perform an important role in determining the clinical outcome of various neurological diseases. The roles of microglia in several neurological diseases are well documented.26, 27, 28
Taken together, the HLA loci are vital for shaping cellular adaptive immune responsiveness, and their impact upon human health and disease has long been appreciated. During the course of four decades, the impact of variation in HLA has been studied with respect to neurological disease. A time‐line of the crucial findings of MHC and KIR loci in relevance to neurological diseases are presented in Fig. 2. In the following sections, we discuss the role of HLA class I and II molecules in these diverse and often debilitating diseases (Table 1).
Figure 2.
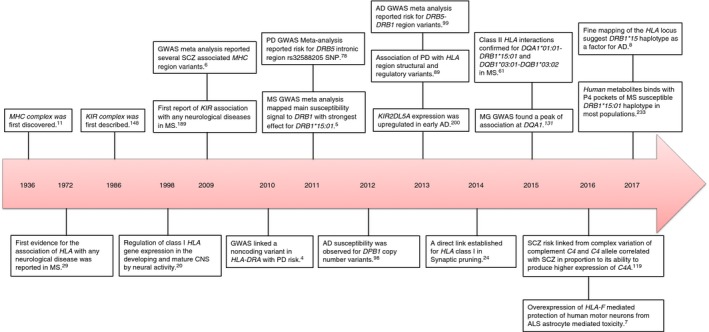
Time‐line of the crucial findings in the immunogenetics of neurological disease. CNS, central nervous system; MS, multiple sclerosis; NMO, neuromyelitis optica; PD, Parkinson's disease; AD, Alzheimer's disease; SCZ, schizophrenia; MG, myasthenia gravis; and ALS, amyotrophic lateral sclerosis.
Table 1.
Summary of HLA class I and II associated susceptible or protective alleles in neurological diseases
| Neurological diseases | MHC class II | MHC class I | References | ||
|---|---|---|---|---|---|
| Predisposing | Protective | Predisposing | Protective | ||
| Multiple sclerosis | DRB1*15:01, DRB1*15, DRB1*08:01, DRB1*04:05, DRB1*03:01; DRB1*13:03; DRB1*13∼DQA1*05:01∼DQB1*03:01 | DRB1*14:01, DRB1*11, DRB1*13‐DQB1*06:03, DQA1*01:01‐DRB1*15:01, DQB1*03:01‐ DQB1*03:02 | A*03, *0301; B*07 | A*02:01; B*44:02, *44, *38:01, *55:01; C*07, *05 | 5, 29, 30, 35, 36, 37, 38, 39, 40, 41, 50, 51, 52, 53, 54, 55, 57, 58, 59, 60, 61, 62, 63, 64 |
| Neuromyelitis optica | DPB1*05:01, DPB1*03:01, DRB1*12, DRB1*16:02, DRB1*03 | DRB1*09:01 | – | – | 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76 |
| Parkinson | DRA, DRB5, DRB1, DRB1*04, DRB1*04:03, DRB1*03, DRB1*03:01 | DRB1*04:06, DRB1*04:04, DQA1*03:01 | B*07:02, *17, *18; C*07:02 | C*03:04 | 4, 77, 78, 84, 86, 87, 89 |
| Alzheimer's | DR1, DR2, DR3, DRB1*03, DPB1, DRB5‐DRB1, DRA | DR4, DR6 | A*02 | B*07:02, A*03:01 | 8, 94, 95, 96, 97, 98, 99, 100, 101, 104 |
| Schizophrenia | DRB1*01:01, DRB1*03:01:01, DRB1*03:01:02, DQA1 | DRB1*03:01, DRB1*04, DRB1*06 | B*08:01, C*01:02 | A*03, *011, *02; B*27, *51 | 107, 108, 111, 114, 117, 118 |
| Myasthenia gravis | DQB1*05:02, DRB1*03, DRB1*04, DQB1*02, DQB1*03, DRB1*09, DRB1*15:01, DQB1*05:02, DRB1*16, DQA1*03:02/DQB1*03:03:02 | DRB1*08, DRB1*13:01, DQA1*05:01 | B*08, C*07:01 | – | 63, 121, 122, 123, 124, 125, 130, 132, 133, 138 |
| Amyotrophic lateral sclerosis | – | – | A*03, A*02, A*28; B*40, B*35, C*04 | A*09, HLA‐F | 7, 140, 141, 142, 145, 146 |
HLA and multiple sclerosis
The neurological disease most clearly and consistently associated with variation in the HLA region is multiple sclerosis (MS). The first evidence for the association of HLA class I antigens with MS was published in 1972,29 with risk for MS initially reported to be associated with HLA‐A*03 and HLA‐B*07 29, 30 on the basis of their serological specificity.29, 30, 31, 32 It later became apparent that these class I alleles were part of an extended class I and class II haplotype, associated with the serological determinant Dw2,33 later renamed DR2.34 The advancement in HLA genotyping approaches and continuous investigation of this region in MS ultimately revealed that the DR2 specificity has two distinctive molecular allotypes, DR*15 and DR*16, and the correlation with MS was pinpointed to DRB1*15:01,35 a subtype of DRB1*15.36 In illustration of the strength and consistency of this association in individuals with European ancestry, a Human Genome Epidemiology (HuGE) report reviewing 72 published studies from 1993 to 2004 observed a significantly increased frequency of DRB1*15:01 among MS patients in the vast majority.37 Reports of non‐association of DRB1*15:01 with MS, in almost every instance, was performed in cohorts of non‐European ancestry. Recently, GWAS performed in collaboration with the International Multiple Sclerosis Genetics Consortium (IMSGC) and the Wellcome Trust Case Control Consortium 2 (WTCCC2) project confirmed that the main susceptibility signal for MS maps to the DRB1 in the class II region of the MHC, and describes up to 10·5% of the genetic variance underlying risk.5 DRB1*15:01 revealed the strongest effect with an average odds ratio (OR) of 3·08, and all additional DRB1 associations emerge to describe less than 2% of the residual variance.5
Similar to other autoimmune diseases, DRB1*15:01 in MS susceptibility adheres to an additive model in a dose‐dependent manner with zero, one or two copies of the causal allele accounting for increased risk, respectively.38, 39 Along with the augmented risks for DRB1*15:01 homozygous genotypes, an epistatic effect for MS risk has been reported for carriers of the DRB1*15:01/*08:01 heterozygous genotype, with an augmented risk compared to other heterozygous DRB1*15:01 genotypes,40 while DRB1*08:01 alone was not observed to be a risk allele. However, a report in an Ashkenazi Jewish cohort suggested an independent association of DRB1*08:01 when considering clinical subgroups, with a weak significant signal observed only in primary progressive patients.41 Moreover, DRB1*15:01 is the most consistently reported MS susceptibility marker of disease severity. An effect of age and gender along with DRB1*15:01 has also been reported, and it was suggested that female MS patients carrying the DRB1*15:01 haplotype have an earlier age of disease onset.42, 43 In an attempt to correlate DRB1*15:01 with disease progression or severity, this allele was associated with the existence of oligoclonal bands and increased immunoglobulin (Ig)G levels in the cerebrospinal fluid (CSF) of MS cases.44, 45 In contrast, there has been no consistent reporting of other MS predisposing DRB1 alleles with respect to disease progression or severity.38, 46
DRB1*15:01 is most often observed in European populations as a segment of an extended haplotype with DQA1*01:02 and DQB1*06:02, and therefore it has been considered challenging to discriminate the main casual allele or locus. Imputation of classical HLA alleles from single nucleotide polymorphism (SNP) data demonstrated DRB1 as a primary risk locus in Europeans, and revealed that the majority of the effect attributed to DQB1*06:02 can be elucidated primarily by association with DRB1*15:01.47 As the HLA region displays varied patterns of linkage disequilibrium between populations, cross‐population analysis can be explanatory in unravelling the predisposing locus from a multilocus association. Examination of the African American MS cohort indicated risk to be strongly attributable to DRB1*15.48 In the same study, the evaluation of alternate DQB1*06:02 haplotypes without DRB1*15 suggested no difference between cases and controls, eliminating DQB1*06:02 as the primary allele of the association signal.48 This observation has been strengthened by a study in a population from Martinique with African ancestry.49
Non‐European studies suggested a correlation of DRB1*15 and DRB1*04:05 with MS in Japanese50 and Asian populations, respectively.50, 51 The same studies have reported the association of DRB1*04:05 with a clinically diverse disease course described by earlier onset age, decreased severity50 and a lack of brain lesions.51 Similarly, in Europeans the detection of a correlation of DRB1*04:05 with MS in Sardinian,52, 53 Sicilian53 and African American54 populations provided a coherent model for DR4 with MS aetiology. Additionally, the correlation of DRB1*03:01 and DRB1*13:03 was first detected in Sardinian55 and Israeli Ashkenazi and non‐Ashkenazi Jewish MS41 patients, respectively, but this allele is extremely common throughout Europe, Africa and Asia. In contrast to DRB1*03:01, DRB1*13:03 has been rarely found at population frequencies higher than 3% worldwide, but the correlation with MS shows effect sizes.56 A study in Canadian multiplex MS families found over‐transmission of the DRB1*13˜DQA1*05:01˜DQB1*03:01 haplotype;57 however, this study did not evaluate the DRB1 locus at high resolution. This haplotype is almost constantly linked with the DRB1*13:03 allele in Europeans, whereas the other common subtypes of DRB1*13 allele, such as of DRB1*13:01 and DRB1*13:02, are usually located on other DQA1˜DQB1 haplotypes; these additional DR*13 haplotypes were not observed to be over‐transmitted in the Canadian study.
Protective effects for HLA have also been observed in some studies. The protective effect of HLA class II alleles was seen for DRB1*14:01 in the European MS cohort,39, 40 while DRB1*11 was protective in both a Brazilian58 and a Canadian MS cohort.59 Similarly, the DRB1*13˜DQB1*06:03 haplotype was protective in Finnish60 and Canadian MS families.57 Another study confirmed interactions involving pairs of HLA class II alleles: DQA1*01:01‐ DRB1*15:01 and DQB1*03:01‐ DQB1*03:02, with a protective effect in MS.61 In the same study, HLA class I‐mediated protection has also been observed for HLA‐A*02:01, HLA‐B*44:02, HLA‐B*38:01 and HLA‐B*55:01.61 More recently, HLA‐B*44‐mediated protection was also reported in MS.62 Similarly, an imputation study revealed protection by the high‐resolution HLA‐B*44:02 genotype in MS.63 Finally, HLA‐B*44:02 was observed in LD with HLA‐C*05, which independently demonstrated protection for MS in the absence of DRB1 risk alleles;64 thus, it is challenging to discriminate whether these observations reflect a single effect through strong LD.
HLA and neuromyelitis optica
Neuromyelitis optica (NMO) was initially considered a variant of MS, but the identification of antibodies for aquaporin 4 (AQP4) or NMO IgG considerably transformed clinical discernment of the disease as an independent entity.65 This led investigators to evaluate the potential role of the HLA region in the aetiology of NMO. Numerous reports in Japanese populations, where the prevalence of NMO is higher than in European populations, suggested DPB1*05:01 as a predisposing allele and DRB1*09:01 with a protective effect.66, 67, 68 These results were confirmed in a replication study on a southern Han Chinese NMO cohort.69 Later, additional HLA class II alleles were found to be predisposing in NMO, including DPB1*03:01,70 DRB1*12 71 and DRB1*16:02.68, 69 Meanwhile, in contrast to Asian populations,69 DPB1*05:01 revealed no association with NMO in a French population, but DRB1*03 was shown to be a susceptibility marker.66, 67, 72 Similarly, DRB1*03 has been also observed with increased risks for NMO among Brazilian mulattos,73 Afro‐Caribbeans74 and Mexican Mestizos,75 but not in Muslim Arabs.76 It is important to consider that as DRB1*03 is comparatively less frequent and DPB1*05:01 is more frequent in Asian population, there may be inadequate power to detect the risk for DRB1*03 in Asian populations and DPB1*05:01 in European populations, yielding these varied results.
