

Abstract
Hepatitis C virus (HCV) is a major cause of liver diseases including liver cirrhosis and hepatocellular carcinoma. Approximately 3% of the world population is infected with HCV. Thus, HCV infection is considered a public healthy challenge. It is worth mentioning, that the HCV prevalence is dependent on the countries with infection rates around 20% in high endemic countries. The review summarizes recent data on HCV molecular biology, the physiopathology of infection (immune-mediated liver damage, liver fibrosis and lipid metabolism), virus diagnostic and treatment. In addition, currently available in vitro, ex vivo and animal models to study the virus life cycle, virus pathogenesis and therapy are described. Understanding of both host and viral factors may in the future lead to creation of new approaches in generation of an efficient therapeutic vaccine.
Keywords: Hepatitis C virus, Transmission, Molecular biology, Pathogenesis, In vitro and ex vivo models of hepatitis C virus infection, Treatment
Core tip: Brief overviews on epidemiology of hepatitis C virus (HCV), virus morphology and the virus life cycle are presented. A special attention was focused on in vitro and in vivo models that are currently used to study the HCV infection. In fact, extensive use of existing models and creating a new ones is a way to reveal important events in the virus-cell interaction. In particular, the models might shed light on the mechanisms behind virus induced pathogenesis and chronicity, and by that contribute to the development of new drugs and prophylactic vaccine. Recently, multiple therapies with a pan-genotypic activity appeared on the market. The new agents (third generation) and new inhibitors (entry inhibitors, release inhibitors) being studied, should allow to cure most of the patients in the mid-term, if they will have equal access to the therapy.
INTRODUCTION
The hepatitis C virus (HCV) is a major blood borne human pathogen. There are approximately 120-130 million or 3% of the total world population that are HCV infected (Figure 1). According to World Health Organization (WHO), annually there are about 3-4 million new cases of infection[1,2]. HCV is considered a major public health issue, since the virus is the etiological factor of chronic hepatitis that frequently progress to a cirrhosis and hepatocellular carcinoma (HCC). In developed countries, the most important route of HCV transmission is intravenous drug abuse, whereas in resource-poor countries invasive procedures or injection-based therapies with contaminated instruments are the predominant source of new infections[3].
Figure 1.

Hepatitis C virus infection in the World. Analysis of seroprevalence[2].
Without treatment, most of the acute infections progress to chronic ones, followed by liver disease, such as cirrhosis and HCC. Alcohol abuse and the metabolic syndrome are the main cofactors influencing the progression to advanced liver disease and HCC[4]. Each year, about a third of liver transplantations are performed on patients with complications associated with the HCV infection, with decompensated cirrhosis or HCC[5]. In the next decade, an increase in the burden of hepatitis C is expected because of aging of the currently infected population[6,7]. During that period, the number of HCV-related cirrhosis cases is estimated to increase by 31% and HCC by approximately 50%[6] with an additive effect due to the occurrence of the metabolic syndrome[8,9]. Thus, HCV infection represents a major public health issue that should be addressed with strong policy interventions to effectively identify and treat HCV infected patients.
There are seven genotypes (gt 1-7) and numerous subtypes of HCV. The rate of infection and subtype prevalence are country depend. The difference between the infection rate in low and high endemic countries is about 20% (Figure 1)[10,11]. For example, North America, Western Europe and North and Australia’s have the lowest HCV prevalence. Conversely, in Asian and African countries the virus prevalence is high. The highest virus prevalence was registered in Egypt, where 22% of population is infected[12]. It has been postulated that the epidemic has been caused by extensive iatrogenic transmission during the era of parenteral-antischistosomal-therapy mass-treatment campaigns before 1985[13].
MOLECULAR BIOLOGY OF HCV
HCV morphology and parameters
HCV is a small, enveloped, positive single-stranded RNA virus that belongs to the Flaviviridae family, genus Hepacivirus. Analysis of viruses from plasma and from cell culture supernatant indicated that enveloped particles are icosahedral and 56-65 nm[14,15] in diameter, while and the viral core is about 45 nm[14,15]. Viral spikes on the membrane of the virion are about 6 nm[14] and they are formed by heterodimers of E1 and E2 glycoproteins. In fact, the population of the extracellular HCV particles is heterogeneous. Particles are pleomorphic and size, buoyant density, and infectivity might differ significantly[14,15]. A large majority of particles is non-infectious. Interestingly, the buoyant densities of infectious particles isolated from serum and from cell culture medium are different. A significant amount of the particles are associated with cellular lipoproteins making that a hallmark mark of HCV[16,17]. The pattern of virus-associated lipoproteins might differ and several of those are associated with HCV most frequently: low density lipoproteins (LDL), very low-density lipoproteins (VLDL) and apolipoproteins (Apo) A1, B, C and E (Figure 2)[18]. The viral particles associated with lipoproteins are called “lipoviral particles (LVP)”. More details on LVP are given in “Virus assembly and release” section. Detailed update on HCV-associated lipoproteins is given in the review by Grassi et al[19].
Figure 2.
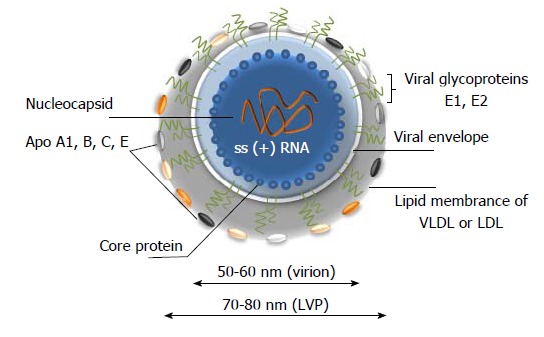
A model of hepatitis C virus lipoviral particle. Lipid membrane formed by low density lipoproteins (LDL) and very low-density lipoproteins (VLDL) on the surface of the virion (given in grey). Viral core is given in blue and viral RNA is shown in orange. Heterodimers of glycoproteins E1 and E2 are partially embedded in the lipid bilayer and are forming 6 nm long spikes (projections) on the surface of the virion[14-19]. As a result of association with LDL and VLDL, the morphology of the virion is not icosahedral. Depending on the viral source, the shape and size of the particles might vary.
Viral genome
The genome of HCV is approximately 9600 nucleotides long (Figure 3A). It contains two highly conserved untranslated regions (UTR) 5’-UTR and 3’-UTR that are flanking a single open reading frame (ORF). Dependent on the genotype, the ORF might contain from 9030 to 9099 nucleotides and it is coding for a single polyprotein precursor of 3010 to 3033 amino acids (aa), respectively (Figure 3B and C)[20,21]. Translation occurs in the endoplasmic reticulum and it is initiated by IRES at the 5’ UTR[22].
Figure 3.

Hepatitis C virus genome, polyprotein precursor and the initial steps of core assembly in endoplasmic reticulum. A: Being structurally identical, the genomes of seven hepatitis C virus (HCV) genotypes demonstrated approximately 30% of sequence diversity[140]. Two non-translated regions (5’- and 3’-NTR) are flanking a single open reading frame (ORF) shown in grey; B: Polyprotein precursor composed of about 3000 amino acids translated from a single ORF. Three structural proteins - a core protein (shown in blue) and two envelope proteins (shown in green) and seven non-structural (NS) proteins NS1, NS3, NS4A, NS4B, NS5A, and NS5B (shown in orange) that are involved in cleavage, assembly, transcription and some other functions are shown. Alternative reading frame protein (ARFP) that overlaps with the core protein sequence is given in blue. Serine protease NS2 is given in pale yellow. Transmembrane fragments in non-structural proteins are shown as staples; p- phosphoprotein; C: A model of initial steps of the virion core assembly in the endoplasmic reticulum (membranous web is given in dark green) on lipid droplet (LD). Polyprotein precursor is cleaved by cellular C-terminal signal peptidase (red arrow) and cellular signal peptidase (blue arrow) to release capsid protein. NS3-NS4A serine protease cleaves the remaining proteins, while NS2-NS3 (grey arrow) protease cleaves itself. Pre-assembled cores are transported to the LD, where the final steps of assembly take place[183].
5’-untranslated region
The 5’-UTR is highly conserved including special site to control the HCV genome replication and the viral polyprotein translation. The region is 341 nucleotides long and it contains four distinct domains (I-IV). The first 125 nucleotides of 5’UTR spanning the domains I and II have been shown to be essential for the viral RNA replication. The domains III-IV composes an internal ribosomal entry site (IRES) involved in ribosome binding which can initiate viral polyprotein translation in a cap-independent manner[20]. The initiation of HCV protein synthesis requires the ordered assembly of ribosomal pre-initiation complexes, beginning with the association of the small (40S) ribosomal subunit with a messenger RNA (mRNA). The cap-independent translation begins with the 40S ribosome binding and the scanning to the initiation codon which is followed by association with the 60S ribosomal subunit to form an active 80S ribosome[21,22]. The HCV IRES is folded into highly structured domains which are called II to IV. The mutational analysis of the IRES domains suggested that a structural integrity was necessary for an efficient protein synthesis both in vitro and in vivo[23-25].
3’-untranslated region
The 400 nucleotides long 3’-UTR region is thought to playing a crucial role in the HCV replication. The region is highly conserved and it is divided into three functionally parts: A variable sequence of 40 nucleotides, a variable internal poly (U/UC) rich tract of 30 to 80 nucleotides (depending on the HCV strains), which is followed by a highly conserved 98-nucleotide X-tail containing three stable stem loop (SL) structures called: 3’SL1, 3’SL2, 3’SL3[25].
Viral proteins
The translation occurs in the endoplasmic reticulum and it is initiated by IRES at the 5’UTR[25]. A single polyprotein precursor is processed by cellular and viral proteases into ten proteins (Figure 3B). Three structural proteins (core, E1, E2) are located at the amino-terminal part of the polyprotein and are essential components of the virions. Seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B) are located in the remaining part of the polyprotein and these proteins are involved in particle morphogenesis, RNA replication and in regulation of cell functions (Table 1). It is worth mentioning, that the structural and non-structural proteins of HCV are multifunctional. A brief characterization of the protein is given below.
Table 1.
Overview of the size of hepatitis C virus proteins
| Protein | No. of aa | aa position n ref. seq. | MW of protein, kDa |
| Core immature | 191 | 1-191 | 23 |
| Core mature | 174 | 1-174 | 21 |
| F protein of ARF protein | 126-161 | Approximately 16-17 | |
| E1 | 192 | 192-383 | 35 |
| E2 | 363 | 384-746 | 70 |
| p7 | 63 | 747-809 | 7 |
| NS2 | 217 | 810-1026 | 21 |
| NS3 | 631 | 1027-1657 | 70 |
| NS4A | 54 | 1658-1711 | 4 |
| NS4B | 261 | 1712-1972 | 27 |
| NS5A | 448 | 1973-2420 | 56 |
| NS5B | 591 | 2421-3011 | 66 |
No. of aa: Number of amino acid; MW: Molecular weight; kDa: KiloDalton; ref.seq.: Reference sequence (HCV strain H77 ; accession number NC_0041).