HLA and Parkinson's disease
The association of HLA with Parkinson's disease (PD) was first reported more than 4 decades ago.77 This study reported an increased risk for PD attributable to the HLA‐B*17 and ‐B*18 antigens.77 However, subsequent studies failed to replicate the association of HLA class I antigens with PD.4, 78 Genomewide association studies (GWAS) provided a new angle for investigating common complex traits such as PD.4, 78 Evaluation of more than a million SNPs in large sample sizes considerably enhanced the statistical power of the associations. Breakthrough discoveries made by two GWAS recognized SNPs in the HLA‐DR region to be associated with PD, confirming the immune component in pathogenesis of PD.4, 78 Hamza et al.4 suggested the association of rs3129882, a non‐coding variant in HLA‐DRA with PD in Americans of European ancestry, while a large‐scale imputation‐based approach applied in a meta‐analysis of five GWAS with data generated from US and European cohorts identified chr6: rs32588205 A/G SNP located in the intronic region of HLA‐DRB5 locus with augmented risk for PD under an additive model.78 Because the HLA‐DRA locus is mostly monomorphic and less often identified in HLA disease association studies, attempts to replicate this observation have generated varied and conflicting results.79, 80, 81, 82, 83, 84, 85 Similarly, the HLA‐DRB5 locus is in strong LD with HLA‐DRB1 and only present in approximately 20% of the population, and thus this association has also been challenging to decipher.
In an attempt to replicate the finding of a GWAS‐reported association of rs3129882 in HLA‐DRA locus, two studies in different populations used a candidate gene approach and confirmed the association of rs3129882 variants with increased risks of PD. The first was conducted in a small cohort of 284 Chinese Han cases and 258 controls from Mainland China,79 and the second in 520 Iranian cases and 520 controls.80 Meanwhile, a meta‐analysis of five case–control studies with a total of 2230 PD cases and 2262 controls from Mainland China, Taiwan, Singapore and Malaysia reported no association of HLA‐DRA rs3129882 variants with PD.85 Subsequently, a study performed in three different European‐ancestry cases and controls from the United States, Ireland and Poland in a comparatively larger cohort of 1313 cases and 1305 controls observed no association of rs3129882 variants with PD under an additive or dominant model.81 However, the same study found a protective effect of GG genotype in the Irish, Polish and combined cohorta under a recessive model.81 Finally, no association with rs3129882 was reported in two GWAS: the first conducted in a relatively homogeneous Ashkenazi Jewish (AJ) population from New York comprised of 2050 cases and 1836 controls;82 and the second in the largest single PD GWAS cohort of 3400 cases and 29 000 controls.83
A French case–control PD study revealed an association of rs660895 within the HLA‐DRB1 locus, which is significantly more polymorphic than HLA‐DRA and, unlike HLA‐DRB5, present in all individuals and is frequently associated with disease.84This study used an imputation approach to infer HLA alleles from SNP data, and suggested a protective effect for DRB1*04.84 Another study reported an association of DRB1*03 with increased risks to PD in individuals with European ancestry.86 Subsequently, these results were confirmed in another study performed on 567 PD Han Chinese patients and 746 controls from Guangdong province of the China, and suggested the strongest association for PD causation with DRB1*03:01, the most common subtype of DRB1*03 allele.87 Meanwhile, the same study has also found a decreased frequency of DRB1*04:06 in PD cases compared to controls, suggesting a protective effect.87 Interestingly, the DRB1*04:06 allele, a subtype of DRB1*04, is rare in European populations; however, it is common in Asian populations (http://www.allelefrequencies.net).88 DRB1*04:03, another subtype of DRB1*04, has been reported to be more frequent among PD cases in Han Chinese.87 Whether DRB1*04:06 displays a susceptible effect in European ancestry populations still needs to be evaluated.
A more recent and large study implicated structural and regulatory variants in the HLA region.89 This study suggested that rs3129882 located in intron‐1 and the closely linked rs9268515 and rs2395163 SNPs positioned in intergenic region remained significant regardless of HLA alleles.89 Further, this study used an imputation approach and suggested an increased risk for B*07:02 ˜ C*07:02 ˜ DRB5*01 ˜ DRB1*15:01 ˜ DQA1*01:02 ˜ DQB1*06:02 haplotype and a protective effect for C*03:04, DRB1*04:04 and DQA1*03:01 alleles.89 However, when they conditioned on the associated SNPs, only C*03:04 and DRB1*04:04 alleles remained significant.89 Finally, this study concluded that rs3129882 and rs2395163 SNPs are in expression quantitative trait loci (eQTLs) for HLA‐DR and HLA‐DQ, and suggested that HLA gene expression might impact PD.89
HLA and Alzheimer's disease
Like MS and PD, the first report of a role for HLA in Alzheimer's disease (AD) was published in the 1970s.90 Since then, multiple studies have evaluated the role of HLA class I91, 92, 93, 94 and class II genes in AD.95, 96, 97, 98, 99 The early findings of an association with HLA‐A*02 in AD were inconsistently replicated. While some studies confirmed a role for this antigen,94, 100 others failed to replicate any association.91 Most of these studies suffered from small sample sizes, but two most well‐powered studies failed to find any association of HLA‐A*02 with AD.92, 93 More recently, a trend for association of SNPs in the HLA‐A locus with atrophy of brain structures has been reported, although the corrected P‐values in this study would be considered marginal.94
The role of HLA class II antigens has also been investigated in the pathophysiology of AD. Curran et al.95 showed that DR1, DR2 and DR3 antigens, in the absence of apolipoprotein E (APOE) risk alleles, are associated with an increased risk for developing late‐onset AD, whereas DR4 or DR6 antigens appear to be associated with a decreased risk of AD. Aisen et al.96 suggested that DR4 might exert a protective influence in AD via modulation of glial activity. A later study also showed that risk for AD in older late‐onset cases is associated with DRB1*03 in APOE4‐negative individuals.97 However, as is the case for HLA‐A*02, these studies have suffered from small sample sizes.
Large‐scale GWAS have also provided evidence for the involvement of HLA class II in AD. Analysis of genomewide copy number variation (CNV) suggested a susceptible association for DPB1 in AD.98 A subsequent GWAS meta‐analysis in 17 008 late‐onset cases and 37 154 controls with European ancestry identified a SNP from the DRB5–DRB1 region to be associated with late‐onset AD risk.99 The association of this SNP was replicated in a Chinese cohort,101 and further work showed that this SNP is associated with cis‐gene expression levels of DRB1 in the temporal cortex and cerebellum.102 Nettiksimmons et al.103 showed an association of the DRB5–DRB1 clusters with cognitive decline at the gene‐level, and more recently methylation of DRB5 in the brain was associated with pathological AD, with another peak of association in DRA.104 Yokoyama et al.105 determined that variants associated with autoimmune disease are also associated with AD and found that a SNP close to DRB5 is associated with AD and psoriasis. The authors also showed that although DRB5 transcript expression is not altered in AD brains, there is an increase in transcript expression for DRA in AD brains compared to control brains.
Finally, an HLA imputation‐based analysis of 5919 European‐ancestry AD Caucasian patients and 5771 controls identified the extended haplotype A*03:01˜B*07:02˜DRB1*15:01˜DQA1*01:02˜DQB1*06:02 as being associated with AD risk (P = 9·6 × 10−4, OR = 1·21)8 in individuals who are negative for APOE 4.8 The authors also found an association of the class I haplotype A*03:01~B*07:02, with higher CSF amyloid levels and a dose‐dependent association of the DR15 haplotype with greater rates of cognitive decline and baseline levels of chemokine CC‐4.8
HLA and schizophrenia
The first evidence for a probable role of HLA in schizophrenia (SCZ) was described in 1974.106 Thereafter, multiple linkage studies provided some evidence for a susceptibility locus on the short arm of chromosome 6.107, 108 These studies correlated numerous class I and class II alleles with SCZ.107, 108 However, subsequent studies failed to replicate these initial findings.107, 108
More recently, publication of the first GWAS and meta‐analysis in SCZ made possible the study of the HLA region at higher resolution.6 A meta‐analysis of three GWAS identified several MHC region variants associated with SCZ in individuals with European ancestry.6, 108, 109, 110 Some of these were consistently replicated or found in other populations.108, 111, 112, 113 However, it is interesting to note that most of the significant variants correlated with SCZ in the meta‐analysis were located in the extended MHC regions, near a cluster of histone genes comprising a position upstream of the class I region, along with a few additional immune genes such as ribonuclease P21 (RPP21) located in class I region and neurogenic locus notch homologue 4 (NOTCH4) located in the extended class II region.109
Subsequently, GWAS analysis performed in SCZ cases from Asia replicated the findings of the European GWAS, and additionally recognized a few novel variants in Chinese112 and Japanese populations.113 Similarly, the rs9272219 and rs9272535 variants in the DQA1 gene revealed a moderate association with SCZ.111 An HLA imputation study showed an association of the risk allele HLA‐C*01:02 in addition to trends for association of the protective alleles DRB1*03:01 and B*08:01.114 A GWAS in Ashkenazi Jews showed supportive evidence for association of the HLA region with SCZ in this population.115 Finally, an eQTL study strengthened these results by providing evidence that the TRIMP26, RNF5 and DRB3 genes, located within the MHC region, are regulated by the top SNPs recognized by meta‐analysis of GWAS data.116 In addition to GWAS results, previous reports suggested an increased frequency of DRB1*01:01 and a decreased frequency of DRB1*04 among SCZ patients.117, 118 However, it is important to note that all HLA association studies performed in SCZ to date have either used low‐resolution genotyping methods or GWAS/SNP imputation approaches. The lack of consistent findings suggest that high‐resolution HLA genotyping approaches will be required to fully appreciate the role of HLA variants in SCZ.
More recently, a well‐powered study associated SCZ risk with complex variation in complement component 4 (C4) genes, also located within the MHC.119 This study found that C4 alleles produced extensively varying levels of C4A and C4B expression in the brain, with each common C4 allele correlating with SCZ in proportion to its ability to produce higher expression of C4A.119 The findings of this study highlight the role of complement genes in pathophysiology of SCZ, and these observations open new frontiers for future investigations of genetic variation in complement genes with SCZ in other ethnic groups in the quest to find a coherent model for SCZ.
HLA and myasthenia gravis
The first report of an HLA association with myasthenia gravis (MG) was published in 1976.120 Thereafter, several studies have reported evidence of association of HLA antigens/alleles with MG. An Italian study identified DQB1*05:02 as being associated with MG,121 while a Tunisian study identified the DRB1*03, DRB1*04, DQB1*02 and DQB1*03 alleles as possible predisposing factors for MG.122 DRB1*03 was then subsequently found to be associated with MG in a Portuguese study.123 Meanwhile, in a northern Han Chinese population, DRB1*09 was associated with risk of MG, while DRB1*08 was protective.124 A GWAS published in 2012 on North Europeans identified the class I SNP rs7750641 as the strongest signal in MG, and further imputation analysis identified HLA‐B*08 as being the major risk allele.125 Similarly, an imputation study observed a risk association for HLA‐C*07:01 with MG.63 There is strong LD between HLA‐C*07:01 and HLA‐B*08, but the latter revealed a marginally weaker association than HLA‐C*07:01 in the same study.63
Examinations of age of onset effects of HLA in MG have yielded mixed results. Although multiple studies have reported the extended HLA haplotype, namely A1‐B8‐DR3‐DQ2, as being associated specifically with early onset of MG (EOMG) in individuals with European ancestry, it is unclear whether the signal maps in class I or class II genes.126, 127, 128 Interestingly, the A allele of the SNP rs1800629 at position 308 nucleotides upstream from the transcription initiation site of tumour necrosis factor‐alpha (TNF‐α) has been linked to higher expression level and higher serum levels of TNF‐α in MG by several studies, and this SNP is known to be in LD with the HLA A1‐B8–DR3 haplotype.129 Confounding interpretation of these results, DRB1*13:01 was found to be protective for EOMG in a Norwegian population,130 while a GWAS performed in a European population by Renton et al.131 found a peak of association for EOMG at DQA1.
The Norwegian study identified DRB1*15:01 as being associated with the risk of late onset of MG (LOMG), while DRB1*13:01 was also found to be protective in LOMG.130 In an Italian cohort, DQB1*05:02 and DRB1*16 have been reported as being associated with LOMG.132 Renton et al.131 found a peak of association for LOMG at HLA‐DQA1, which was distinct from that observed in the same GWAS in EOMG. Another GWAS showed three distinct and largely independent association peaks for LOMG corresponding to MHC class II, HLA‐A and MHC class III SNPs, while imputation of HLA alleles showed a protective effect of DQA1*05:01.133
Additional studies have sought to elucidate an association of HLA with specific subtypes of MG. Four studies found an association of DQ5 with the specific subgroup of muscle‐specific kinase (MuSK) antibody‐positive (Ab+) MG patients.134, 135, 136, 137 A Turkish study also found that DRB1*14 and DRB1*16 were associated with this specific subgroup,136 whereas in a Serbian cohort, DRB1*13 seems to be completely absent in this specific patient population.137 It has been hypothesized that childhood‐onset ocular MG in southern Han Chinese may present a particular subgroup of distinct genetic background, correlating with the haplotype DQA1*03:02/DQB1*03:03:02.138 Later, the haplotype HLA‐B*46:01‐DRB1*09:01 was found to be associated with juvenile ocular MG in the same population.139 However, it is important to note that all studies involved cohorts of, at most, a few hundred individuals, making it difficult to fully elucidate the role of HLA in MG.
HLA and amyotrophic lateral sclerosis
A very limited number of genetic association studies have evaluated the HLA region in amyotrophic lateral sclerosis (ALS). During the 1980s, a few studies with low‐resolution genotyping sought to examine HLA in ALS.140, 141, 142 Initial studies found no correlation between HLA antigens and ALS in patients from California143 and Guam.144 Later, a significantly increased frequency of HLA‐A*03 was reported in an ALS cohort from the greater Boston area140 and Israel.141 Similarly, HLA‐A*02 and ‐A*28 have been shown to be more frequent in ALS cases recruited from Glasgow and Scotland,145 while an increased frequency of HLA‐B*40 was found in an ALS cohort from Finland.142 A study from the greater New York area observed HLA‐Bw35 and ‐Cw4 more frequently in ALS cases, and a trend towards decreased frequency was also found for HLA‐A*09.146 These initial findings were marked by substantial inconsistency in identification of a link between a particular HLA antigen and ALS across study populations, suggesting perhaps that HLA determinants may not play a major role in susceptibility to this diseases.
Thereafter, almost three decades passed without HLA association studies conducted in ALS. However, a recent study demonstrating that overexpression of a single non‐classical HLA class I molecule, HLA‐F, resulted in protection of human motor neurons from ALS astrocyte‐mediated toxicity, coupled with a role for the killer cell immunoglobulin‐like receptor KIR3DL2,7 clearly indicated an immune component in ALS pathogenesis. Finally, an association study published in 2017 in a Chinese Han population indicated a role for HLA class II in ALS.147 While inconclusive, these more recent investigations suggest an immunogenetic component to ALS, warranting further study.
The killer‐immunoglobulin‐like receptor (KIR) complex: a new horizon in the immunogenetics of neurological disease
The KIR complex was first defined in 1986,148 and was initially recognized as KIR inhibitory receptors. The family of the KIR proteins are mainly expressed on natural killer (NK) cells149 and a small percentage of T‐cells.150 The KIR complex maps on the long arm of human chromosome 19q13·4, and is considered as a crucial component of innate and adaptive immunity. Although KIR and HLA are members of two different gene families, the interaction of KIR with their cognate HLA class I ligands serves as a functional bridge in the regulation of NK cell functions and maintenance of immune homeostasis. KIR are inhibitory and stimulatory surface receptors that regulate NK cell function and responsiveness.151 All these receptors consist of either two (2D) or three (3D) extracellular immunoglobulin domains (D). The transmembrane and cytoplasmic domains govern the functional characteristics of these receptors. The inhibitory receptors consist of long (L) cytoplasmic tails comprising immunoreceptor tyrosine‐based inhibitory motifs (ITIMs), whereas stimulating receptors possess short (S) cytoplasmic tails and link to the stimulating adaptor DAP12 through a charged residue in the transmembrane domain. However, KIR2DL4 is an exception, and despite having a long cytoplasmic tail with an ITIM transmits a positive signal through its interaction with the stimulating adaptor FceR1γ.152, 153 Specific KIR molecules recognize one or more of four epitopes of HLA class I molecules. In contrast to the T‐cell receptor, KIR bind to the upper face of the HLA class I molecule, creating contact with the N‐terminal part of the α1 helix, the C‐terminal part of the α1 helix, and the bound peptide.154 Genetic variation in the class I α1 helix governs the three major epitopes perceived by KIR, HLA‐C1, ‐C2 and ‐Bw4. The inhibitory KIR2DL1 and KIR2DL2/3 and the stimulating KIR2DS1, KIR2DS2 and KIR2DS4 interact divergently with the reciprocally unique C1 or C2 epitopes carried by all HLA‐C allotypes and a small subset of HLA‐B molecules.155, 156, 157 KIR3DL2, KIR2DS2 and KIR2DS4 recognize a subset of HLA‐A allotypes transmitting the A*03/A*11 epitope (e.g. A*11:01).56 Finally, KIR3DL1/S1 binds subsets of HLA‐A and ‐B allotypes that carry the Bw4 epitope (e.g. A*24:02).56 In contrast, HLA‐B alleles with the Bw6 epitope do not bind with any KIRs. Adding further complexity, these receptor interactions are further tuned by allelic variations of KIR and HLA class I and by the sequence of the bound peptide.158, 159, 160, 161
The KIR gene complex exhibits extensive heterogeneity in gene content at both intra‐ and interpopulation levels. KIR haplotypes comprise from four to 14 genes and, based on their genomic structure, are divided into two groups, termed A and B.162 The group A haplotype is characterized by a single configuration of seven genes that express predominantly inhibitory KIR, and all remaining configurations are termed B haplotypes. As an indication of probable functional differences between them, B haplotypes typically express more activating KIR than A haplotypes.163, 164, 165 The haplotypes are formed from combinations of unique centromeric and telomeric gene‐content motifs, which also belong to the A or B groups. Although a huge number of unique haplotypes are described, a few comparatively common haplotypes repeatedly account for greater than 90% of the KIR haplotypic variation detected within a specific population, and are observed throughout major ethnic groups.166, 167 Our recent work and that of others has shown that the prevalence of KIR haplotypes and specific combinations of cognate KIR and HLA allotypes are associated in autoimmune162, 168, 169, 170 and infectious diseases such as human immunodeficiency virus (HIV) and hepatitis C,171, 172, 173, 174 cancer,175, 176 and are critical to the success of solid organ and haematopoietic stem cell transplant (HCT)177, 178, 179, 180 and pregnancy.181, 182, 183, 184, 185
Although the correlation of HLA variation with neurological disease has been well documented, there is a paucity of studies aiming to evaluate the impact of NK cells or their receptors, including KIR in these diseases. As HLA class I molecules function as the primary ligand for several KIRs, it is possible that the class I association signals perceived for various diseases is, in fact, related to KIR function. In various neurological diseases, such as MS,29, 30, 63, 186 myasthenia gravis,126, 127, 128 schizophrenia,107, 108, 114 Parkinson's disease,77, 89 Alzheimer's disease8, 94, 100 and amyotrophic lateral sclerosis,140, 141, 145, 146 the alleles of HLA‐A, ‐B and ‐C that are recognized to function as cognate ligands for their respective KIR genes have been linked with disease (Table 2). Here, it is important to note that the majority of the identified HLA class I association with various neurological diseases described used an imputation approach from data obtained through GWAS rather than direct assay. Meanwhile, a direct link of KIR allele variations with neurological diseases has not been observed in GWAS, very possibly because of a limited number of markers in the KIR region on all common available GWAS platforms. An insufficiency of appropriate reference alignments has traditionally impeded incorporation of KIR exclusive SNPs on the available GWAS platforms, and the large diversification of gene‐content in KIR haplotypes is characteristically discordant with standard quality thresholds. Finally, the Immunochip, which is exclusively enriched for markers in the KIR chromosomal region, predominantly recognizes non‐coding variants on the common group A haplotype that mainly comprises inhibitory KIRs.187 To date, therefore, the majority of described KIR correlations with immune diseases,188 including multiple sclerosis189, 190, 191 and schizophrenia,192 used approaches which determine only KIR gene content variation. KIR genotyping approaches that determine gene content are usually impotent to discriminate copy number, but rather assess only presence/absence. As copy number has repercussions on the immune reactions,193 this further hinders the capacity to detect any locus level associations with disease. Additionally, strong LD within gene content haplotypes166, 194 creates another hurdle in the determination of the causative locus.
Table 2.
HLA class I associations and putative KIR receptor involvement in neurological diseases
| Neurological disease | HLA class I associations | Potential KIR receptors | References |
|---|---|---|---|
| Multiple sclerosis | HLA‐B*07, *44, *44:02, *37:01, *38:01; HLA‐C*07, *05; HLA‐A*02:01, *03, *0301 | KIR3DL1S1; KIR2DL1, KIR2DL2/3, KIR2DS1, KIR2DS2, KIR2DS4, KIR3DL2 | 29, 30, 60, 61, 62, 63, 64 |
| Parkinson's disease | HLA‐B *17, *07:02, HLA‐C*07:02, *03:04 | KIR3DL1S1, KIR2DL2/3, KIR2DS2, KIR2DS4, KIR3DL2 | 77, 89 |
| Alzheimer's disease | HLA‐B*07:02, HLA‐A*03:01, *02 | KIR3DL2, KIR2DS4 | 8, 94, 100 |
| Schizophrenia | HLA‐B*27, *51; HLA‐C*01:02, HLA‐A*03, *011, *02 | KIR3DL1S1; KIR2DL2/3, KIR2DS2, KIR2DS4; KIR3DL2 | 107, 108, 114 |
| Myasthenia gravis | HLA‐B*08; HLA‐C*07:01 | KIR2DL2/3, KIR2DS2, KIR2DS4 | 63, 125 |
| Amyotrophic lateral sclerosis | HLA‐A*09, *02, *03, HLA‐C*04, HLA‐F | KIR3DL1S1, KIR2DL1, KIR2DS1, KIR2DS4, KIR3DL2 | 7, 140, 141, 145, 146 |
A limited number of studies has examined the association of KIR gene content variation to date with neurological diseases, the majority of them in multiple sclerosis189, 190, 191 and one in schizophrenia.192 A study conducted on a relatively small sample size of 200 schizophrenia patients and 561 controls in European Polish populations have found no correlation of either KIR gene frequency or KIR gene ligands with disease.192 There could be two probable reasons for the non‐association of KIR variation with SCZ. First, the genotyping method for KIR varied considerably, and the differential accuracy of genotyping approaches due to the strong homology between KIR gene or possible amplification biases contingent upon sample quality makes it difficult to calculate KIR gene frequencies precisely. The second limitation is that this study genotyped only for epitopes HLA‐A Bw4+, HLA‐B Bw4 Ile, Bw4 Thr, HLA‐C1 and ‐C2 but did not genotype for particular HLA‐A, ‐B and ‐C alleles, limiting the ability to analyse the interaction of KIR with specific HLA allotypes. Numerous reports of KIR gene content studies in MS from European populations have suggested a role for KIR loci in disease predisposition. Lack of the inhibitory KIR2DL3 has been suggested in MS susceptibility,195 implicating either KIR2DL2 (which segregates as the alternate allele of the same locus) or the closely associated KIR2DS2 in disease. Subsequently, a study has observed the increased incidence of KIR2DL5 and KIR3DS1 in MS cases compared to controls.196 Finally, two other reports in Portuguese and Italian MS cohorts determined a diverse telomeric locus, KIR2DS1, as protective.197, 198 Similarly, our study in an African American MS cohort revealed a strong protective effect for KIR3DL1 in combination with HLA‐A and ‐B alleles bearing the Bw4 motif.191 Finally, the up‐regulated expression of KIR2DL5A was observed in early Alzheimer's disease.199 Although these initial observations are encouraging, an extensive assessment of KIR allele‐level variation in a set of established and well‐characterized cohorts encompassing a wide range of neurological diseases in several different ethnicities, and their correlation with KIR expression, is needed to fully appreciate the role of these critical immune receptors in disease susceptibility and prognosis.