Core protein (p22)
The core protein of HCV is translated as an immature protein of 22 kDa, that it is composed of 191 amino acids (aa). It is excised from the polyprotein in the endoplasmic reticulum (ER) by a cellular signal peptidase (SP)[26]. An additional cleavage of the immature protein results in the mature 21 kDa core protein. The core protein contains three domains. The first domain spans the N-terminal region of 117 aa (aa 1-117). It contains mostly basic residues and there are two short hydrophobic regions. This domain is involved in binding to the viral RNA. The second domain between the aa 118 and 174 is more hydrophobic and less basic. This domain is engaged in the creation of links with the lipid droplet (LD). LDs are intracellular structures that are used for the lipid storage (Figure 3C)[18] . The third domain is localized between the aa 175 and aa 191 and it contains the signal sequence for the ER membrane translocation of E1 ectodomain[27]. In vivo, the mature core proteins are believed to form homo-multimers that are accumulated mainly at the ER membrane and can be self-assembly into the HCV-like particles[28]. During the viral capsid assembly, the protein also interacts with the HCV RNA. Besides that, the core protein poses regulatory functions during the RNA translation. Analyses of HCV core protein expression indicate that additionally it may be involved in several processes in cells such as apoptosis, lipid metabolism [HCV-related (mainly gt 3) fatty liver] and the development of HCC[29]. Interestingly, the core protein can induce the redistribution of LDs by the regeneration and the regression of these organelles in specific intracellular domains[30]. This phenomenon is also linked to the assembly of HCV particles. LDs are associated with the HCV core protein at the second domain and the interaction seems to trigger steatosis[30]. It has been shown, that if the core protein is mutated, the association with LDs might be disrupted. A decrease of this association has a negative effect on the HCVcc production. The HCV core protein was also shown to be involved in the Ca2+ regulation[31]. Thus, it is not only a basic structural element of HCV, but it is an active player in a significant amount of additional processes, including signaling pathways regulation[17,30,31].
Envelope proteins E1 (gp35) and E2 (gp70)
Glycoproteins E1 (gp35 kDa, 192 aa long) and E2 (gp70 kDa, 363 aa long) are type 1 transmembrane proteins that are forming the envelope of the virus particle (Figures 2 and 3B). E1 an E2 are cleaved from the precursor in ER by the cellular SP. The glycoproteins contained large hydrophilic ectodomains and 30 aa long transmembrane domains (TMD). The TMD is responsible for an anchoring of the envelope proteins in the membrane of the ER and their ER retention[32]. The ectodomains of both glycoproteins are heavily glycosylated. The E1 has 4-5 and the E2 - 11 putative N-glycosylation sites, respectively. Interestingly, the glycosylation sites are rather conserved in each genotype[33], but the number of glycosylation sites varies between the genotypes. It’s worth mentioning, that some of the glycan’s are engaged in folding and formation of the E1-E2 heterodimer complexes on the surface of the virion. These complexes are essential for the interaction with cellular receptors and helps to promote the virus-to-cell fusion[34].
The HCV E2 contains two hypervariable regions HVR 1 and HVR2 which are the most mutable parts of the HCV genome. The first 27 amino acids of E2 are attributed to the HVR1 sequence. It is suggested that a prominent heterogeneity of the region could help the virus to evade from the immune system pressure and to develop a chronic infection[35]. It is worth mentioning, that an infectious clone lacking the HVR1 was still capable to infect chimpanzee, but with a largely reduced efficiency, thus supporting the HVR1 engagement in the cell infection[36]. The HVR2 includes seven aa (91-97) and this region was shown to contribute to the HCV E2 receptor binding[37]. The high variability of the HVRs reflects the exposure of these domains to HCV-specific antibodies. In fact, the E2-HVR1 is the most frequent target for neutralizing antibodies[38,39]. However, the combination of a viral mutation with the selective pressure of the humoral immune response leads to the viral escape via epitope alterations[40]. Moreover, the association of virions with lipoproteins and the presence of a glycan shield (possessed by the E1-E2 heterodimers), reduce the antibody access to the “sensitive” neutralizing epitopes[24,41,42]. This protection is representing a serious obstacle on the way to obtain broadly neutralizing antibodies.
Non-structural protein NS1 (p7)
The NS1 (also called “p7”) is a small 63 aa long protein. It is located between E2 and NS2 proteins and it is linked to both structural and non-structural proteins. Its cleavage is mediated by the cellular SP. Interestingly, the E2-p7-NS2 precursor can also be detected[43,44]. It is suggested, that such a precursor may regulate the kinetic of the HCV infection; however, up to now the precise role of this protein in the HCV life cycle remains obscure. NS1 has two transmembrane domains (TMDS) embedded in the ER membrane and the C-terminal TMD of NS1 can act as a signal sequence to promote the translocation of NS2 to the ER lumen[45]. The protein can also form the ion channel which has an important role in the HCV infection. This channel can be blocked by the antiviral drug[45]. The recent studies demonstrate that NS1 acts to prevent the acidification which is required for the production of HCV particles[46]. In addition, the protein appears to be essential during assembly and release of infectious particles as shown on different genotypes[47]. Based on that, NS1 may be considered as a potential target for new antivirals[48,49].
Non-structural protein NS2 (p23)
The NS2 is a hydrophobic transmembrane protein of 23 kDa composed of 217 aa. It is a cysteine protease which is required for the HCV infectivity[50]. The N-terminal residues of NS2 can form 3 or 4 transmembrane helices which are inserted in the ER membrane. The crystal structure has been determined. The C-terminal residues of NS2 with the 181 aa long N-terminal domain of NS3 play an important role in the function of the NS2/3 cysteine protease[51]. The NS2/3 protease is spanning from the amino acid 810 to amino acid 1206. It has been shown that the NS3 zinc-binding domain could stimulate the activity of the NS2 protease[52]. The well-known function of NS2 is the auto-cleavage at the NS2/3 site[53]. Oem et al[53] suggested that the expression of HCV NS2 results in the up-regulation of the fatty acid synthase transcription. In fact, this may implicate the role of NS2 in the prompting HCV induced steatosis[54]. There data are in favor of the protein engagement in the viral assembly and release[54].
Non-structural protein NS3 (p70)
The NS3 is a 70 kDa protein composed of 631 aa. It is cleaved at its N-terminus by the viral NS2/NS3 autoprotease. The C-terminal part of NS3 (442 aa) has an ATPase/helicase activity, and it catalyses the binding and unwinding of the viral RNA genome during the viral replication[55,56]. The N-terminal part (189 aa) of the NS3 protein has both serine protease and NTPase activities[56]. The NS3 along with the non-structural protein NS4 is involved in cleavage of the NS3/4A, NS4A/4B, NS4B/5A and NS5A/5B junctions[57,58]. A direct interaction between NS3 and NS5B is mediated through the protease domain of NS3. It is suggested, that these proteins may act together during the HCV replication[59]. The HCV protease NS3/4A can also cleave some cellular targets involved in the innate immunity, such as MAVS (an antiviral signaling protein), which can indirectly activate the nuclear factor-kappa B (NF-κB) and the IFN regulatory factor 3 to induce type I interferon[60,61]. The protein NS3/NS4A can interfere with the adaptive immunity. In addition, the NS3/NS4A protease is essential for the viral infectivity, thus, it is a promising target for antivirals[61-64]. In 2011, two potent NS3/NS4A inhibitors, boceprevir[61] and telaprevir[62] were approved by FDA and EMA and were used in combination with IFN α and ribavirin. However, several resistance-associated mutations within the NS3/NS4A coding region have been observed.
Non-structural protein NS4A (p8)
The NS4A is an 8 kDa proteins that is composed of 54 aa. As indicated above, it is a co-factor required for the NS3 protease activity. The N-amino-terminal domain of NS4A is hydrophobic and the deletion analysis shows that NS4A was required for the ER targeting of NS3[65-68]. In addition the NS4A interaction with NS5A is essential for the phosphorylation of NS5A[68]. Besides its important role in HCV replication, NS4A can also contribute to the viral pathogenesis by influencing some cellular function[66]. Interestingly, NS4A was detected not only on the ER, but also on the mitochondria either alone or together with NS3 in the form of NS3/4A polyprotein. It has been shown, that the NS4 expression altered the distribution of mitochondria and caused damage which leads to the host cell apoptosis[66].
Non-structural protein NS4B (p27)
The NS4B is a 27 kDa protein of 217 aa. It has four membrane spanning domains which are important in recruitment of the other non-structural viral proteins[68]. By electron microscopy, it has been shown, that NS4B induced a tight structure termed “membranous web”. All viral proteins were found to be associated with this membranous web that formed a viral replication complex (“factory”) in HCV infected cells[67]. The polymerization activity and lipid modification of NS4B are important for the induction of the specialized membrane structure involved in the viral RNA replication[68,69]. NS4B is likely not engaged in the virus replication, but may contribute to the assembly and the release of the virus particles[69].
Non-structural protein NS5A (p56/p58)
The NS5A is a 56 kDa phosphoprotein of 485 aa (Figure 3). In HCV infected cells, the protein is present in two differently phosphorylated forms: phosphorylated (56 kDa) and hyper phosphorylated (58 kDa). NS5A is multifunctional and it contributes to the HCV replication, virus pathogenesis, modulation of cell signaling pathways, virus propagation and the interferon response[70,71]. The N-terminal amphipathic helix of the NS5A protein is conserved in different genotypes and it is necessary for the membrane localization[72-76]. Mutations in NS5A enhanced the capacity of the sub genomic HCV RNA replication in cell culture systems[77]. It was shown that the N-terminal part of NS5A contained a new zinc-coordination motif which affected the HCV replication[70]. NS5A has a potential role in mediating the IFN response. It contains a so-called “interferon-α sensitivity-determining region” (ISDR, aa 237-276)[78]. ISDR can be used to predict the resistance and the sensitivity of HCV to the IFN treatment. NS5A was also showed to be associated with a variety of cellular signaling pathways including apoptosis[70]. NS5A is already used as a target for the direct acting antivirals.
NS5B protein (p66-68)
The NS5B is a 591 aa long protein and it is located at the C-terminus of the precursor. NS5B is a RNA-dependent RNA polymerase containing the GDD motif in its active site[79]. The NS5B initiates synthesis of the HCV negative-strand RNA. The crystal structure of NS5B showed a typical ‘right hand’ polymerase shape with finger, palm and thumb sub domain. Since NS5B lacks the “proof-reading’’ function[79], numerous mutants might be generated during transcription. Because of its key role in the virus replication, the NS5B protein is considered as a potential target for the antiviral drug[80].
F protein, ARFP
In addition to ten proteins described above, the frameshift (F) or alternate reading frame protein (ARFP), or “core+1” protein has been reported[81-83]. The ARFP is the result of a -2/+1 ribosomal frameshift between codons 8 and 14 of the adenosine-rich region encoding the core protein. ARFP ends have different stop codons depending on the genotype. Thus, its length may vary from 126 to 161 amino acids. In the case of genotype 1a, the protein contains 161 amino acids. However, the situation with ARFP is more complicated, since alternative forms of this protein such as ARFP/DF (double-frame shift) in genotype 1b, and ARFP/S (short form) were recently described[83]. ARFP is a short-living protein located in the cytoplasm[84] in associated with the endoplasmic reticulum[85]. Detection of anti-ARFP antibodies in sera of HCV-positive subjects indicates that the protein is expressed during infection[86], but is likely not involved in the virus replication. Some findings suggested engagement of ARFP in the modulation of dendritic cells function and stimulation of the T cell responses[87]. The implication of ARFP in the viral life cycle remain to be elucidated.