Notably, both immunoregulatory dysfunction and activated inflammatory mediator pathways have been suggested in the pathophysiology of neurological diseases, particularly PD,200,201 MS202 and MG,203 as well as many other neurological diseases.204 The reported disease association of HLA variations bolsters this notion. KIR, through the NK cell, regulates the production of cytokine and chemokines.205 As cytokine and chemokines regulate neuroinflammation,206 it remains a plausible hypothesis that KIR allelic variation may influence the course of various neurological diseases through neuroinflammatory pathways.
Accumulating evidence suggests a role for NK cells in various neurological diseases, such as MS,207, 208, 209 NMO,210 PD,211, 212 AD,213 SCZ,214 myasthenia gravis215 and ALS,216 strengthening the notion that KIR variation may be important in disease predisposition and/or development. NK cells are a key component of innate immunity and act as a first line of defence in resisting infections, but may also be involved in the induction of neurological diseases, and accumulate in specific neuronal cells or tissues in some diseases.215, 216, 217, 218, 219 In the MS murine model, experimental autoimmune encephalomyelitis (EAE), studies suggested a role for NK cells in down‐regulation of disease progression.207, 208, 209 In the meantime, enhanced predisposition and disease severity in EAE has been linked with NK cells in concurrence with individual cytokines.218, 219 Studies in humans suggested an immunoregulatory role for NK cells in MS, causing an abatement of the inflammatory pathways.220, 221 In contrast, in‐vitro studies demonstrated that NK cells can straightforwardly lyse neural tissue, and may consequently contribute to tissue injury in MS.222, 223 While the immunobiology of NK cells in certain neurological diseases such as MS has been explored, comparatively less is known about the specific role of NK cells in other neurological diseases, such as NMO, PD, AD, SCZ, MG and ALS. The results of NK cell studies in MS continue to be controversial, and fail to point to a coherent model. Thus, understanding the precise role of KIR variation in immunopathogenesis of neurological diseases may open new horizons for identification of biomarkers or could pave the way for new therapeutic approaches.
Consideration of HLA and KIR regulatory region variation in neurological disease
The emergence of next‐generation sequencing enhanced our ability to determine the HLA and KIR sequences at a very high‐resolution level. This provides the opportunity to determine the role of both coding and non‐coding HLA and KIR region variations in a variety of neurological diseases, given that the non‐coding regions of the human genome including HLA and KIR regions contain regulatory elements, such as promoters, enhancers and untranslated regions (UTRs); these are the strong candidate regions for pathogenic variation and participate directly in the determination of the abundance of expressed genes. In current laboratory practice almost all the reported HLA alleles, either in the disease association studies or in the databases, have used genotyping approaches that only sequenced through exons 2 (class I and class II) and 3 (class I), and this limits our ability to analyse HLA non‐coding variations. HLA non‐coding variations such as SNPs or small insertion/deletions (indels), as well as larger‐scale copy number variants (CNV) present in regulatory regions, could impact the course of neurological diseases through alteration of gene expression. Non‐coding variation in HLA has already been associated clearly with disease. For example, variation in the 3′UTR of HLA‐DPB1 is linked with spontaneous clearance of hepatitis B virus in both Japanese and US populations.224, 225 The proposed mechanism for enabling viral clearance might be linked to the rs9277534 A/G SNP, which describes HLA‐DP cell‐surface expression.225 Similarly, rs2281389 is a non‐coding region variant in HLA‐DP linked with acute graft‐versus‐host disease (GVHD).226 However, rs2281389 variants are not detectable through standard genotyping approaches. Finally, a promoter region SNP of HLA‐C has been reported to be linked with control of HIV infection, and cell surface expression of the HLA molecule was identified to be in LD with a 3′UTR variant that regulates binding of micro‐RNA, the putative source of the expression variation.227 Distorted patterns of gene expression are a characteristic of many neurological diseases, such as MS, Alzheimer's disease and schizophrenia, and in various cases these altered gene expressions can be correlated straightforwardly to genomic/pathogenic variations.228, 229, 230 These findings support the hypothesis of a robust association between anomalous gene expression and neurological diseases, suggesting that variants in non‐coding regulatory elements are outstanding candidates for some of the observed missing heritability in neurological diseases. The identification of many null or expression variants of common HLA alleles will improve our understanding of their role in immune functions. Recently, a variant of the multiple sclerosis‐linked allele HLA‐B*44:02 has been determined that produces only a soluble, rather than cell surface, molecule; a point mutation at the end of intron 4 alters the exon 5 splice site.231 Meanwhile, this variant is not appreciable through standard genotyping approaches, and hence these alleles are usually genotyped as B*44:02; similarly, the actual population‐level frequency of the marginal allele is not recognized. Moreover, if the non‐surface‐expressed variant of this allele is common, this might elucidate the link to disease. Taken together, these data recommend that the genotyping of regulatory regions variants may improve our understanding about the role of HLA and KIR in neurological disease.
Concluding remarks and future perspective
Taken together, the findings of HLA and KIR association studies are consistent with a polygenic model of inheritance in the heterogeneous and multifactorial nature of complex traits in various neurological diseases. The majority of the neurological diseases, such as MS, NMO, PD, AD, SCZ, MG and ALS, are considerably more common among individuals transmitting specific HLA alleles. This further strengthens the decades‐long contention of a strong immune component in the determination of clinical outcomes of neurological diseases.
Looking to the future of immunogenetics in neurological diseases, we recommend focus upon high‐resolution genotyping for both HLA and KIR. Investigating both coding and non‐coding region variation in these immunogenetic loci using high throughput high‐resolution technologies in groups with diverse ancestries will almost certainly be required to fully appreciate their role in neurological diseases. There are only limited examinations of HLA and KIR variations at transcriptomics and proteomics levels; therefore, the functional assessment of both allelic and regulatory regional variation is highly desirable. The impact of micro‐RNA on diverse HLA and KIR alleles in regulatory regions also needs to be evaluated in neurological diseases in order to recognize the significance of epigenetic factors in disease pathophysiology. Finally, our recent observation, that certain human metabolites occupy the P4 pocket of MS‐susceptible DRB1*15:01 haplotype in most populations and could be implicated in autoimmunity,232 suggest that similar investigations of both HLA class I and class II molecules in an allele‐specific manner could be undertaken. This approach might be advantageous to weight or group together various HLA genes and alleles that are involved in predisposition across diseases. The functional assessment of binding of human metabolites with HLA class I and class II molecules, and investigation of their impact upon T‐cell proliferation and responsiveness, could pave the way for designing novel therapies, leading to a step closer to reaching the goal of personalized medicine.
Disclosures
None to declare.
Acknowledgements
The authors are supported by grants from National Institutes of Health (U19NS095774, R01AI128775).
References
- 1. Cottler LB, Zunt J, Weiss B, Kamal AK, Vaddiparti K. Building global capacity for brain and nervous system disorders research. Nature 2015; 527:S207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chin JH, Vora N. The global burden of neurologic diseases. Neurology 2014; 83:349–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foo JN, Liu JJ, Tan EK. Whole‐genome and whole‐exome sequencing in neurological diseases. Nat Rev Neurol 2012; 8:508–17. [DOI] [PubMed] [Google Scholar]
- 4. Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D et al Common genetic variation in the HLA region is associated with late‐onset sporadic Parkinson's disease. Nat Genet 2010; 42:781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium , Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA et al Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature 2011; 476:214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. International Schizophrenia Consortium , Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC et al Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460:748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Song S, Miranda CJ, Braun L, Meyer K, Frakes AE, Ferraiuolo L et al Major histocompatibility complex class I molecules protect motor neurons from astrocyte‐induced toxicity in amyotrophic lateral sclerosis. Nat Med 2016; 22:397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steele NZ, Carr JS, Bonham LW, Geier EG, Damotte V, Miller ZA et al Fine‐mapping of the human leukocyte antigen locus as a risk factor for Alzheimer disease: a case‐control study. PLOS Med 2017; 14:e1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet 2009; 54:15–39. [DOI] [PubMed] [Google Scholar]
- 10. Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK et al Gene map of the extended human MHC. Nat Rev Genet 2004; 5:889–99. [DOI] [PubMed] [Google Scholar]
- 11. Gorer P. The detection of a hereditary antigenic difference in the blood of mice by means of human group A serum. J Genet 1936; 32:17–31. [Google Scholar]
- 12. Complete sequence and gene map of a human major histocompatibility complex. The MHC sequencing consortium. Nature 1999; 401:921–3. [DOI] [PubMed] [Google Scholar]
- 13. Bailey A, Dalchau N, Carter R, Emmott S, Phillips A, Werner JM et al Selector function of MHC I molecules is determined by protein plasticity. Sci Rep 2015; 5:14928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Holling TM, Schooten E, van Den Elsen PJ. Function and regulation of MHC class II molecules in T‐lymphocytes: of mice and men. Hum Immunol 2004; 65:282–90. [DOI] [PubMed] [Google Scholar]
- 15. Lidman O, Olsson T, Piehl F. Expression of nonclassical MHC class I (RT1‐U) in certain neuronal populations of the central nervous system. Eur J Neurosci 1999; 11:4468–72. [DOI] [PubMed] [Google Scholar]
- 16. Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science 2000; 290:2155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neumann H, Schmidt H, Wilharm E, Behrens L, Wekerle H. Interferon gamma gene expression in sensory neurons: evidence for autocrine gene regulation. J Exp Med 1997; 186:2023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Linda H, Hammarberg H, Piehl F, Khademi M, Olsson T. Expression of MHC class I heavy chain and beta2‐microglobulin in rat brainstem motoneurons and nigral dopaminergic neurons. J Neuroimmunol 1999; 101:76–86. [DOI] [PubMed] [Google Scholar]