HCV TRANSMISSION ROUTES
The HCV in blood and blood products is the main source of infection. However, the transmission routes of HCV might be different and ountry dependent. The iatrogenic transmissions are: the blood transfusion of unscreened products[88], the transfusion of clotting factors or other blood products[89,90], the organ transplantation, the reuse of medical instruments used in invasive settings (e.g., needles, infusion sets, syringes, catheters) in Egypt before 1985, hemodialysis, endoscopy, intravenous drug use[91-95]. The sexual way of transmission is controversial. Nevertheless, the risk may increase when favoring conditions such as sexually transmitted infections, the frequencies and the type of sexual activity are taking place[94-101].
The rate of HCV mother-to-child transmission is about 4.3%, but it is much higher, 22.1% among the human immunodeficiency virus (HIV) co-infected mothers[102,103]. The mother-to-child transmission generally occurs at delivery, but also in utero, if associated with high risk factors, such as high maternal HCV RNA levels and/or HIV co-infection. Interestingly, vaginal vs cesarean section delivery, amniocentesis and breast-feeding did seem to increase the transmission risk[102-108]. Because of a passive transfer of anti-HCV antibodies at, or after 18 mo of ages, perinatal transmission diagnosis as serum HCV RNA and/or anti-HCV antibodies should be performed twice in infants (6 mo and 1 year of age)[109] during this period, as recommended by the European pediatric HCV network[110]. It should be emphasized, that the INFL3 CC (IL28B) genotype is likely associated with a spontaneous clearance of the HCV genotype 1(gt1) infection[104].
According to the World Health Organization, thousands of new cases of the HCV infection (as a result of an occupational exposure via skin injury), are registered annually[111]. Most of these cases occurred during surgery in emergency departments, and routine medical procedures[112]. Seroconversion rates in infected individuals are ranging from 0% to 10.3% (+/- 0.75%)[113,114]. For instance, after accidental needle stick, seroconversion rate was reported as 1.8%[115]. During a 5-year period (2008-2012), a total of 16 HCV outbreaks resulting in 160 outbreak-associated cases and more than 90000 at-risk persons notified for screening were reported by the Centers for Disease Control (CDC) and Prevention[115]. Among dentists[116] and surgeons[117], an eye protection should be reinforced because of possible HCV transmission by splashes of blood and other body fluids[117]. Cosmetic procedures and/or acupuncture, as well as circumcision are extensively associated with HCV transmission, but the risk seems to be small[118,119]. Lacking of sterile techniques and/or nonprofessionally performed tattoos or piercings, especially before the mid-1980s, raised the HCV transmission rate significantly[120-126].
HCV GENOTYPES/SUBTYPES DEPENDENT TRANSMISSION
The HCV genome demonstrated a prominent genetic diversity. Seven genotypes (gt 1-7) and 67 subtypes (a, b, c, etc.) have been described. The genotypes are characterized by a distinct geographic distribution and clinical manifestations[127,128]. For example, the genotype 1 is prevalent in Americas, Japan and Europe[129]. Indeed, the HCV genotype prevalence also differs according to the transmission route and the age of infected individuals. For instance, the HCV gt 3a and 1a are highly represented among intravenous drug users and the HCV gt 1b is frequent among patients who received blood transfusions[130-134]. In Japan, the HCV gt 1b is the most prevalent[1] while, the infected population is generally older than in the United States[2], and an iatrogenic transmission is a predominant risk factor for the HCV acquisition.
HCV LIFE CYCLE
Binding and entry
The HCV entry initiates the viral replication cycle. Virus that is a complex and multi-step process that is not completely understood. However, the main principal steps and the principal cell surface interaction partners are known. At least, four cellular entry factors and two specific receptors are required for a successful virus entry (Figure 4). Since many interactions should take place, the binding to the cell surface is a relatively slow process which should be well coordinated[135-141]. The initiation of HCV entry seems to be similar to that of many other viruses, that utilize for attachment the glycosaminoglycans (GAGs)[141]. Earlier, it was proposed that E1 and E2 are involved in these interactions[141]. However, the HCV particle interacts with lipoproteins and in this regard, it is more likely that the apolipoprotein E is responsible for the interaction with GAGs[141]. Furthermore, due to the nature of the viral lipoprotein particle, it was proposed that the LDL receptor is required in the initial stage of attachment. However, this interaction leads the particle to a degradation pathway[141]. After the completion of docking, the viral particle interacts with specific cellular receptors. It was shown that the envelope glycoprotein E2 interacts with two receptors a scavenger receptor B1 (SR-B1), and with CD81 (tetraspanin family protein -TSPAN28)[142-144]. It is likely, that interaction of HCV with SR-B1 comes first[142]. In this regard, it remains to be determined whether SR-B1 and CD81 form a complex that facilitates the entry. It is noteworthy that the interaction with CD81 alone can activate signaling pathways that may be important for the viral infectious cycle[141] and the interaction between CD81 and the E2 glycoprotein appears to be essential for initiating the adsorption[141]. Next, the receptor complex with attached virion is moving to the tight junction, where the interaction with the proteins claudin-1 (Cldn1) and occludin (OCLN) is likely taking place. However, there is no experimental evidence indicating a direct interaction between the viral particle and the tight junction proteins. Thus, these proteins cannot be considered as classical receptors. Finally, other cellular factors such as epidermal growth factor receptor[145] and the Niemann-Pick C1-like 1 cholesterol uptake receptor[146] are likely involved in the HCV entry. It has been shown that CD81 and Cldn1 proteins may interact with the surface of hepatocytes[147], suggesting that CD81-Cldn1 form a complex that is implicated in the HCV entry. Although the tight junction proteins are involved in the HCV entry, the role of cell polarization in viral entry remains controversial. Interestingly, a live imaging of the fluorescent HCV particle in Huh7.5 cells showed that the particle was first attached on filopodia and then migrated towards the main body of the cell using presumably an actin-dependent transport mechanism[148]. It is important to note that the hepatocyte is a polarized cell with multiple apical and basolateral poles presenting a unique organization that is observed only in the liver. Another characteristic of HCV is its ability to spread by cell-to-cell contacts[149]. The essential role of the E1, E2 transmembrane domains in the viral entry[150] and the involvement of different regions of the ectodomain of E2 in the viral assembly were demonstrated[151]. It has been shown that glycans on the envelope proteins may be involved in different stages of the viral infectious cycle[42,152,153]. Furthermore, the N-linked glycan shield of the viral glycoproteins is essential to protect against the neutralization of the virus by antibodies[154].
Figure 4.
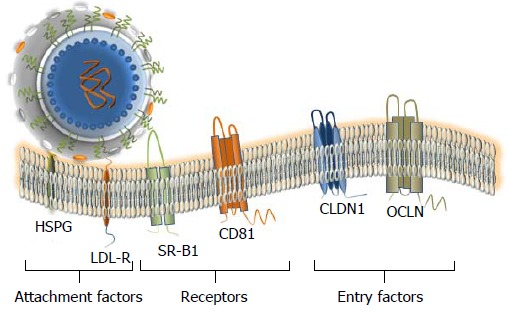
Attachment factors, receptor and entry factors utilized by hepatitis C virus. At least six membrane proteins are essential for the virus attachment and entry. Heparan sulfate proteoglycan (HSPG) - a type of glycosaminoglycan (GAG) and low density lipoproteins (LDL)-receptor that is promoting the LDL endocytosis are considered as binding factors for hepatitis C virus (HCV). Scavenger Receptor class B member 1 (SR-B1) and CD81 that are ubiquitously expressed on the cell surface, are considered as true receptors. The receptor–viral cargo complex is then moving to the cell-cell contacts (not shown), where the interaction with the tight-junction proteins Claudin 1 (CLDN1) and Occludin (OCLN) takes place. Both proteins are required at the late stage of the entry. Additional interaction partners that are likely engaged in the HCV entry are described in the text. The docking of the LVP to the cellular membrane is shown.
An additional interaction partner of the CD81 receptor, called “EWI-2wint” was identified[155]. It was demonstrated that EWI-2wint is a natural inhibitor of the HCV entry, which by an interaction with the CD81 receptor can prevent CD81-E2 interaction. Later on, the regions involved in the interaction between CD81 and EWI-2wint were identified[156] and studies in living cells at a single molecule level proved that EWI-2wint reduces the mobility of CD81 at the plasma membrane[157,158]. Finally, it was reported that the enrichment of the plasma membrane in ceramides has an inhibitory effect on the viral entry[159].
Post-entry events
It was shown by a traffic monitoring that after binding to the cells surface, the virus particles enter into the cells using clathrin dependent endocytic pathway[160,161]. The viral particles are transported to the early endosomes expressing RAB5A where the merger took place. This process requires an acidification of the compartment[162]. The virus capsid is then released and destroyed, while the viral RNA is released to the cytoplasm. The RNA is used for both processes the replication and the polyprotein translation (Figure 5). The RNA translation occurs in the endoplasmic reticulum (ER) and it is initiated by a binding of the 5’UTR IRES to the ribosome. The primary translation product is approximately 3000 amino acid long polyprotein precursor which contains structural and non-structural proteins of the HCV. Then, the polyprotein is cleaved by the host and viral proteases into structural and non-structural proteins.
Figure 5.
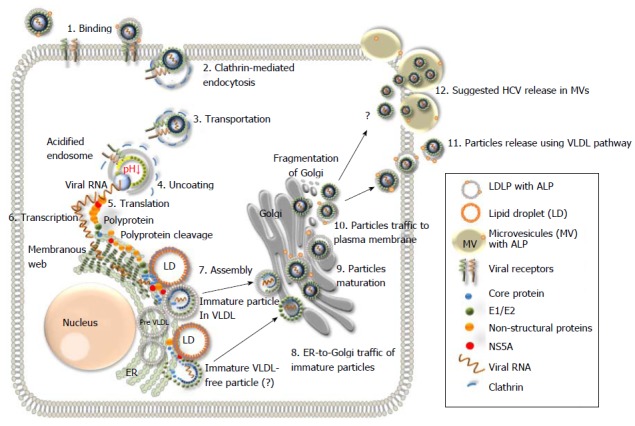
Schematic representation of the hepatitis C virus life cycle. (1) The lipoviral particles (LVP) binds to entry factors and receptors on the surface of hepatocyte; (2) The virus enters into the cell by a clathrin-mediated endocytosis; (3) Transportation of the virus in endosome; (3) Acidification of endosome, un-coating of the virion and dissociation of the viral core; (4) Release of the viral RNA; (5 and 6) Translation and replication of the viral RNA in the ER in the convoluted membrane structure called the membranous web (shown in dark green); (7) Cleavage of the protein precursor by cellular and viral proteases, assembly of the core on the surface of the lipid droplet (LD) and recruitment of the newly synthesized viral RNA to the viral core during the formation. The mechanism of the viral RNA recruitment to the site of the assembly is not known. It is also unclear whether the core maturation is finalized at this stage; (8) The viral nucleocapsid egresses into the lumen side of the ER, likely interacted with very low-density lipoproteins (VLDL) and translocated to Golgi. It is not clear if some VLDL-free particles can also be produced (?). Details of traffic machinery are not well defined; (9) Final maturation of viruses in Golgi and virus induced partial fragmentation of Golgi; (10) The hepatitis C virus (HCV) complex with VLDL is directed to the plasma membrane using the VLDL secretory pathway; (11) Release of particles from the cell (adapted from Ref.[16,19,135]); and (12) The HCV release in MV cannot be excluded as another pathway for the virus release during active replication.