- 19. Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science 1995; 269:549–52. [DOI] [PubMed] [Google Scholar]
- 20. Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 1998; 21:505–20. [DOI] [PubMed] [Google Scholar]
- 21. Loconto J, Papes F, Chang E, Stowers L, Jones EP, Takada T et al Functional expression of murine V2R pheromone receptors involves selective association with the M10 and M1 families of MHC class Ib molecules. Cell 2003; 112:607–18. [DOI] [PubMed] [Google Scholar]
- 22. Ishii T, Hirota J, Mombaerts P. Combinatorial coexpression of neural and immune multigene families in mouse vomeronasal sensory neurons. Curr Biol 2003; 13:394–400. [DOI] [PubMed] [Google Scholar]
- 23. Linda H, Hammarberg H, Cullheim S, Levinovitz A, Khademi M, Olsson T. Expression of MHC class I and beta2‐microglobulin in rat spinal motoneurons: regulatory influences by IFN‐gamma and axotomy. Exp Neurol 1998; 150:282–95. [DOI] [PubMed] [Google Scholar]
- 24. Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB et al Synapse elimination and learning rules co‐regulated by MHC class I H2‐Db. Nature 2014; 509:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron 2009; 64:40–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noristani HN, Sabourin JC, Gerber YN, Teigell M, Sommacal A, Vivanco M et al Brca1 is expressed in human microglia and is dysregulated in human and animal model of ALS. Mol Neurodegener 2015; 10:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N et al Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co‐expression meta‐analysis. Acta Neuropathol Commun 2015; 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK et al A unique microglia type associated with restricting development of Alzheimer's disease. Cell 2017; 169:1276–90 e1217. [DOI] [PubMed] [Google Scholar]
- 29. Naito S, Namerow N, Mickey MR, Terasaki PI. Multiple sclerosis: association with HL‐A3. Tissue Antigens 1972; 2:1–4. [DOI] [PubMed] [Google Scholar]
- 30. Compston DA, Batchelor JR, McDonald WI. B‐lymphocyte alloantigens associated with multiple sclerosis. Lancet 1976; 2:1261–5. [DOI] [PubMed] [Google Scholar]
- 31. Bertrams J, Kuwert E, Liedtke U. HL‐A antigens and multiple sclerosis. Tissue Antigens 1972; 2:405–8. [DOI] [PubMed] [Google Scholar]
- 32. Jersild C, Fog T, Hansen GS, Thomsen M, Svejgaard A, Dupont B. Histocompatibility determinants in multiple sclerosis, with special reference to clinical course. Lancet 1973; 2:1221–5. [DOI] [PubMed] [Google Scholar]
- 33. Hauser SL, Fleischnick E, Weiner HL, Marcus D, Awdeh Z, Yunis EJ et al Extended major histocompatibility complex haplotypes in patients with multiple sclerosis. Neurology 1989; 39:275–7. [DOI] [PubMed] [Google Scholar]
- 34. Haines JL, Terwedow HA, Burgess K, Pericak‐Vance MA, Rimmler JB, Martin ER et al Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The Multiple Sclerosis Genetics Group. Hum Mol Genet 1998; 7:1229–34. [DOI] [PubMed] [Google Scholar]
- 35. Barcellos LF, Oksenberg JR, Green AJ, Bucher P, Rimmler JB, Schmidt S et al Genetic basis for clinical expression in multiple sclerosis. Brain 2002; 125:150–8. [DOI] [PubMed] [Google Scholar]
- 36. Olerup O, Hillert J. HLA class II‐associated genetic susceptibility in multiple sclerosis: a critical evaluation. Tissue Antigens 1991; 38:1–15. [DOI] [PubMed] [Google Scholar]
- 37. Schmidt H, Williamson D, Ashley‐Koch A. HLA‐DR15 haplotype and multiple sclerosis: a HuGE review. Am J Epidemiol 2007; 165:1097–109. [DOI] [PubMed] [Google Scholar]
- 38. Barcellos LF, Oksenberg JR, Begovich AB, Martin ER, Schmidt S, Vittinghoff E et al HLA‐DR2 dose effect on susceptibility to multiple sclerosis and influence on disease course. Am J Hum Genet 2003; 72:710–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barcellos LF, Sawcer S, Ramsay PP, Baranzini SE, Thomson G, Briggs F et al Heterogeneity at the HLA‐DRB1 locus and risk for multiple sclerosis. Hum Mol Genet 2006; 15:2813–24. [DOI] [PubMed] [Google Scholar]
- 40. Dyment DA, Herrera BM, Cader MZ, Willer CJ, Lincoln MR, Sadovnick AD et al Complex interactions among MHC haplotypes in multiple sclerosis: susceptibility and resistance. Hum Mol Genet 2005; 14:2019–26. [DOI] [PubMed] [Google Scholar]
- 41. Kwon OJ, Karni A, Israel S, Brautbar C, Amar A, Meiner Z et al HLA class II susceptibility to multiple sclerosis among Ashkenazi and non‐Ashkenazi Jews. Arch Neurol 1999; 56:555–60. [DOI] [PubMed] [Google Scholar]
- 42. Hensiek AE, Sawcer SJ, Feakes R, Deans J, Mander A, Akesson E et al HLA‐DR 15 is associated with female sex and younger age at diagnosis in multiple sclerosis. J Neurol Neurosurg Psychiatry 2002; 72:184–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Celius EG, Harbo HF, Egeland T, Vartdal F, Vandvik B, Spurkiand A. Sex and age at diagnosis are correlated with the HLA‐DR2, DQ6 haplotype in multiple sclerosis. J Neurol Sci 2000; 178:132–5. [DOI] [PubMed] [Google Scholar]
- 44. Goris A, Pauwels I, Gustavsen MW, van Son B, Hilven K, Bos SD et al Genetic variants are major determinants of CSF antibody levels in multiple sclerosis. Brain 2015; 138:632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mero IL, Gustavsen MW, Saether HS, Flam ST, Berg‐Hansen P, Sondergaard HB et al Oligoclonal band status in Scandinavian multiple sclerosis patients is associated with specific genetic risk alleles. PLOS ONE 2013; 8:e58352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Okuda DT, Srinivasan R, Oksenberg JR, Goodin DS, Baranzini SE, Beheshtian A et al Genotype–phenotype correlations in multiple sclerosis: HLA genes influence disease severity inferred by 1HMR spectroscopy and MRI measures. Brain 2009; 132:250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patsopoulos NA, Barcellos LF, Hintzen RQ, Schaefer C, van Duijn CM, Noble JA et al Fine‐mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non‐HLA effects. PLOS Genet 2013; 9:e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oksenberg JR, Barcellos LF, Cree BA, Baranzini SE, Bugawan TL, Khan O et al Mapping multiple sclerosis susceptibility to the HLA‐DR locus in African Americans. Am J Hum Genet 2004; 74:160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quelvennec E, Bera O, Cabre P, Alizadeh M, Smadja D, Jugde F et al Genetic and functional studies in multiple sclerosis patients from Martinique attest for a specific and direct role of the HLA‐DR locus in the syndrome. Tissue Antigens 2003; 61:166–71. [DOI] [PubMed] [Google Scholar]
- 50. Yoshimura S, Isobe N, Yonekawa T, Matsushita T, Masaki K, Sato S et al Genetic and infectious profiles of Japanese multiple sclerosis patients. PLOS ONE 2012; 7:e48592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matsuoka T, Matsushita T, Osoegawa M, Kawano Y, Minohara M, Mihara F et al Association of the HLA‐DRB1 alleles with characteristic MRI features of Asian multiple sclerosis. Mult Scler 2008; 14:1181–90. [DOI] [PubMed] [Google Scholar]
- 52. Marrosu MG, Muntoni F, Murru MR, Spinicci G, Pischedda MP, Goddi F et al Sardinian multiple sclerosis is associated with HLA‐DR4: a serologic and molecular analysis. Neurology 1988; 38:1749–53. [DOI] [PubMed] [Google Scholar]
- 53. Brassat D, Salemi G, Barcellos LF, McNeill G, Proia P, Hauser SL et al The HLA locus and multiple sclerosis in Sicily. Neurology 2005; 64:361–3. [DOI] [PubMed] [Google Scholar]
- 54. Isobe N, Gourraud PA, Harbo HF, Caillier SJ, Santaniello A, Khankhanian P et al Genetic risk variants in African Americans with multiple sclerosis. Neurology 2013; 81:219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marrosu MG, Murru MR, Costa G, Cucca F, Sotgiu S, Rosati G et al Multiple sclerosis in Sardinia is associated and in linkage disequilibrium with HLA‐DR3 and ‐DR4 alleles. Am J Hum Genet 1997; 61:454–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hollenbach JA, Oksenberg JR. The immunogenetics of multiple sclerosis: a comprehensive review. J Autoimmun 2015; 64:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lincoln MR, Ramagopalan SV, Chao MJ, Herrera BM, Deluca GC, Orton SM et al Epistasis among HLA‐DRB1, HLA‐DQA1, and HLA‐DQB1 loci determines multiple sclerosis susceptibility. Proc Natl Acad Sci USA 2009; 106:7542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaimen‐Maciel DR, Reiche EM, Borelli SD, Morimoto HK, Melo FC, Lopes J et al HLA‐DRB1* allele‐associated genetic susceptibility and protection against multiple sclerosis in Brazilian patients. Mol Med Rep 2009; 2:993–8. [DOI] [PubMed] [Google Scholar]
- 59. Ramagopalan SV, Morris AP, Dyment DA, Herrera BM, DeLuca GC, Lincoln MR et al The inheritance of resistance alleles in multiple sclerosis. PLOS Genet 2007; 3:1607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Laaksonen M, Pastinen T, Sjoroos M, Kuokkanen S, Ruutiainen J, Sumelahti ML et al HLA class II associated risk and protection against multiple sclerosis‐a Finnish family study. J Neuroimmunol 2002; 122:140–5. [DOI] [PubMed] [Google Scholar]
- 61. Moutsianas L, Jostins L, Beecham AH, Dilthey AT, Xifara DK, Ban M et al Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet 2015; 47:1107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harbo HF, Isobe N, Berg‐Hansen P, Bos SD, Caillier SJ, Gustavsen MW et al Oligoclonal bands and age at onset correlate with genetic risk score in multiple sclerosis. Mult Scler 2014; 20:660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. International MHC and Autoimmunity Genetics Network , Rioux JD, Goyette P, Vyse TJ, Hammarstrom L, Fernando MM et al Mapping of multiple susceptibility variants within the MHC region for 7 immune‐mediated diseases. Proc Natl Acad Sci USA 2009; 106:18680–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yeo TW, De Jager PL, Gregory SG, Barcellos LF, Walton A, Goris A et al A second major histocompatibility complex susceptibility locus for multiple sclerosis. Ann Neurol 2007; 61:228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364:2106–12. [DOI] [PubMed] [Google Scholar]
- 66. Matsushita T, Matsuoka T, Isobe N, Kawano Y, Minohara M, Shi N et al Association of the HLA‐DPB1*0501 allele with anti‐aquaporin‐4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 2009; 73:171–6. [DOI] [PubMed] [Google Scholar]
- 67. Yamasaki K, Horiuchi I, Minohara M, Kawano Y, Ohyagi Y, Yamada T et al HLA‐DPB1*0501‐associated opticospinal multiple sclerosis: clinical, neuroimaging and immunogenetic studies. Brain 1999; 122:1689–96. [DOI] [PubMed] [Google Scholar]
- 68. Yoshimura S, Isobe N, Matsushita T, Yonekawa T, Masaki K, Sato S et al Distinct genetic and infectious profiles in Japanese neuromyelitis optica patients according to anti‐aquaporin 4 antibody status. J Neurol Neurosurg Psychiatry 2013; 84: 29–34. [DOI] [PubMed] [Google Scholar]
- 69. Wang H, Dai Y, Qiu W, Zhong X, Wu A, Wang Y et al HLA‐DPB1 0501 is associated with susceptibility to anti‐aquaporin‐4 antibodies positive neuromyelitis optica in southern Han Chinese. J Neuroimmunol 2011; 233:181–4. [DOI] [PubMed] [Google Scholar]
- 70. Fukazawa T, Kikuchi S, Miyagishi R, Miyazaki Y, Yabe I, Hamada T et al HLA‐dPB1*0501 is not uniquely associated with opticospinal multiple sclerosis in Japanese patients. Important role of DPB1*0301. Mult Scler 2006; 12:19–23. [DOI] [PubMed] [Google Scholar]
- 71. Isobe N, Matsushita T, Yamasaki R, Ramagopalan SV, Kawano Y, Nishimura Y et al Influence of HLA‐DRB1 alleles on the susceptibility and resistance to multiple sclerosis in Japanese patients with respect to anti‐aquaporin 4 antibody status. Mult Scler 2010; 16:147–55. [DOI] [PubMed] [Google Scholar]
- 72. Zephir H, Fajardy I, Outteryck O, Blanc F, Roger N, Fleury M et al Is neuromyelitis optica associated with human leukocyte antigen? Mult Scler 2009; 15:571–9. [DOI] [PubMed] [Google Scholar]
- 73. Brum DG, Barreira AA, dos Santos AC, Kaimen‐Maciel DR, Matiello M, Costa RM et al HLA‐DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler 2010; 16:21–9. [DOI] [PubMed] [Google Scholar]
- 74. Deschamps R, Paturel L, Jeannin S, Chausson N, Olindo S, Bera O et al Different HLA class II (DRB1 and DQB1) alleles determine either susceptibility or resistance to NMO and multiple sclerosis among the French Afro‐Caribbean population. Mult Scler 2011; 17:24–31. [DOI] [PubMed] [Google Scholar]
- 75. Alonso VR, de Jesus Flores Rivera J, Garci YR, Granados J, Sanchez T, Mena‐Hernandez L et al Neuromyelitis Optica (NMO IgG+) and Genetic Susceptibility, Potential Ethnic Influences. Cent Nerv Syst Agents Med Chem 2016; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 76. Brill L, Mandel M, Karussis D, Petrou P, Miller K, Ben‐Hur T et al Increased occurrence of anti‐AQP4 seropositivity and unique HLA Class II associations with neuromyelitis optica (NMO), among Muslim Arabs in Israel. J Neuroimmunol 2016; 293:65–70. [DOI] [PubMed] [Google Scholar]
- 77. Emile J, Truelle JL, Pouplard A, Hurez D. Association of Parkinson's disease with HLA‐B17 and B18 antigens. Nouv Presse Med 1977; 6:4144. [PubMed] [Google Scholar]
- 78. International Parkinson Disease Genomics Consortium , Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM et al Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet 2011; 377:641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guo Y, Deng X, Zheng W, Xu H, Song Z, Liang H et al HLA rs3129882 variant in Chinese Han patients with late‐onset sporadic Parkinson disease. Neurosci Lett 2011; 501:185–7. [DOI] [PubMed] [Google Scholar]
- 80. Jamshidi J, Movafagh A, Emamalizadeh B, Zare Bidoki A, Manafi A, Ghasemi Firouzabadi S et al HLA‐DRA is associated with Parkinson's disease in Iranian population. Int J Immunogenet 2014; 41:508–11. [DOI] [PubMed] [Google Scholar]
- 81. Puschmann A, Verbeeck C, Heckman MG, Soto‐Ortolaza AI, Lynch T, Jasinska‐Myga B et al Human leukocyte antigen variation and Parkinson's disease. Parkinsonism Relat Disord 2011; 17:376–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu X, Cheng R, Verbitsky M, Kisselev S, Browne A, Mejia‐Sanatana H et al Genome‐wide association study identifies candidate genes for Parkinson's disease in an Ashkenazi Jewish population. BMC Med Genet 2011; 12:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U et al Web‐based genome‐wide association study identifies two novel loci and a substantial genetic component for Parkinson's disease. PLOS Genet 2011; 7:e1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ahmed I, Tamouza R, Delord M, Krishnamoorthy R, Tzourio C, Mulot C et al Association between Parkinson's disease and the HLA‐DRB1 locus. Mov Disord 2012; 27:1104–10. [DOI] [PubMed] [Google Scholar]
- 85. Ma ZG, Liu TW, Bo YL. HLA‐DRA rs3129882 A/G polymorphism was not a risk factor for Parkinson's disease in Chinese‐based populations: a meta‐analysis. Int J Neurosci 2015; 125:241–6. [DOI] [PubMed] [Google Scholar]
- 86. Saiki M, Baker A, Williams‐Gray CH, Foltynie T, Goodman RS, Taylor CJ et al Association of the human leucocyte antigen region with susceptibility to Parkinson's disease. J Neurol Neurosurg Psychiatry 2010; 81:890–1. [DOI] [PubMed] [Google Scholar]
- 87. Sun C, Wei L, Luo F, Li Y, Li J, Zhu F et al HLA‐DRB1 alleles are associated with the susceptibility to sporadic Parkinson's disease in Chinese Han population. PLOS ONE 2012; 7:e48594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gonzalez‐Galarza FF, Christmas S, Middleton D, Jones AR. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res 2011; 39:D913–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wissemann WT, Hill‐Burns EM, Zabetian CP, Factor SA, Patsopoulos N, Hoglund B et al Association of Parkinson disease with structural and regulatory variants in the HLA region. Am J Hum Genet 2013; 93:984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Henschke PJ, Bell DA, Cape RD. Alzheimer's disease and HLA. Tissue Antigens 1978; 12:132–5. [DOI] [PubMed] [Google Scholar]
- 91. Harris JM, Cumming AM, Craddock N, St Clair D, Lendon CL. Human leucocyte antigen‐A2 increases risk of Alzheimer's disease but does not affect age of onset in a Scottish population. Neurosci Lett 2000; 294:37–40. [DOI] [PubMed] [Google Scholar]
- 92. Small GW, Scott WK, Komo S, Yamaoka LH, Farrer LA, Auerbach SH et al No association between the HLA‐A2 allele and Alzheimer disease. Neurogenetics 1999; 2:177–82. [DOI] [PubMed] [Google Scholar]
- 93. Araria‐Goumidi L, Lambert JC, Cottel D, Amouyel P, Chartier‐Harlin MC. No association of the HLA‐A2 allele with Alzheimer's disease. Neurosci Lett 2002; 335:75–8. [DOI] [PubMed] [Google Scholar]
- 94. Wang ZX, Wang HF, Tan L, Sun FR, Tan MS, Tan CC et al HLA‐A2 alleles mediate Alzheimer's disease by altering hippocampal volume. Mol Neurobiol 2017; 54:2469–76. [DOI] [PubMed] [Google Scholar]
- 95. Curran M, Middleton D, Edwardson J, Perry R, McKeith I, Morris C et al HLA‐DR antigens associated with major genetic risk for late‐onset Alzheimer's disease. NeuroReport 1997; 8:1467–9. [DOI] [PubMed] [Google Scholar]
- 96. Aisen PS, Luddy A, Durner M, Reinhard JF Jr, Pasinetti GM. HLA‐DR4 influences glial activity in Alzheimer's disease hippocampus. J Neurol Sci 1998; 161:66–9. [DOI] [PubMed] [Google Scholar]
- 97. Neill D, Curran MD, Middleton D, Mawhinney H, Edwardson JA, McKeith I et al Risk for Alzheimer's disease in older late‐onset cases is associated with HLA‐DRB1*03. Neurosci Lett 1999; 275:137–40. [DOI] [PubMed] [Google Scholar]
- 98. Swaminathan S, Shen L, Kim S, Inlow M, West JD, Faber KM et al Analysis of copy number variation in Alzheimer's disease: the NIALOAD/NCRAD Family Study. Curr Alzheimer Res 2012; 9:801–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lambert JC, Ibrahim‐Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013; 45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Payami H, Kaye J, Becker W, Norman D, Wetzsteon P. HLA‐A2, or a closely linked gene, confers susceptibility to early‐onset sporadic Alzheimer's disease in men. Neurology 1991; 41:1544–8. [DOI] [PubMed] [Google Scholar]
- 101. Jiao B, Liu X, Zhou L, Wang MH, Zhou Y, Xiao T et al Polygenic analysis of late‐onset Alzheimer's disease from Mainland China. PLOS ONE 2015; 10:e0144898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Allen M, Kachadoorian M, Carrasquillo MM, Karhade A, Manly L, Burgess JD et al Late‐onset Alzheimer disease risk variants mark brain regulatory loci. Neurol Genet 2015; 1:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nettiksimmons J, Tranah G, Evans DS, Yokoyama JS, Yaffe K. Gene‐based aggregate SNP associations between candidate AD genes and cognitive decline. Age 2016; 38:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yu L, Chibnik LB, Srivastava GP, Pochet N, Yang J, Xu J et al Association of Brain DNA methylation in SORL1, ABCA7, HLA‐DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol 2015; 72:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yokoyama JS, Wang Y, Schork AJ, Thompson WK, Karch CM, Cruchaga C et al Association between genetic traits for immune‐mediated diseases and Alzheimer disease. JAMA Neurol 2016; 73:691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cazzullo CL, Smeraldi E, Penati G. The leucocyte antigenic system HL‐A as a possible genetic marker of schizophrenia. Br J Psychiatry 1974; 125:25–7. [DOI] [PubMed] [Google Scholar]
- 107. Wright P, Nimgaonkar VL, Donaldson PT, Murray RM. Schizophrenia and HLA: a review. Schizophr Res 2001; 47:1–12. [DOI] [PubMed] [Google Scholar]
- 108. Debnath M, Cannon DM, Venkatasubramanian G. Variation in the major histocompatibility complex [MHC] gene family in schizophrenia: associations and functional implications. Prog Neuropsychopharmacol Biol Psychiatry 2013; 42:49–62. [DOI] [PubMed] [Google Scholar]
- 109. Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe'er I et al Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009; 460:753–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D et al Common variants conferring risk of schizophrenia. Nature 2009; 460:744–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Schizophrenia Psychiatric Genome‐Wide Association Study Consortium . Genome‐wide association study identifies five new schizophrenia loci. Nat Genet 2011; 43:969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ikeda M, Aleksic B, Kinoshita Y, Okochi T, Kawashima K, Kushima I et al Genome‐wide association study of schizophrenia in a Japanese population. Biol Psychiatry 2011; 69:472–8. [DOI] [PubMed] [Google Scholar]
- 113. Yue WH, Wang HF, Sun LD, Tang FL, Liu ZH, Zhang HX et al Genome‐wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11.2. Nat Genet 2011; 43:1228–31. [DOI] [PubMed] [Google Scholar]
- 114. Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium . Genome‐wide association study implicates HLA‐C*01:02 as a risk factor at the major histocompatibility complex locus in schizophrenia. Biol Psychiatry 2012;72: 620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Goes FS, McGrath J, Avramopoulos D, Wolyniec P, Pirooznia M, Ruczinski I et al Genome‐wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet B Neuropsychiatr Genet 2015; 168:649–59. [DOI] [PubMed] [Google Scholar]
- 116. de Jong S, van Eijk KR, Zeegers DW, Strengman E, Janson E, Veldink JH et al Expression QTL analysis of top loci from GWAS meta‐analysis highlights additional schizophrenia candidate genes. Eur J Hum Genet 2012; 20:1004–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Akaho R, Matsushita I, Narita K, Okazaki Y, Okabe Y, Matsushita M et al Support for an association between HLA‐DR1 and schizophrenia in the Japanese population. Am J Med Genet 2000; 96:725–7. [DOI] [PubMed] [Google Scholar]
- 118. Arinami T, Otsuka Y, Hamaguchi H, Itokawa M, Aoki J, Shibuya H et al Evidence supporting an association between the DRB1 gene and schizophrenia in Japanese. Schizophr Res 1998; 32:81–6. [DOI] [PubMed] [Google Scholar]
- 119. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N et al Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Pirskanen R. Genetic associations between myasthenia gravis and the HL‐A system. J Neurol Neurosurg Psychiatry 1976; 39:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Baggi F, Antozzi C, Andreetta F, Confalonieri P, Ciusani E, Begovich AB et al Identification of a novel HLA class II association with DQB1*0502 in an Italian myasthenic population. Ann NY Acad Sci 1998; 841:355–9. [DOI] [PubMed] [Google Scholar]
- 122. Fekih‐Mrissa N, Klai S, Zaouali J, Gritli N, Mrissa R. Association of HLA‐DR/DQ polymorphism with myasthenia gravis in Tunisian patients. Clin Neurol Neurosurg 2013; 115:32–6. [DOI] [PubMed] [Google Scholar]
- 123. Santos E, Bettencourt A, da Silva AM, Boleixa D, Lopes D, Bras S et al HLA and age of onset in myasthenia gravis. Neuromuscul Disord 2017; 27:650–4. [DOI] [PubMed] [Google Scholar]