Several studies evidenced that the HCV infection results in ER stress and autophagy responses and that HCV can regulate the autophagy pathway. In fact, the autophagy machinery is required to initiate the HCV replication and suppression of autophagy inhibits this process[163-171]. Interestingly, it has been demonstrated that HCV induces autophagosomes via a Class IIIPI3K-independent pathway and uses autophagosomal membranes as sites for its own RNA replication[171]. For the HCV RNA replication, polarized positive HCV RNA genome synthesizes a negative strand by the NS5B RNA-dependent RNA polymerase. The newly synthesized negative RNA strand may further act as a template to synthesize the positive strand of the viral RNA[172,173].
Viral assembly and release
A assembly of the virion is another multi-step process and certain steps of it remains obscure. It is known that the particle assembly occurs within the ER in a close proximity or directly on the surface to LD[170] (Figure 5). After the proteolytic cleavage of the polyprotein precursor, NS5A initiates the early phase of the viral particle formation by the interaction with the core protein and its C-terminal serine cluster determines the NS5A-core protein interaction[174].
The viral RNA released from the viral core is recruited to the replication complex within the membranous web. The HCV RNA-dependent RNA polymerase (NS5B) has no proofreading mechanism to correct errors during the strand synthesis. This propensity for error during replication results in an accumulation of the HCV quasispecies, those are closely related, but genetically somehow distinct. The newly synthesized positive-sense viral RNA is transported to the site of the core assembly and is encapsulated. However, the mechanism that engaged in RNA delivery to the capsid during assembly is not known. The nucleocapsids with RNA are presumably enveloped by budding into the lumen of the ER. So, after the nucleocapsid formation, it is associated with the envelope protein forming an immature particle and secreted from cell through the cytoplasmic membrane[173,174]. However, it remains unclear how the coupling with lipoproteins and apolipoproteins is regulated, since the pattern of lipoproteins on the virus particle might differ significantly. Next, the virions are transported through the Golgi compartment to finalize the maturation. Recently, it has been shown, that HCV can induce the fragmentation of Golgi[175]. Thus, it might be another important indication of the virus-induced pathogenesis. However, if the fragmentation of Golgi is significant, it may cause not only destructions of Golgi, but also provoke the cell death. Definitely, it might be not advantageous for the virus. It cannot be excluded, that this phenomenon might take place predominantly during the active replication of the virus. In this regard, it might be useful to investigate the HCV-induced Golgi destructions, as a possible trigger of liver necrosis.
Virus release and extracellular particles
The detail mechanism of the HCV release requires more comprehensive investigation. It is well established that secretion of the HCV particle depends on both the VLDL and the apolipoprotein (apoB) presence[176-187]. So, when the maturation is finished, the virions are transported to the plasma membrane using the VLDL pathway[145]. Particles isolated from cell-cultures and from patient sera are pleomorphic, immature and most of them are non-infectious[170]. In this regard, in the virus preparations from human plasma examined by electronic microscopy (EM), it was difficult to reveal “perfect” mature particle[170]. The association with fragments of cellular membranes (or other cellular debris) and/or an irregular lipoprotein content might be the reasons of the problem. Anyhow, it is evident, that a classical “assembly-maturation-release” processing line of HCV is facing problems. A diverse buoyant density of infectious particles is reflecting the problems. The analysis of LVP from plasma by sucrose gradient centrifugation demonstrated, that the buoyant density of the virus-contained fractions is diverse (1.10 g/mL to 1.16 g/mL)[170]. Positioning of virus-containing fraction on the gradient depends on the initial material that was used for the virus isolation. The infectious particles isolated by isopicnic centrifugation from cell-culture were mostly present in the fraction with a density 1.14 g/mL and examined by cryo EM, when were about 60 nm in diameter[170]. Significantly more virus-associated cellular impurities including lipoproteins, are associated with viruses isolated from patient sera and that influence the virus density and the particles are significantly less dense[177,178]. It is considered that the observed density fluctuation is a result of irregular content of lipoproteins tightly associated with virion. In fact, this diversity might have an additional reason. It cannot be excluded that in vivo the virus particles might in addition be transported by extracellular vesicles. In fact, the size of putative transporters might be large enough to carry not a single, but numerous viral particles. For example, membrane derived microvesicles (MVs) can be 500-1000 nm in diameter, so as large as lipid droplets[184,185]. MVs are present in the fractions of different densities, indicating a diversity of diameter and/or cargo[186].
Analysis of cell supernatant by gradient density centrifugation demonstrated that the MVs subpopulations are different, depending on the cell line. It is known, that MVs are intensively released by primary hepatocytes and immortalized cells of hepatic origin[186]. The release from hepatocytes might be increased by an undergoing lipotoxicity[187]. Thus, MVs are actively used by the hepatocytes as delivery system and the amounts of MVs might be increased in response to lipotoxicity and likely other complications. In this regard, we speculate that during acute phase of infection (or high rate of replication) the virus can trigger the MV release and that might be an additional pathway for the virus. How MVs containing virus particles can influence HCV distribution along the density gradient? In fact, MV containing different amount of viruses may contribute to observed diversity of buoyant densities of HCV, especially when plasma (or serum) is investigated.
It is known, that the diffusion of spherical particle through a viscous liquid is dependent on the particle diameter. It is described by Stokes-Einstein equation:
Math 1
Math 1.
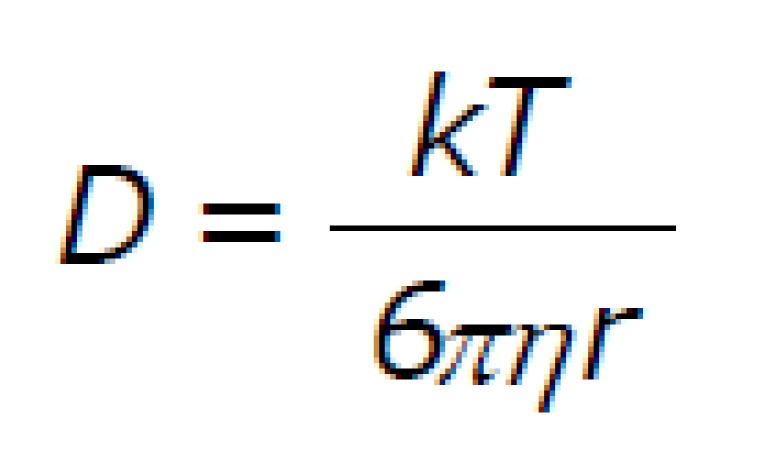
Math(A1).
D = diffusion constant, k = Boltzmann constant, T = temperature (K), η = solvent viscosity, r = radius of spherical particle.
Another parameter that influences the diffusion is the mass of spherical particle and the equation counting these parameters looks as follows:
Math 1
Math 1.

Math(A1).
where: D = diffusion; α = radius of the spherical particle; M = mass of the particle; η = viscosity of the medium.
So, the diffusion of a spherical particle is decreased with the increase of diameter and mass. Thus, if MVs besides the regular cargo (mRNA, miRNA, modulators, peptides, proteins ) have in addition a different particle load, it might explain why MVs of the same sizes might be recovered in fractions with different densities, and vice versa. Thus, when supernatants (or plasma) are examined by density gradient centrifugation, MVs of different size and different virus loaded, can increase the amount of virus-containing fractions. The expected distribution of lipoproteins, LVP from human plasma in a sucrose gradient and the suggested positioning of MVs with different virus load are given below (Figure 6, adapted from[170] with modifications).
Figure 6.
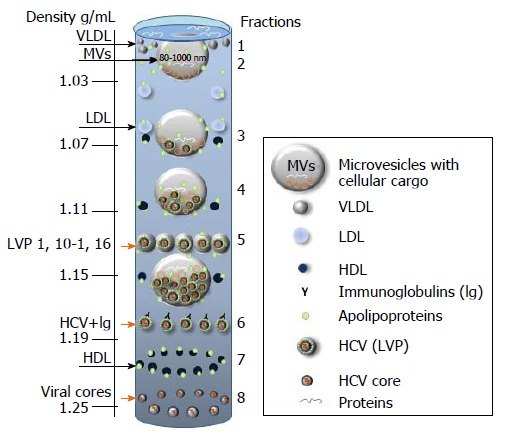
Putative distribution of lipoproteins (LDL, VLDL, HDL), lipoviral particles and viral cores after density gradient centrifugation of plasma from infected individual. The picture is based on the known buoyant densities of analyzed elements[15,170,184-187]. The density values (g/mL) are given on the left. The initial material that has been used for the virus isolation, contributes greatly to the associated lipid content of the virions and, as a consequence, it influences the buoyant density[177,183-187]. Because of an irregular protein to cholesterol amounts, high density lipoproteins (HDL) of 5-15 nm in diameter can be detected in the different fractions (1.06-1.21 g/mL). The family of HDL is given as dark blue spheres. The buoyant density of microvesicles (MVs) may vary from 1.02 g/mL to 1.16 g/mL, depending on MVs diameter and cargo (miRNA, tRNA, fragments of mRNA and proteins). If hepatitis C virus (HCV) is released in MVs with similar diffusion parameters (size and mass), the buoyant density of vesicle should be viral cargo-dependent. Fractions 1: Single cellular or viral proteins “floating” on the surface, very low-density lipoproteins (VLDL, diameter 50-75 nm, density 0.95-1.006 g/mL); Fraction 2: Low density lipoproteins (LDL, diameter 18-25 nm, density 1.019-1.06 g/mL) and virus-free MVs with a low cellular cargo content; Fraction 3: MVs that carry few viral particles, possible overlap with LDL and HDL; Fraction 4: MVs with a higher viral load, possible overlaps with HDL and with densely loaded virus-free MVs; Fraction 5: Lipoviral particles (LVP, diameter 60-80 nm, density 1.10-1.16 g/mL) and MVs with a significant viral and/or mixed load. Overlaps with HDL, virus-loaded MVs and “dense” virus-free MVs are possible; Fraction 6: Lipid-free virions with attached immunoglobulins, possible overlap with HDL; Fractions 7: HDL with a high protein load; Fraction 8: Non-enveloped viral cores that might overlap with dense HDL. Small grey spheres - VLDL. Light blue spheres - LDL. Orange arrows indicated fractions of the gradient that are most likely to contain viruses and viral cores.