- 124. Xie YC, Qu Y, Sun L, Li HF, Zhang H, Shi HJ et al Association between HLA‐DRB1 and myasthenia gravis in a northern Han Chinese population. J Clin Neurosci 2011; 18:1524–7. [DOI] [PubMed] [Google Scholar]
- 125. Gregersen PK, Kosoy R, Lee AT, Lamb J, Sussman J, McKee D et al Risk for myasthenia gravis maps to a (151) Pro–>Ala change in TNIP1 and to human leukocyte antigen‐B*08. Ann Neurol 2012; 72:927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Janer M, Cowland A, Picard J, Campbell D, Pontarotti P, Newsom‐Davis J et al A susceptibility region for myasthenia gravis extending into the HLA‐class I sector telomeric to HLA‐C. Hum Immunol 1999; 60:909–17. [DOI] [PubMed] [Google Scholar]
- 127. Vandiedonck C, Beaurain G, Giraud M, Hue‐Beauvais C, Eymard B, Tranchant C et al Pleiotropic effects of the 8.1 HLA haplotype in patients with autoimmune myasthenia gravis and thymus hyperplasia. Proc Natl Acad Sci USA 2004; 101:15464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Giraud M, Beaurain G, Eymard B, Tranchant C, Gajdos P, Garchon HJ. Genetic control of autoantibody expression in autoimmune myasthenia gravis: role of the self‐antigen and of HLA‐linked loci. Genes Immun 2004; 5:398–404. [DOI] [PubMed] [Google Scholar]
- 129. Avidan N, Le Panse R, Berrih‐Aknin S, Miller A. Genetic basis of myasthenia gravis – a comprehensive review. J Autoimmun 2014; 52:146–53. [DOI] [PubMed] [Google Scholar]
- 130. Maniaol AH, Elsais A, Lorentzen AR, Owe JF, Viken MK, Saether H et al Late onset myasthenia gravis is associated with HLA DRB1*15:01 in the Norwegian population. PLOS ONE 2012; 7:e36603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Renton AE, Pliner HA, Provenzano C, Evoli A, Ricciardi R, Nalls MA et al A genome‐wide association study of myasthenia gravis. JAMA Neurol 2015; 72:396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Testi M, Terracciano C, Guagnano A, Testa G, Marfia GA, Pompeo E et al Association of HLA‐DQB1 *05:02 and DRB1 *16 alleles with late‐onset, nonthymomatous, AChR‐Ab‐positive myasthenia gravis. Autoimmune Dis 2012; 2012:541760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Seldin MF, Alkhairy OK, Lee AT, Lamb JA, Sussman J, Pirskanen‐Matell R et al Genome‐wide association study of late‐onset myasthenia gravis: confirmation of TNFRSF11A, and identification of ZBTB10 and three distinct HLA associations. Mol Med 2015; 21:769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Niks EH, Kuks JB, Roep BO, Haasnoot GW, Verduijn W, Ballieux BE et al Strong association of MuSK antibody‐positive myasthenia gravis and HLA‐DR14‐DQ5. Neurology 2006; 66:1772–4. [DOI] [PubMed] [Google Scholar]
- 135. Bartoccioni E, Scuderi F, Augugliaro A, Chiatamone Ranieri S, Sauchelli D, Alboino P et al HLA class II allele analysis in MuSK‐positive myasthenia gravis suggests a role for DQ5. Neurology 2009; 72:195–7. [DOI] [PubMed] [Google Scholar]
- 136. Alahgholi‐Hajibehzad M, Yilmaz V, Gulsen‐Parman Y, Aysal F, Oflazer P, Deymeer F et al Association of HLA‐DRB1 *14, ‐DRB1 *16 and ‐DQB1 *05 with MuSK‐myasthenia gravis in patients from Turkey. Hum Immunol 2013; 74:1633–5. [DOI] [PubMed] [Google Scholar]
- 137. Nikolic AV, Andric ZP, Simonovic RB, Rakocevic Stojanovic VM, Basta IZ, Bojic SD et al High frequency of DQB1*05 and absolute absence of DRB1*13 in muscle‐specific tyrosine kinase positive myasthenia gravis. Eur J Neurol 2015; 22:59–63. [DOI] [PubMed] [Google Scholar]
- 138. Zhu WH, Lu JH, Lin J, Xi JY, Lu J, Luo SS et al HLA‐DQA1*03:02/DQB1*03:03:02 is strongly associated with susceptibility to childhood‐onset ocular myasthenia gravis in Southern Han Chinese. J Neuroimmunol 2012; 247:81–5. [DOI] [PubMed] [Google Scholar]
- 139. Feng HY, Yang LX, Liu WB, Huang X, Qiu L, Li Y. The HLA‐B*4601‐DRB1*0901 haplotype is positively correlated with juvenile ocular myasthenia gravis in a southern Chinese Han population. Neurol Sci 2015; 36:1135–40. [DOI] [PubMed] [Google Scholar]
- 140. Antel JP, Arnason BG, Fuller TC, Lehrich JR. Histocompatibility typing in amyotrophic lateral sclerosis. Arch Neurol 1976; 33:423–5. [DOI] [PubMed] [Google Scholar]
- 141. Kott E, Livni E, Zamir R, Kuritzky A. Cell‐mediated immunity to polio and HLA antigens in amyotrophic lateral sclerosis. Neurology 1979; 29:1040–4. [DOI] [PubMed] [Google Scholar]
- 142. Jokelainen M, Tiilikainen A, Lapinleimu K. Polio antibodies and HLA antigens in amyotrophic lateral sclerosis. Tissue Antigens 1977; 10:259–66. [DOI] [PubMed] [Google Scholar]
- 143. Terasaki PI, Mickey MR. HL‐A haplotypes of 32 diseases. Transplant Rev 1975; 22:105–19. [DOI] [PubMed] [Google Scholar]
- 144. Hoffman PM, Robbins DS, Gibbs CJ Jr, Gajdusek DC, Garruto RM, Terasaki OI. Histocompatibility antigens in amyotrophic lateral sclerosis and parkinsonism‐dementia on Guam. Lancet 1977; 2:717. [DOI] [PubMed] [Google Scholar]
- 145. Behan PO, Durward WF, Dick H. Histocompatibility antigens associated with motor‐neuron disease. Lancet 1976; 2:803. [DOI] [PubMed] [Google Scholar]
- 146. Bartfeld H, Pollack MS, Cunningham‐Rundles S, Donnenfeld H. HLA frequencies in amyotrophic lateral sclerosis. Arch Neurol 1982; 39:270–1.7073543 [Google Scholar]
- 147. Yang X, Zheng J, Tian S, Chen Y, An R, Zhao Q et al HLA‐DRA/HLA‐DRB5 polymorphism affects risk of sporadic ALS and survival in a southwest Chinese cohort. J Neurol Sci 2017; 373:124–8. [DOI] [PubMed] [Google Scholar]
- 148. Harel‐Bellan A, Quillet A, Marchiol C, DeMars R, Tursz T, Fradelizi D. Natural killer susceptibility of human cells may be regulated by genes in the HLA region on chromosome 6. Proc Natl Acad Sci USA 1986; 83:5688–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol 2002; 20:217–51. [DOI] [PubMed] [Google Scholar]
- 150. Bjorkstrom NK, Beziat V, Cichocki F, Liu LL, Levine J, Larsson S et al CD8 T cells express randomly selected KIRs with distinct specificities compared with NK cells. Blood 2012; 120:3455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Parham P. Killer cell immunoglobulin‐like receptor diversity: balancing signals in the natural killer cell response. Immunol Lett 2004; 92:11–3. [DOI] [PubMed] [Google Scholar]
- 152. Kikuchi‐Maki A, Catina TL, Campbell KS. Cutting edge: KIR2DL4 transduces signals into human NK cells through association with the Fc receptor gamma protein. J Immunol 2005; 174:3859–63. [DOI] [PubMed] [Google Scholar]
- 153. Kikuchi‐Maki A, Yusa S, Catina TL, Campbell KS. KIR2DL4 is an IL‐2‐regulated NK cell receptor that exhibits limited expression in humans but triggers strong IFN‐gamma production. J Immunol 2003; 171:3415–25. [DOI] [PubMed] [Google Scholar]
- 154. Saunders PM, Vivian JP, O'Connor GM, Sullivan LC, Pymm P, Rossjohn J et al A bird's eye view of NK cell receptor interactions with their MHC class I ligands. Immunol Rev 2015; 267:148–66. [DOI] [PubMed] [Google Scholar]
- 155. Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol 2013; 13:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Colonna M, Moretta A, Vely F, Vivier E. A high‐resolution view of NK‐cell receptors: structure and function. Immunol Today 2000; 21:428–31. [DOI] [PubMed] [Google Scholar]
- 157. Moretta L, Bottino C, Pende D, Castriconi R, Mingari MC, Moretta A. Surface NK receptors and their ligands on tumor cells. Semin Immunol 2006; 18:151–8. [DOI] [PubMed] [Google Scholar]
- 158. Fadda L, Borhis G, Ahmed P, Cheent K, Pageon SV, Cazaly A et al Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci USA 2010; 107:10160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Moesta AK, Norman PJ, Yawata M, Yawata N, Gleimer M, Parham P. Synergistic polymorphism at two positions distal to the ligand‐binding site makes KIR2DL2 a stronger receptor for HLA‐C than KIR2DL3. J Immunol 2008; 180:3969–79. [DOI] [PubMed] [Google Scholar]
- 160. Thananchai H, Gillespie G, Martin MP, Bashirova A, Yawata N, Yawata M et al Cutting edge: allele‐specific and peptide‐dependent interactions between KIR3DL1 and HLA‐A and HLA‐B. J Immunol 2007; 178:33–7. [DOI] [PubMed] [Google Scholar]
- 161. Cassidy SA, Cheent KS, Khakoo SI. Effects of peptide on NK cell‐mediated MHC I recognition. Front Immunol 2014; 5:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Parham P. Influence of KIR diversity on human immunity. Adv Exp Med Biol 2005; 560:47–50. [DOI] [PubMed] [Google Scholar]
- 163. Middleton D, Meenagh A, Gourraud PA. KIR haplotype content at the allele level in 77 Northern Irish families. Immunogenetics 2007; 59:145–58. [DOI] [PubMed] [Google Scholar]
- 164. Uhrberg M, Parham P, Wernet P. Definition of gene content for nine common B haplotypes of the Caucasoid population: KIR haplotypes contain between seven and eleven KIR genes. Immunogenetics 2002; 54:221–9. [DOI] [PubMed] [Google Scholar]
- 165. Hsu KC, Chida S, Dupont B, Geraghty DE. The killer cell immunoglobulin‐like receptor (KIR) genomic region: gene‐order, haplotypes and allelic polymorphism. Immunol Rev 2002; 190:40–52. [DOI] [PubMed] [Google Scholar]
- 166. Hollenbach JA, Nocedal I, Ladner MB, Single RM, Trachtenberg EA. Killer cell immunoglobulin‐like receptor (KIR) gene‐content variation in the HGDP‐CEPH populations. Immunogenetics 2012; 64:719–37. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hollenbach JA, Augusto DG, Alaez C, Bubnova L, Fae I, Fischer G et al 16(th) ihiw: population global distribution of killer immunoglobulin‐like receptor (KIR) and ligands. Int J Immunogenet 2013; 40:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Hollenbach JA, Ladner MB, Saeteurn K, Taylor KD, Mei L, Haritunians T et al Susceptibility to Crohn's disease is mediated by KIR2DL2/KIR2DL3 heterozygosity and the HLA‐C ligand. Immunogenetics 2009; 61:663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Khakoo SI, Carrington M. KIR and disease: a model system or system of models? Immunol Rev 2006; 214:186–201. [DOI] [PubMed] [Google Scholar]
- 170. Williams AP, Bateman AR, Khakoo SI. Hanging in the balance. KIR and their role in disease. Mol Interventions 2005; 5:226–40. [DOI] [PubMed] [Google Scholar]
- 171. Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F et al Innate partnership of HLA‐B and KIR3DL1 subtypes against HIV‐1. Nat Genet 2007; 39:733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Li Y, Zhang T, Ho C, Orange JS, Douglas SD, Ho WZ. Natural killer cells inhibit hepatitis C virus expression. J Leukoc Biol 2004; 76:1171–9. [DOI] [PubMed] [Google Scholar]
- 173. Khakoo SI, Chloe LT, Martin MP, Brooks CR, Gao X, Astemborski J et al HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 2004; 305:872–4. [DOI] [PubMed] [Google Scholar]
- 174. Bashirova AA, Thomas R, Carrington M. HLA/KIR restraint of HIV: surviving the fittest. Annu Rev Immunol 2011; 29:295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Misra MK, Prakash S, Moulik NR, Kumar A, Agrawal S. Genetic associations of killer immunoglobulin like receptors and class I human leukocyte antigens on childhood acute lymphoblastic leukemia among north Indians. Hum Immunol 2016; 77:41–6. [DOI] [PubMed] [Google Scholar]