What might be the advantage, for the virus to use the MVs secretory pathway? Since the MV are released by budding from the plasma membrane, it allows the virus to acquire additional protection from the “rough environment”. Second, if MV is loaded with numerous particles, it might be the way to increase the probability of a successful infection of distant cells by creating a high “local MOI” after MV distruction. That effect might be even more prominent, if the MV loaded with viruses would be endocytosed by the cell. We suggest, that the MV pathway might be used during active virus replication of the virus. The possible implication of MVs for the virus delivery might be interesting to investigate using in vitro models.
MODELS TO STUDY HCV
The progress in HCV research is completely dependent on model systems[188]. The most frequently used models (Figure 7) for investigation of HCV are described below.
Figure 7.

In vitro, ex vivo and in vivo models to study hepatitis C virus.
HCV replicon systems
The HCV genome was identified in 1989 by cloning it from infected chimpanzee, while in humans the amounts were too low for detection[189]. The first complete full-length HCV cDNA clone was constructed from the HCV strain H77 (genotype 1a). The HCV RNA transcribed from this clone was found to be infectious after intrahepatic injection in a chimpanzee. The HCV viremia was detected at week 1 and increased from 1 × 102 genomes/mL to 1 × 106 genomes/mL at week 8[190,191]. Then, several full-length HCV RNAs were synthesized and were shown to be infectious in chimpanzees[190-193]. However, these HCV clones were found to replicate inefficiently in vitro. This limitation was resolved by the group of Prof. Ralf Bartenschlager, when subgenomic HCV replicon, cloned from the HCV genome was constructed[73]. After transfection of this subgenomic clone into the Huh7 cells, it was found that in drug-resistant cells a high-level of the HCV RNA replication occurred. The replication of the HCV subgenomic replicon was confirmed in several cell lines, but the hepatocarcinoma cell line Huh7 was the most permissive. The interferon treatment of the replicon inoculated Huh7 cell clones, made them more permissive to support both subgenomic and full-length HCV replication, (these so-called cured cell lines are Huh7.5 or Huh 7.5.1 cells)[15,194]. Afterwards, several studies demonstrated that the virus and host factors were important for the HCV replication in cells. Some mutations in the wide-type (wt) consensus sequence efficiently enhanced the HCV replicon replication, some mutations in the non-structural protein efficiently contributed to the replication and the adaptation to the host cells[195-198]. The mechanism of improved replication caused by adaptive mutations is still unknown. Although these replicons with adaptive mutations could replicate with a high efficiency, they were not able to produce infectious particles in vitro.
This problem was solved with development of the genotype 2a infectious clone JFH-1. A selectable HCV replicon was constructed containing the full-length HCV cDNA and showed to produced infectious particles in vitro and in vivo[199]. Based on this infectious clone, several different genotypes chimeric clones (genotype 1a, 1b, 2a and 3b) where the non-structural genes have been replaced by those of JFH-1 were constructed and were shown to be infectious in Huh-7 cells. The most efficient construct is the genotype 2a/2a clone which consists of J6CF and JFH-1 derived sequence[200]. The HCV replicon is remarkably valuable for studying the HCV replication and for testing of new antiviral drugs. The HCV subgenomic replicons containing reporter genes (luciferase, secreted alkaline phosphatase and chloramphenicol transferase) facilitated the study of the HCV infection. This high-throughput screening assay allowed the visualization and tracking of the HCV replication complex in living host cells without affecting the HCV replication[47,201].
HCV pseudotype virus particles
The HCV pseudotyped particles were constructed with chimeric genes expressing HCV (genotype 1a) envelope E1 and E2 proteins (HCVpp) and the transmembrane and cytoplasmic tail of vesicular stomatitis virus G protein. The pseudotyped particles allowed a detailed study of the role of HCV receptors in the early steps of HCV infection (adsorption, and viral entry) in Huh7 cells and primary human hepatocytes[202-204]. The system is useful for testing of new antiviral drugs[203].
The HCV subgenomic replicon and the HCV pseudotyped particles (HCVpp) have markedly improved studies on the HCV infection. However, the high replication rate in cells was not correlated with amount of released infectious virions. However, the main disadvantage of these in vitro systems is that the viruses released from these cells are not infectious. The major reason of that might be that the adaptive mutations enhancing HCV replication rates are deleterious for HCV particles assembly and release. The problem was solved when a genotype 2a subgenomic replicon was established in the Huh-7 cell lines. It contained a full-length genotype 2a clone JFH-1 derived from serum of a Japanese patient with fulminant hepatitis. The replication rate was about 20 times greater than Con1 (gt1b) when transfected into Huh-7 cells. In addition, this replicon could replicate efficiently without amino acid mutations[47]. In 2005, Wakita et al[200] continued to transfect this JFH-1 clone into Huh-7 cell lines and found high levels of intracellular HCV RNA replication and protein expression. By means of passages in infected cells, they demonstrated that JFH-1 transfected cells had continuous HCV replication. In this study, it was also shown, that the HCV viral particles (HCVcc) secreted in the culture supernatant had a density of approximately 1.15-1.17 g/mL. The released particles have a spherical morphology and a diameter of about 55 nm. The JFH-1 infectivity could be neutralized by anti-CD81 antibody, suggesting the important role of CD81 in HCV entry. In addition it was found that the secreted particles could also infect chimpanzee[191]. Later, several groups observed that JFH-1 could efficiently infect and replicate without adaptive gene mutation in different cell types and HCV infectious particles could be produced in the culture supernatant[25,199,205-209]. Zhong et al[192] established a robust highly infectious in vitro system with JFH-1 and Huh-7.5.1 cells. The advantage of this model, compared to that established by the group of Prof. Wakita, was the possibility of HCV to undergo serial passages without losing of infectivity. Although JFH-1 in vitro system provides a powerful tool to study anti-viral drugs and vaccines, it has some limitations: HCV JFH-1 derived from an exceptional case of HCV-related fulminant hepatitis belongs to genotype 2a which is not the dominant genotype worldwide. That is why since 2005, different chimeric JFH-1 clones were constructed to transfect Huh-7.5 cells, based on intergenotypic genotype replicons such as J6/JFH-1 (genotype 2a/2a), S52/JFH1 (genotype 3a/2a) and sa13/JFH-1 (genotype 5a/2a)[209,210], but they are less infectious than HCV JFH-1. The reason why JFH-1 is more permissive to Huh-7.5 cell lines than other genotypes is still not fully understood. In summary, the HCVcc model allowed to understand the entire life cycle of HCV and that serves as a background for development of numerous antivirals[211].
Animal models
Chimpanzee could be a good model to study the HCV infection[194,212]. However, these animals are rare, difficult to handle, very costly and limited in their use by ethical issues[212]. Few data obtained using chimpanzee model indicated a specific immune response at the acute phase of infection, resulting in inconsistent specific neutralization and viral eradication.
Tupaia (Treeshrew, Anathana ellioti) is potentially a good model to study the HCV infection, but the instability of the infection and its low level limit the use of these animals.
The immunotolerized rat model supports the HCV replication. Histological and biochemical evidence of infection were present, but viremia was relatively low as compared with viral load in humans[188].
Mouse models compared with other animal models, have some advantages, such as producing animals in a short time (short gestation period), lower breeding cost and their small size making them easy to manipulate[188,213-217]. Heterotopic liver graft mouse model seemed to be suitable to evaluate the putative effect of anti-HCV drugs, but the short and low viremia and loss of the liver graft were limiting factors. Hepatic repopulation mouse model as the chimeric urokinase-type plasminogen activator/severe combined immunodeficient disorder mouse model has many similarities to human systems, and so is the most successful small animal model for HCV infection. The main limitation of the model is the absence of a normal immune system, which may be overcome by combining the model with a human hemato-lymphoid system, the value and reproducibility of which has yet to be established[218].
A mouse combined human immune system/human liver chimeric Rag2-g(c) model was developed in order to study immune system and liver pathogens. This ‘Hu-HEP’ model was used to study hepatocytes derived from human induced pluripotent stem cells. The main limitations of these models are the low number of animals that can be generated and the high cost[219-221].
Recent study demonstrated that hepatocyte-like cells differentiated from human embryonic stem cells and patient-derived induced pluripotent stem cells could be engrafted in the liver parenchyma of immune-deficient transgenic mice carrying the urokinase-type plasminogen activator gene driven by the major urinary protein promoter. This efficient engraftment and in vivo HCV infection of human stem cell-derived hepatocytes provide a model to study chronic HCV infection in patient-derived hepatocytes, action of antiviral therapies, and the biology of HCV infection[222-225].
In vitro models
Established human hepatocarcinoma cell line Huh-7 and its derivatives support the HCV replication. However, as every transformed cell line, they resemble the primary cells only partially. Thus, results of experiments performed on these cells might not be always appropriated[222].
The primary human hepatocytes and human fetal hepatocytes are more clinically and physiologically relevant. Thus, they are used to test the susceptibility of HCV to drugs and drug-metabolizing enzymes. Another in vitro model, the micropatterned co-cultures of primary human hepatocytes surrounded by a supportive stroma that expressed all known HCV entry factors. The cells could be infected by HCV pseudotyped particles and HCVcc, albeit with low viral titers. Using this method in combination with the highly sensitive luminescence-based and fluorescence reporter systems, the efficiency of anti-HCV therapeutics has been evaluated[222].
Recent studies show that both embryonic[223] and induced pluripotent[224,225] stem cells can be differentiated into hepatocytes, that are phenotypically similar to human fetal liver. Study of genetic defects that impact the HCV infection could be performed in a human iPS-derived hepatocyte-like cell-based model[225]. Induced human liver-like cells supported the entire life cycle of HCV genotype 2a reporter virus. Produced infectious particles were able to infect HuH-7.5 cells and secretion of TNF-α, and IL-28B/IL-29 was detected in cell culture supernatants. Thus, the evaluation of antiviral drugs using these cells was possible[222].
Ex vivo model
Analysis of precision-cutting adult human liver slices from infected or non-infected individuals represents another promising model. In fact, this model allowes to maintain the tridimensional structure of liver and analyse gene and protein expression.It is worth mentioning, that using this model, it was demonstrated for the first time, the ability of primary isolates (as well as JFH-1, H77/C3, Con1/C3) to undergo de novo viral replication with the production of high titer infectious virus. Thus, this approach allowed to validate the efficiency of the new antiviral drugs[226,227].
PATHOGENESIS
Immune-mediated liver damage
The HCV causes damage to the liver cells, but the exact mechanism of this phenomenon is unknown.It is believed, that the damage is largely mediated by the host immune response. In immunocompetent and immunocompromised patients with little, or no intrahepatic damage, including inflammation, high levels of the HCV replication have been reported[133,228,229]. In about 30% of HCV liver transplanted patients, despite the high levels of HCV replication, a recurrent hepatitis is developed one year after transplantation [230]. However, high levels of an intrahepatic HCV replication are usually tolerated by the host immune system. A lympho-mononuclear infiltrate represented mainly by CD8+ T cells expected to play a major role in the viral containment, though other subsets, such as CD4+ T and natural killer (NK) cells, and regulatory T cells (Treg) are considered[231]. The intrahepatic CD4+ and CD8+ T cells can recognize HCV structural and nonstructural antigens[232,233]. However, why in most patients the immune response cannot resolve the infection remains obscure. In fact, cytotoxic CD8+ T cell-mediated killing could be blunt by a predominant Treg response[234].
The liver fibrosis is caused by inflammatory cells of the intrahepatic infiltrate secreting cytokines and chemokines to activate hepatic stellate cells (HSC) to secrete collagen[235]. HSCs may exist as several different phenotypes with distinct molecular and cellular functions and features, each of which contributes significantly to the liver homeostasis and the disease. The quiescent stellate cells are critical to the normal metabolic functioning of the liver. The liver injury provokes the transdifferentiation of quiescent stellate cells to their activated phenotype, leading to a metabolic reprogramming. That increases the autophagy (to fuel the metabolic demands), the amplification of parenchymal injury and the development of ‘classic’ phenotypic features of activated HSCs/myofibroblasts. Through these changes, the activated stellate cells drive the fibrotic response to injury and the development of cirrhosis. As liver injury subsides, the activated stellate cells can be eliminated by one of three pathways: Apoptosis, senescence or reversion to an inactivated phenotype. The senescent stellate cells are more likely to be cleared by the NK cell-mediated cell death while the inactivated stellate cells remain ‘primed’ to respond to further liver injury. Reduction in the number of activated stellate cells contributes to the regression of fibrosis or cirrhosis and the liver repair in most, but not all patients. The relative inputs of these three pathways on the fibrosis regression are not clearly defined[233]. The HCV-specific CD8+ T cells expressing PD-1, a marker for exhaustion, were found at the time of the acute phase[236]. The acquisition of a memory phenotype and the recovery of an efficient CD8+ T cell function declined with HCV infections PD-1 expression, whereas, when HCV persisted and the HCV-specific CD8+ cells remained dysfunctional, high levels of PD-1 were maintained, the PD-1 ligation likely provides an overall inhibitory signal to Tregs cells shown in a study of PD-1 expression on Tregs in humans during the chronic HCV infection. In response to HCV antigens, the PD-1 blockade enhanced an interleukin-2 (IL-2)–dependent proliferation of intrahepatic Tregs and enhanced the overall ability of Tregs to inhibit T-effector cells. An increase in Treg proliferation with PD-1 blockade was linked to this effect[237]. A typical model of the wound-healing response to a persistent liver injury is the hepatic fibrosis occurring in the chronic hepatitis C[238]. Cytokines and chemokines capable of activating hepatic stellate cells to secrete collagen are secreted by the inflammatory cells of the intrahepatic infiltrate[239,240]. Thus, the fibrogenesis seems to be linked to the HCV expression through indirect mechanisms, mediated by a virally driven inflammation, but the direct role of viral factors in the disease progression should be investigated in more details. The cell injury, such as an oxidative stress and a steatosis, may be induced specifically by several viral proteins alone which could directly activate the hepatic stellate cells[241,242]. These observations may explain why some patients with chronic hepatitis C with normal liver enzymes and a minimal/mild inflammation may present a significant liver fibrosis as shown by the histology of liver biopsies. Overall, up to 70% of patients with a HCV cirrhosis will demonstrate a reversibility on follow-up biopsies[243-248]. Moreover, a reduced portal pressure and a decreased all-cause mortality are improved when the reversal occurs[245]. It is to point out that approximately 10% of HCV patients present a persistent or even progressive fibrosis following SVR, which might reflect other concurrent underlying liver diseases, especially a nonalcoholic fatty liver disease (NAFLD)[248]. The stellate cells can respond to cytokines and growth factors after priming stimuli. Then the proliferation, contractility, fibrogenesis, matrix degradation and proinflammatory signaling are enhanced. Now, it is clear that there are disease-specific pathways of fibrosis, without all activated cytokine pathways[249,250]. This is especially relevant to NAFLD, where there are many convergent pathogenic routes[248-250]. Importantly, different families of inflammatory cell types and their subsets may either promote or inhibit fibrosis[249-252].
Alterations of lipid metabolism
Lipids are required for the HCV replication and particles assembly. As mentioned above, HCV can modify the host serum lipid profile and this(ese) modification(s) can provoke the steatosis[253]. The steatosis is more frequent and more severe in patients with HCV gt 3 and it is correlated with a high HCV RNA levels. On one hand, in HCV-infected patients, the steatosis can be considered as a marker of the liver disease progression[254] and, on the other hand, as an indication of the reduced response to therapy[254]. However, if it is not metabolic or alcoholic steatosis, an efficient antiviral therapy is capable to reduce it[255,256].
Extrahepatic replication of HCV
The apparent presence of the HCV genomes in extrahepatic sites of patients infected with HCV has been shown. Using sensitive PCR assays, the HCV was revealed in leukocytes and there are evidences that these cells may represent a reservoir of the virus after treatment[257,258]. Interestingly, the pool of the HCV quasi-species differs between the plasma and peripheral blood monocytes, suggesting an independent spread of HCV within different cell types[259,260]. The infection of B-cells from non-Hodgkin’s lymphoma by HCV has also been demonstrated. A cell line established from transformed lymphocytes supported the HCV replication and also enable production of infectious viral particles capable to infect peripheral blood B cells[261]. The significance of these extrahepatic HCV reservoirs is not well understood, although one could speculate that the leukocyte compartments might represent an additional route by which HCV can directly manipulate the immune system and also another means by which the virus avoids the eradication.
Extrahepatic manifestations associated with the HCV infection
At least, one clinically significant extrahepatic manifestation occurs in 38% to 76% of HCV infected patients with chronic HCV[262,263]. The most frequent associated pathology is a mixed cryoglobulinemia. It is detected in 19% to 50% of HCV infected patients, while only 15% of them are symptomatic. The cryoglobulins are immunoglobulins which precipitate at a temperature below 37 °C. They are produced by HCV activated B cells. The cryoglobulins deposed in small and medium vessels are the cause of systemic vasculitis which can manifest in level joint, skin, renal or peripheral nerves[262]. Other observed extrahepatic manifestations are the following: lymphoma, thyroid disorders, diabetes, xerostomia and xerophthalmia[263-265]. The HCV infection cure leads to a gradual decrease of the cryoglobulin level in serum, followed by the remission of cryoglobulin-related symptoms and pathologic lesions[265]. Interestingly, as a result of treatment the incidence of type 2 diabetes is also reduced by approximately two thirds[264,265].
CLINICAL MANIFESTATION OF HCV INFECTION
Acute hepatitis
The acute hepatitis C is asymptomatic in 90% of infected people[266]. In some cases, asthenia, fever and muscle, and joint pain can appear. While, signs of jaundice are not frequent. Acute hepatitis C is characterized by a transient increase in the rate of serum transaminases. The first detectable virus marker is viral RNA that appears one to two weeks after exposure. Then seven-eight weeks later, the anti-HCV IgG response can be detected[266-268]. In 20% of cases, hepatitis C is resolved spontaneously through the innate and adaptive immunity[269]. The viral RNA becomes undetectable within three to four months after infection. Various factors could promote the viral clearance. Similarly, hepatitis acute symptoms would reflect a significant immune response of the host. The gene polymorphism of interleukin (IL) 28B also influences the host immune response[268,269]. The fulminant hepatitis C is exceptional[268].
Chronic infection
In about 80% of cases[268], the immune system is not capable to eradicate the HCV during the acute phase of infection. When the viral replication persists for more than six months after acute infection, the hepatitis is considered chronic. At the stage of chronic hepatitis, most patients are asymptomatic and may have no non-specific symptoms such as fatigue, arthralgia or myalgia. The transaminase levels may be moderately increased or even normal[270]. The long-term evolution of chronic infection is variable. The factors that accelerate the disease progression are the following: acquisition of more than 40 years, male gender, co-infection by HIV, higher body mass index, fatty liver and alcohol consumption[269]. After 10 to 30 years, about 20 to 30% of patients develop a cirrhosis. The cirrhosis may be associated with a liver failure, as a decompensation following a portal hypertension (ascites, gastrointestinal bleeding, etc.). In cirrhotic patients, the risk of death from complications is 4% per year, and their risk of developing an hepatocellular carcinoma (HCC) is 1 to 5% per year. Thirty-three percent of patients with HCC die within one year after diagnosis[269,270]. Overall, among patients with cirrhosis, the 5-year survival rate is 50%. In fact, the decompensated cirrhosis is the leading cause of liver transplantation[268,269] (Figure 8).
Figure 8.
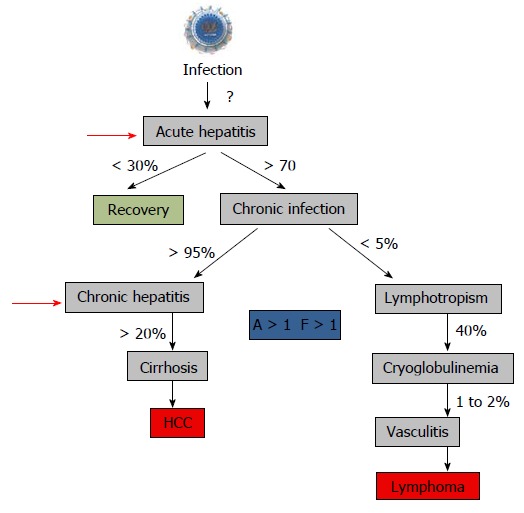
Natural history of hepatitis C virus infection and start to treat. The recommendation to treat chronic hepatitis is usually a “significant” fibrosis as defined by a Child–Pugh score (A to C) and a fibrosis grade (F) greater than 1 by the Metavir scoring system, with usually a significant necrotic-inflammation as defined by an activity stage greater than 1 by the Metavir scoring system[288,289]. The Child–Pugh score employs five clinical measures of liver disease: Total bilirubin, Serum albumin, Prothrombin time, Ascites, Hepatic encephalopathy. The letter F refers to the scars of the liver caused by the aggression. It is classified from F0 to F4: F1, F2 are minimal to moderate fibrosis, F3 corresponds to a pre-cirrhotic stage and F4 corresponds to cirrhosis. Red arrows indicated the time to start treatment[290].
DIAGNOSTIC
For HCV diagnosis both serologic and nucleic acid-based tests were developed[270,271]. Serologic tests are sufficient when chronic hepatitis C is expected, with a sensitivity of more than 99% if using the 3rd generation assays. Positive serologic results require additional HCV RNA or (with slightly reduced sensitivity) HCV core antigen measurements in order to differentiate between chronic hepatitis C and resolved HCV infection from the past. When an acute hepatitis C is considered, a serologic screening alone is insufficient, because mature anti-HCV antibodies are developed late after transmission of the virus.
Morphological methods like immunohistochemistry, in situ hybridization or PCR from liver specimens play no relevant role in the diagnosis of hepatitis C because of their low sensitivity, poor specificity and low efficacy compared to serologic and nucleic acid-based approaches.