- 176. Boudreau JE, Giglio F, Gooley TA, Stevenson PA, Le Luduec JB, Shaffer BC et al KIR3DL1/HL A‐B subtypes govern acute myelogenous leukemia relapse after hematopoietic cell transplantation. J Clin Oncol 2017; 35:2268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT et al Donors with group B KIR haplotypes improve relapse‐free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood 2009; 113:726–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Marsh SG et al Donor killer cell Ig‐like receptor B haplotypes, recipient HLA‐C1, and HLA‐C mismatch enhance the clinical benefit of unrelated transplantation for acute myelogenous leukemia. J Immunol 2014; 192:4592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A et al Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295:2097–100. [DOI] [PubMed] [Google Scholar]
- 180. Kunert K, Seiler M, Mashreghi MF, Klippert K, Schonemann C, Neumann K et al KIR/HLA ligand incompatibility in kidney transplantation. Transplantation 2007; 84:1527–33. [DOI] [PubMed] [Google Scholar]
- 181. Hiby SE, Apps R, Chazara O, Farrell LE, Magnus P, Trogstad L et al Maternal KIR in combination with paternal HLA‐C2 regulate human birth weight. J Immunol 2014; 192:5069–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Hiby SE, Walker JJ, O'shaughnessy KM, Redman CW, Carrington M, Trowsdale J et al Combinations of maternal KIR and fetal HLA‐C genes influence the risk of preeclampsia and reproductive success. J Exp Med 2004; 200:957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Moffett A, Hiby SE. How does the maternal immune system contribute to the development of pre‐eclampsia? Placenta 2007; 28:S51–6. [DOI] [PubMed] [Google Scholar]
- 184. Moffett‐King A. Natural killer cells and pregnancy. Nat Rev Immunol 2002; 2:656–63. [DOI] [PubMed] [Google Scholar]
- 185. Lanier LL. Natural killer cells fertile with receptors for HLA‐G? Proc Natl Acad Sci USA 1999; 96:5343–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Harbo HF, Lie BA, Sawcer S, Celius EG, Dai KZ, Oturai A et al Genes in the HLA class I region may contribute to the HLA class II‐associated genetic susceptibility to multiple sclerosis. Tissue Antigens 2004; 63:237–47. [DOI] [PubMed] [Google Scholar]
- 187. Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther 2011; 13:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Kulkarni S, Martin MP, Carrington M. The yin and yang of HLA and KIR in human disease. Semin Immunol 2008; 20:343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Lorentzen AR, Karlsen TH, Olsson M, Smestad C, Mero IL, Woldseth B et al Killer immunoglobulin‐like receptor ligand HLA‐Bw4 protects against multiple sclerosis. Ann Neurol 2009; 65:658–66. [DOI] [PubMed] [Google Scholar]
- 190. Kaur G, Trowsdale J, Fugger L. Natural killer cells and their receptors in multiple sclerosis. Brain 2013; 136:2657–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Hollenbach JA, Pando MJ, Caillier SJ, Gourraud PA, Oksenberg JR. The killer immunoglobulin‐like receptor KIR3DL1 in combination with HLA‐Bw4 is protective against multiple sclerosis in African Americans. Genes Immun 2016; 17:199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Wisniewski A, Frydecka D, Nowak I, Majorczyk E, Senitzer D, Piotrowski P et al Are KIR and HLA class I genes associated with schizophrenia? Tissue Antigens 2014; 84:503–4. [DOI] [PubMed] [Google Scholar]
- 193. Pelak K, Need AC, Fellay J, Shianna KV, Feng S, Urban TJ et al Copy number variation of KIR genes influences HIV‐1 control. PLOS Biol 2011; 9:e1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Gourraud PA, Gagne K, Bignon JD, Cambon‐Thomsen A, Middleton D. Preliminary analysis of a KIR haplotype estimation algorithm: a simulation study. Tissue Antigens 2007; 69(Suppl 1):96–100. [DOI] [PubMed] [Google Scholar]
- 195. Jelcic I, Hsu KC, Kakalacheva K, Breiden P, Dupont B, Uhrberg M et al Killer immunoglobulin‐like receptor locus polymorphisms in multiple sclerosis. Mult Scler 2012; 18:951–8. [DOI] [PubMed] [Google Scholar]
- 196. Garcia‐Leon JA, Pinto‐Medel MJ, Garcia‐Trujillo L, Lopez‐Gomez C, Oliver‐Martos B, Prat‐Arrojo I et al Killer cell immunoglobulin‐like receptor genes in Spanish multiple sclerosis patients. Mol Immunol 2011; 48:1896–902. [DOI] [PubMed] [Google Scholar]
- 197. Bettencourt A, Silva AM, Carvalho C, Leal B, Santos E, Costa PP et al The role of KIR2DS1 in multiple sclerosis–KIR in Portuguese MS patients. J Neuroimmunol 2014; 269:52–5. [DOI] [PubMed] [Google Scholar]
- 198. Fusco C, Guerini FR, Nocera G, Ventrella G, Caputo D, Valentino MA et al KIRs and their HLA ligands in remitting‐relapsing multiple sclerosis. J Neuroimmunol 2010; 229:232–7. [DOI] [PubMed] [Google Scholar]
- 199. Chong MS, Goh LK, Lim WS, Chan M, Tay L, Chen G et al Gene expression profiling of peripheral blood leukocytes shows consistent longitudinal downregulation of TOMM40 and upregulation of KIR2DL5A, PLOD1, and SLC2A8 among fast progressors in early Alzheimer's disease. J Alzheimers Dis 2013; 34:399–405. [DOI] [PubMed] [Google Scholar]
- 200. Deleidi M, Gasser T. The role of inflammation in sporadic and familial Parkinson's disease. Cell Mol Life Sci 2013; 70:4259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Blandini F, Armentero MT. Animal models of Parkinson's disease. FEBS J 2012; 279:1156–66. [DOI] [PubMed] [Google Scholar]
- 202. Hartung HP, Aktas O, Menge T, Kieseier BC. Immune regulation of multiple sclerosis. Handb Clin Neurol 2014; 122:3–14. [DOI] [PubMed] [Google Scholar]
- 203. Le Panse R, Berrih‐Aknin S. Autoimmune myasthenia gravis: autoantibody mechanisms and new developments on immune regulation. Curr Opin Neurol 2013; 26:569–76. [DOI] [PubMed] [Google Scholar]
- 204. Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D et al Inflammation in neurodegenerative diseases – an update. Immunology 2014; 142:151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK‐cell cytokine and chemokine production by target cell recognition. Blood 2010; 115:2167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol 2017; 17:49–59. [DOI] [PubMed] [Google Scholar]
- 207. Xu W, Fazekas G, Hara H, Tabira T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J Neuroimmunol 2005; 163:24–30. [DOI] [PubMed] [Google Scholar]
- 208. Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar‐Mather TP et al The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J 2006; 20:896–905. [DOI] [PubMed] [Google Scholar]
- 209. Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI et al Central nervous system (CNS)‐resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med 2010; 207:1907–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol 2012; 123:861–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Solerte SB, Cravello L, Ferrari E, Fioravanti M. Overproduction of IFN‐gamma and TNF‐alpha from natural killer (NK) cells is associated with abnormal NK reactivity and cognitive derangement in Alzheimer's disease. Ann N Y Acad Sci 2000; 917:331–40. [DOI] [PubMed] [Google Scholar]
- 212. Mihara T, Nakashima M, Kuroiwa A, Akitake Y, Ono K, Hosokawa M et al Natural killer cells of Parkinson's disease patients are set up for activation: a possible role for innate immunity in the pathogenesis of this disease. Parkinsonism Relat Disord 2008; 14:46–51. [DOI] [PubMed] [Google Scholar]
- 213. Solerte SB, Fioravanti M, Pascale A, Ferrari E, Govoni S, Battaini F. Increased natural killer cell cytotoxicity in Alzheimer's disease may involve protein kinase C dysregulation. Neurobiol Aging 1998; 19:191–9. [DOI] [PubMed] [Google Scholar]
- 214. Yovel G, Sirota P, Mazeh D, Shakhar G, Rosenne E, Ben‐Eliyahu S. Higher natural killer cell activity in schizophrenic patients: the impact of serum factors, medication, and smoking. Brain Behav Immun 2000; 14:153–69. [DOI] [PubMed] [Google Scholar]
- 215. Shi FD, Wang HB, Li H, Hong S, Taniguchi M, Link H et al Natural killer cells determine the outcome of B cell‐mediated autoimmunity. Nat Immunol 2000; 1:245–51. [DOI] [PubMed] [Google Scholar]
- 216. Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK et al T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci USA 2008; 105:17913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Dalakas MC, Illa I. Common variable immunodeficiency and inclusion body myositis: a distinct myopathy mediated by natural killer cells. Ann Neurol 1995; 37:806–10. [DOI] [PubMed] [Google Scholar]
- 218. Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL‐18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN‐gamma by NK cells. J Immunol 2000; 165:3099–104. [DOI] [PubMed] [Google Scholar]
- 219. Vollmer TL, Liu R, Price M, Rhodes S, La Cava A, Shi FD. Differential effects of IL‐21 during initiation and progression of autoimmunity against neuroantigen. J Immunol 2005; 174:2696–701. [DOI] [PubMed] [Google Scholar]
- 220. Takahashi K, Miyake S, Kondo T, Terao K, Hatakenaka M, Hashimoto S et al Natural killer type 2 bias in remission of multiple sclerosis. J Clin Invest 2001; 107:R23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Takahashi K, Aranami T, Endoh M, Miyake S, Yamamura T. The regulatory role of natural killer cells in multiple sclerosis. Brain 2004; 127:1917–27. [DOI] [PubMed] [Google Scholar]
- 222. Backstrom E, Chambers BJ, Kristensson K, Ljunggren HG. Direct NK cell‐mediated lysis of syngenic dorsal root ganglia neurons in vitro . J Immunol 2000; 165:4895–900. [DOI] [PubMed] [Google Scholar]
- 223. Backstrom E, Chambers BJ, Ho EL, Naidenko OV, Mariotti R, Fremont DH et al Natural killer cell‐mediated lysis of dorsal root ganglia neurons via RAE1/NKG2D interactions. Eur J Immunol 2003; 33:92–100. [DOI] [PubMed] [Google Scholar]
- 224. Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N et al A genome‐wide association study identifies variants in the HLA‐DP locus associated with chronic hepatitis B in Asians. Nat Genet 2009; 41:591–5. [DOI] [PubMed] [Google Scholar]
- 225. Thomas R, Thio CL, Apps R, Qi Y, Gao X, Marti D et al A novel variant marking HLA‐DP expression levels predicts recovery from hepatitis B virus infection. J Virol 2012; 86:6979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Petersdorf EW, Malkki M, Gooley TA, Spellman SR, Haagenson MD, Horowitz MM et al MHC‐resident variation affects risks after unrelated donor hematopoietic cell transplantation. Sci Transl Med 2012; 4:144ra101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, Bass SE et al Differential microRNA regulation of HLA‐C expression and its association with HIV control. Nature 2011; 472:495–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Maver A, Lavtar P, Ristic S, Stopinsek S, Simcic S, Hocevar K et al Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci Rep 2017; 7:3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Karch CM, Ezerskiy LA, Bertelsen S, Alzheimer's Disease Genetics Consortium (ADGC) , Goate AM. Alzheimer's disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 Loci. PLOS ONE 2016; 11:e0148717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Hwang Y, Kim J, Shin JY, Kim JI, Seo JS, Webster MJ et al Gene expression profiling by mRNA sequencing reveals increased expression of immune/inflammation‐related genes in the hippocampus of individuals with schizophrenia. Transl Psychiatry 2013; 3:e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Hoarau JJ, Cesari M, Caillens H, Cadet F, Pabion M. HLA DQA1 genes generate multiple transcripts by alternative splicing and polyadenylation of the 3’ untranslated region. Tissue Antigens 2004; 63:58–71. [DOI] [PubMed] [Google Scholar]
- 232. Misra MK, Damotte V, Hollenbach JA. Structure based selection of Human metabolite binding P4 pocket of DRB1*15:01 and DRB1*15:03, with implications for multiple sclerosis. Genes Immun 2017; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]