HCV core antigen assay
Recently, a new quantitative HCV core antigen assay (Architect HCV Ag, Abbott Diagnostics) was approved by the EMA. This assay comprises 5 different antibodies targeted the HCV core. The test is highly specific (99.8%), equally effective for different HCV genotypes, and shows a relatively high sensitivity for the determination of chronic hepatitis C (corresponding to 600-1000 IU/mL HCV RNA). However, HCV core antigen correlated well, but not fully linearly, with HCV RNA serum levels, and false-negative results might be obtained in patients with an impaired immunity[271-273]. Another study has shown that the HCV core antigen quantification could be an alternative to the HCV RNA quantification for on-treatment antiviral response monitoring[274]. Here, a HCV core antigen below the limit of quantification at treatment 1 wk was strongly predictive of RVR, whereas patients with a less than 1 log10 decline in HCV core antigen at treatment 12 wk had a high probability of achieving nonresponse. The new HCV core antigen assay could be a cheaper, though somewhat less sensitive, alternative for nucleic acid testing.
Nucleic acid testing for HCV
Since the HCV RNA is detectable within a few days of infection; the nucleic acid-based tests are efficient in an early diagnostic of acute hepatitis C and should be considered as mandatory. The HCV RNA measurement is furthermore important in determination of the HCV genotype, selection of treatment strategy, therapy duration and evaluation of the treatment success[274]. For a number of antiviral combination therapies, the HCV RNA follow-up studies are essential to define the outcome of the treatment and further therapeutic strategies, if necessary. Traditionally, the tests should be repeated 24 wk after treatment completion to assess whether a sustained virologic response (SVR) has been achieved. However, as the probability of a virologic relapse is similar after 12 and 24 wk, the new time point for assessment of final virological treatment outcome is 12 wk after the end-of-treatment[275,276]. Both qualitative and quantitative PCR-based detection assays are available. Qualitative PCR tests are sensitive and are used for initial diagnostic of hepatitis C, for screening of blood and organ donations and for confirming SVR after treatment completion (Table 2). Quantitative reverse transcriptase (RT) real-time PCR-based assays can detect and quantify the HCV RNA over a very wide range, from approximately 10 IU/mL to 10 million IU/mL. The measurements are essential in the treatment monitoring when the virus load is gradually reducing.
Table 2.
Commercially available hepatitis C virus RNA detection assays
| Assay | Distributor | Technology | Detection limits (IU/mL) | Genotypes | Approval status |
| Qualitative HCV RNA detection assays | |||||
| AmplicorTM HCV 2.0 | Roche molecular Systems | PCR | 50 | All | FDA, CE |
| VersantTM HCV | Siemens Medical Solutions Diagnostics | TMA | 5-10 | All | FDA, CE |
| Quantitative HCV RNA detection | |||||
| AmplicorTM HCV Monitor 2.0 | Roche molecular Systems | PCR | 500 to approximately 5.105 | All | CE |
| HCV SuperQuantTM | National Genetics institute | PCR | 30 to 107 | All | U.S only |
| VersantR HCV RNA 3.0 | Siemens Medical Solutions Diagnostics | bDNA | 615 to 8.106 | All | FDA, CE |
| CobasR AmpliPrep/High pure system/CobasR TaqManR | Roche molecular Systems | Real-time PCR | 15 to 107 | All | FDA, CE |
| Abbot RealTimeTM HCV | Abbot Diagnostics | Real-time PCR | 10 to 107 | All | FDA, CE |
| Artus HCV QS-RGQ assay | Qiagen | Real-time PCR | 34 to 108 | All | CE |
| VersantRR HCV 1.0 kPCR assay | Siemens | Real-time PCR | 10 to approximately 107 | - | CE |
HCV: hepatitis C virus; TMA: Transcription-mediated amplification of RNA; bDNA: Branched DNA hybridization assay.
HCV genotyping
HCV genotyping is mandatory for every patient who considers antiviral therapy[277]. For DAA-based therapies, the determination of HCV genotypes and even subtypes is important because of significantly distinct barriers to resistance on the HCV subtype level. However, the importance for the HCV genotyping may decline with the availability of highly and broadly effective all oral combination therapies in the future. Both direct sequence analysis and reverse hybridization technology allow the HCV genotyping. Initial assays were designed to analyze exclusively the 5’UTR, which was burdened with a high rate of misclassification especially on the subtype level. Current assays were improved by additionally analyzing the coding regions, in particular the genes encoding core protein and the NS5B, both of which provide non-overlapping sequence differences between the genotypes and subtypes[278,279].
TREATMENT
All people with confirmed chronic HCV infection should be offered a high-quality care as soon as possible. Simultaneously, the screening and management of alcohol use is essential to prevent the progression to cirrhosis[278,280]. The modelling suggests that the treatment early in the course of HCV disease for all or specific populations could prevent the disease progression and onward transmission[281-283]. In fact, not all people with chronic HCV infection will progress to fibrosis[284]. So, medical authorities of a country to should decide when to start anti-HCV treatment. It should be emphasized that the transmission has been documented among people recently cured who lacked access to prevention services[285]. More research is needed to determine cost-effective eligibility criteria for both key and other populations that maximize reductions in the HCV-related morbidity, mortality, and transmission in different epidemiological contexts.
The WHO guidelines recommend the prioritizing treatment among people with an advanced fibrosis or cirrhosis in order to prevent a liver cancer[286]. The HCV co-infection in people living with HIV increases mortality while HIV has been shown to accelerate the progression of HCV disease[287,288]. Therefore, people with HIV/HCV co-infection may also warrant treatment prioritization. The stage of HCV disease could be estimated through liver biopsy (METAVIR system) identifying individuals needing an immediate treatment[288]. However, the liver biopsy test is expensive and can lead to complications such as infections, excessive bleeding, pain, or accidental injury to other organs. The use of liver function tests and platelet counts to determine the degree of liver fibrosis, such as the aminotransferase-to-platelet ratio index and the Fibrosis-4 score could be a non-invasive alternative and a more useful approach for gauging treatment eligibility across different tiers of health systems[289,290]. Monitoring systems should be put in place for people who do not initiate treatment immediately.
In the course of the last two decades, treatment of the HCV infection has significantly improved. For nearly 15 years, the combination of pegylated interferon alfa and ribavirin (PR) allowed a moderate sustain virologic response (SVR). Cure of the infection was achieved in 45% of genotype 1 and 65% of genotype 3 and around 85% of genotype 2 infected patients[291]. Development of the direct-acting antiviral drugs (DAAs) targeting viral proteins (NS3/4A protease, NS5B polymerase with nucleotide and non-nucleotide inhibitors, NS5A viral replication complex) was achieved due to the better understanding of the HCV life cycle. It revolutionized the treatment of chronic hepatitis C[292,293]. The classes of oral anti-HCV inhibitors and their characteristics are shown (Table 3). The combination of PR with the first-generation protease inhibitors demonstrated a high antiviral effectiveness (75% of SVR, but restricted to genotypes 1) with substantial adverse effects for the first-generation protease inhibitors. In 2011, telaprevir and boceprevir obtained a market approval. In 2012, the recommendations were approved for their use in HCV mono- and in 2013 for HCV/HIV co-infected infected patients. Then, SVR rates increased from 75% to 90% while reducing treatment duration, adverse effects, and the number of pills, thanks to the combination of PR with second-generation protease inhibitors[63,294,295]. From 2015 the standard of care is a combination of DAAs. Sofosbuvir, daclatasvir and the sofosbuvir/ledipasvir combination are part of the preferred regimens in the WHO guidelines, and can achieve cure rates above 95%[63]. These medicines are much more effective, safer and better-tolerated than the older therapies. In the first studies, excellent results were obtained on a group of so-called “easy-to-treat patients” and in small groups of patients have been confirmed in the phase III studies and in ”difficult-to-treat patients” [such as in check of previous regimens (including protease inhibitors), cirrhotic patients, liver or kidney transplanted patients, HIV infected patients or multi-drug treated patients, at an increased risk of the drug interaction][63]. Nevertheless, viral variants resistant to DAAs can emerge during the antiviral therapy[295-298] (Table 4). The estimation of a putative virus resistance profile prior to an antiviral therapy can help to select the optimal treatment regimen for individual patients[295,296].
Table 3.
Selected directly acting antiviral agents and host targeting agents in the pipeline[63]
| Drugs name | Company | Target/active site | Phase |
| NS3/4A protease inhibitors | |||
| Vaniprevir (MK-7009) | Merck | Active site/macrocyclic | III |
| Voxilaprevir GS-9857 | Gilead | Active site | III |
| Glecaprevir (ABT-493) | Abbvie | Active site | III |
| IDX21437 | Idenix | Active site | II |
| Sovaprevir (ACH-1625) | Achillion | Active site/macrocyclic? | II |
| Nucleoside analog NS5B polymerase inhibitors (NI) | |||
| MK-3682 (formerly IDX20963) | Merck | Active site | II |
| ACH-3422 | Achillion/Janssen | Active site | II |
| Non- Nucleoside analog NS5B polymerase inhibitors (NNI) | |||
| Beclabuvir (BMS-791325) | Bristol-Myers Squib | NNI site 1/Thumb 1 | III |
| Setrobuvir (ANA598) | Anadys/Roche | NNI site 4?/palm 1 | II |
| NS5A inhibitors | |||
| BMS-824393 | Bristol-Myers Squibb | NS5A protein | II |
| PPI-461 | Presidio | NS5A protein | II |
| PPI-668 | Presidio | NS5A protein | II |
| Pibrentasvir (ABT-530) | Abbvie | NS5A protein | III |
| ACH-2928 | Achillion | NS5A protein | I |
| Ruzasvir (MK-8408) | Merck | NS5A protein | II |
| Host targeting agents | |||
| SCY-635 | Scynexis | Cyclophilin inhibitor | II |
| Miravirsen | Santaris | miRNA122 antisense NA | II |
| RG-101- | Regulus | miRNA122 antisense NA | II |
| TT-0034 | Tacere Therapeutics | RNA interference with HCV | II |
Table 4.
Selected directly acting antiviral agents and host targeting agents whose development has been stopped or temporarily halted[63]
| Drugs name Company Target/Active site | ||
| NS3 /4A protease inhibitors | ||
| Ciluprevir BILN 2061 | Boehringer Ingelheim | Active site/macrocyclic |
| Narlaprevir (SCH900518) | Schering-Plough | Active site/linear |
| PHX1766 | Pheromix | Active site |
| Danoprevir (R7227) | Roche/InterMune | Active site/macrocyclic |
| Faldaprevir (BI201335) | Boehringer Ingelheim | Active site/linear |
| Telaprevir (VX-950) | Vertex | Withdrawn |
| Boceprevir (SCH503034) | Merck | Withdrawn |
| Nucleoside analogue NS5B polymerase inhibitors (NI) | ||
| Valopicitabine (NM283) | Idenix/Novartis | Active site |
| R1626 | Roche | Active site |
| Mericitabine (R7128) | Roche/Pharmasset | Active site |
| GS-938 | Gilead | Active site |
| IDX184 | Idenix | Active site |
| Non-nucleoside NS5B polymerase inhibitors (NNI) | ||
| BILB 1941 | Boehringer Ingelheim | NNI site 1/thumb |
| MK-3281 | Merck | NNI site 1/thumb 1 |
| VX-759 | Vertex | NNI site 2/thumb 2 |
| VX-222 | Vertex | NNI site 2/thumb 2 |
| VX-916 | Vertex | NNI site 2/thumb 2 |
| ABT-072 | AbbVie | NNI site 3/palm 1 |
| HCV-796 | ViroPharma/Wyeth | NNI site 4/palm 2 |
| Filibuvir (PF-00868554) | Pfizer | NNI site 2/thumb 2 |
| IDX375 | Idenix | NNI site 4/palm 2 |
| Tegobuvir (GS-9190) | Gilead | NNI site 4/palm 2 |
| GS-9669 | Gilead | NNI site 3/palm 1 |
| Deleobuvir | Böhringer | NNI site 3/palm 1 |
| Host targeting agents | ||
| NIM811 | Novartis | Cyclophilin inhibitor |
| Alisporivir (Debio-025) | Novartis | Cyclophilin inhibitor |
CONCLUSION
A prominent genetic diversity of HCV in combination with non-trivial replication cycle poses serious problems in virus research and therapy of virus-associated diseases. However, during the last several years a significant progress in understanding of the HCV molecular biology and pathogenesis has been achieved. In many aspects that allows to making a great step forward towards antivirals design, that result in DAA therapy. However, even with the new drugs the HCV-associated problems, of both fundamental and applied origin, are not solved. In fact, the resolution of these problems is going hand in hand.
First, in the absence of direct cytopathic effect, the mechanism of lipid deregulation, that results in liver pathologies, is not completely understood. Second, the system of the HCV control during latency and putative triggers of virus active replication were poorly investigated. Third, design of prophylactic vaccine is still in its infancy. Finally, the access to drugs and therapies for the people leaving in the third world is very restricted. Even if SVR rates > 90% are reached, nearly 10 million people will be living without treatment options[63]. Indeed, an easy access to diagnosis and treatment is still missing to drug users, people in difficult socio-economic situations, migrants, prisoners, because of themselves or health policy.
Therefore, fundamental studies on virus-cell interactions and studies directing towards development of the prophylactic vaccine should be intensified.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest to declare.
Manuscript source: Invited manuscript
Peer-review started: December 5, 2017
First decision: December 11, 2017
Article in press: February 7, 2018
Specialty type: Gastroenterology and hepatology
Country of origin: France
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Chetty R, He ST, Savopoulos CG S- Editor: Wang JL L- Editor: A E- Editor: Li RF
Contributor Information
Vladimir Alexei Morozov, Center for HIV and Retrovirology, Department of Infectious Diseases, Robert Koch Institute, Berlin 13353, Germany.
Sylvie Lagaye, Department of Immunology, Institut Pasteur, INSERM U1223, Paris 75015, France.
References
- 1.World Health Organization. Hepatitis C fact sheet. Available from: http://www.who.int/mediacentre/factsheets/fs164/en. Updated April 2014. Accessed February 18, 2015.
- 2.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 3.Hauri AM, Armstrong GL, Hutin YJ. The global burden of disease attributable to contaminated injections given in health care settings. Int J STD AIDS. 2004;15:7–16. doi: 10.1258/095646204322637182. [DOI] [PubMed] [Google Scholar]
- 4.Alberti A. What are the comorbidities influencing the management of patients and the response to therapy in chronic hepatitis C? Liver Int. 2009;29 Suppl 1:15–18. doi: 10.1111/j.1478-3231.2008.01945.x. [DOI] [PubMed] [Google Scholar]
- 5.Ponziani FR, Gasbarrini A, Pompili M, Burra P, Fagiuoli S. Management of hepatitis C virus infection recurrence after liver transplantation: an overview. Transplant Proc. 2011;43:291–295. doi: 10.1016/j.transproceed.2010.09.102. [DOI] [PubMed] [Google Scholar]
- 6.Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138:513–521, 521.e1-521.e6. doi: 10.1053/j.gastro.2009.09.067. [DOI] [PubMed] [Google Scholar]
- 7.Razavi H, Waked I, Sarrazin C, Myers RP, Idilman R, Calinas F, Vogel W, Mendes Correa MC, Hézode C, Lázaro P, et al. The present and future disease burden of hepatitis C virus (HCV) infection with today’s treatment paradigm. J Viral Hepat. 2014;21 Suppl 1:34–59. doi: 10.1111/jvh.12248. [DOI] [PubMed] [Google Scholar]
- 8.Kanwal F, Hoang T, Kramer JR, Asch SM, Goetz MB, Zeringue A, Richardson P, El-Serag HB. Increasing prevalence of HCC and cirrhosis in patients with chronic hepatitis C virus infection. Gastroenterology. 2011;140:1182–1188.e1. doi: 10.1053/j.gastro.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arase Y, Suzuki F, Suzuki Y, Akuta N, Kobayashi M, Kawamura Y, Yatsuji H, Sezaki H, Hosaka T, Hirakawa M, et al. Sustained virological response reduces incidence of onset of type 2 diabetes in chronic hepatitis C. Hepatology. 2009;49:739–744. doi: 10.1002/hep.22703. [DOI] [PubMed] [Google Scholar]
- 10.Centers for Disease Control. Disease burden from viral hepatitis A, B, and C in the United States. Available from: http://www.cdc.gov/hepatitis/Statistics/index.htm. Accessed February 18, 2015.
- 11.Centers for Disease Control and Prevention. CDC Health Information for International Travel 2014. New York: Oxford University Press; 2014. Available from: http://wwwnc.cdc.gov/travel/yellowbook/2014/chapter-3-infectious-diseases-related-to-travel/hepatitis-c. Accessed February 18, 2015. [Google Scholar]
- 12.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 13.Mohamoud YA, Mumtaz GR, Riome S, Miller D, Abu-Raddad LJ. The epidemiology of hepatitis C virus in Egypt: a systematic review and data synthesis. BMC Infect Dis. 2013;13:288. doi: 10.1186/1471-2334-13-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaito M, Watanabe S, Tsukiyama-Kohara K, Yamaguchi K, Kobayashi Y, Konishi M, Yokoi M, Ishida S, Suzuki S, Kohara M. Hepatitis C virus particle detected by immunoelectron microscopic study. J Gen Virol. 1994;75(Pt 7):1755–1760. doi: 10.1099/0022-1317-75-7-1755. [DOI] [PubMed] [Google Scholar]
- 15.Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, Chisari FV. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol. 2010;84:10999–11009. doi: 10.1128/JVI.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moriishi K, Matsuura Y. Exploitation of lipid components by viral and host proteins for hepatitis C virus infection. Front Microbiol. 2012;3:54. doi: 10.3389/fmicb.2012.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannini C, Bréchot C. Hepatitis C virus biology. Cell Death Differ. 2003;10 Suppl 1:S27–S38. doi: 10.1038/sj.cdd.4401121. [DOI] [PubMed] [Google Scholar]
- 18.Vercauteren K, Mesalam AA, Leroux-Roels G, and Meuleman P. Impact of lipids and lipoproteins on hepatitis C virus infection and virus neutralization. World J Gastroenterol. 2014;20:15975–15991. doi: 10.3748/wjg.v20.i43.15975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grassi G, Di Caprio G, Fimia GM, Ippolito G, Tripodi M, Alonzi T. Hepatitis C virus relies on lipoproteins for its life cycle. World J Gastroenterol. 2016;22:1953–1965. doi: 10.3748/wjg.v22.i6.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma SD. Hepatitis C virus: molecular biology & current therapeutic options. Indian J Med Res. 2010;131:17–34. [PubMed] [Google Scholar]
- 21.Fraser CS, Doudna JA. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat Rev Microbiol. 2007;5:29–38. doi: 10.1038/nrmicro1558. [DOI] [PubMed] [Google Scholar]
- 22.Fukushi S, Katayama K, Kurihara C, Ishiyama N, Hoshino FB, Ando T, Oya A. Complete 5’ noncoding region is necessary for the efficient internal initiation of hepatitis C virus RNA. Biochem Biophys Res Commun. 1994;199:425–432. doi: 10.1006/bbrc.1994.1246. [DOI] [PubMed] [Google Scholar]
- 23.Targett-Adams P, Hope G, Boulant S, McLauchlan J. Maturation of hepatitis C virus core protein by signal peptide peptidase is required for virus production. J Biol Chem. 2008;283:16850–16859. doi: 10.1074/jbc.M802273200. [DOI] [PubMed] [Google Scholar]
- 24.Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S. An overview of HCV molecular biology, replication and immune responses. Virol J. 2011;8:161. doi: 10.1186/1743-422X-8-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 26.Ait-Goughoulte M, Hourioux C, Patient R, Trassard S, Brand D, Roingeard P. Core protein cleavage by signal peptide peptidase is required for hepatitis C virus-like particle assembly. J Gen Virol. 2006;87:855–860. doi: 10.1099/vir.0.81664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roingeard P, Hourioux C. Hepatitis C virus core protein, lipid droplets and steatosis. J Viral Hepat. 2008;15:157–164. doi: 10.1111/j.1365-2893.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- 28.Roingeard P, Depla M. The birth and life of lipid droplets: learning from the hepatitis C virus. Biol Cell. 2011;103:223–231. doi: 10.1042/BC20100119. [DOI] [PubMed] [Google Scholar]
- 29.Dubuisson J, Cosset FL. Virology and cell biology of the hepatitis C virus life cycle: an update. J Hepatol. 2014;61:S3–S13. doi: 10.1016/j.jhep.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 30.Williamson CD, Colberg-Poley AM. Access of viral proteins to mitochondria via mitochondria-associated membranes. Rev Med Virol. 2009;19:147–164. doi: 10.1002/rmv.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV Jr, Ott M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat Med. 2010;16:1295–1298. doi: 10.1038/nm.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goffard A, Callens N, Bartosch B, Wychowski C, Cosset FL, Montpellier C, Dubuisson J. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J Virol. 2005;79:8400–8409. doi: 10.1128/JVI.79.13.8400-8409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slater-Handshy T, Droll DA, Fan X, Di Bisceglie AM, Chambers TJ. HCV E2 glycoprotein: mutagenesis of N-linked glycosylation sites and its effects on E2 expression and processing. Virology. 2004;319:36–48. doi: 10.1016/j.virol.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 34.von Hahn T, Rice CM. Hepatitis C virus entry. J Biol Chem. 2008;283:3689–3693. doi: 10.1074/jbc.R700024200. [DOI] [PubMed] [Google Scholar]
- 35.Sandres K, Dubois M, Pasquier C, Payen JL, Alric L, Duffaut M, Vinel JP, Pascal JP, Puel J, Izopet J. Genetic heterogeneity of hypervariable region 1 of the hepatitis C virus (HCV) genome and sensitivity of HCV to alpha interferon therapy. J Virol. 2000;74:661–668. doi: 10.1128/jvi.74.2.661-668.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forns X, Thimme R, Govindarajan S, Emerson SU, Purcell RH, Chisari FV, Bukh J. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc Natl Acad Sci USA. 2000;97:13318–13323. doi: 10.1073/pnas.230453597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roccasecca R, Ansuini H, Vitelli A, Meola A, Scarselli E, Acali S, Pezzanera M, Ercole BB, McKeating J, Yagnik A, et al. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J Virol. 2003;77:1856–1867. doi: 10.1128/JVI.77.3.1856-1867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, Shimizu Y, Shapiro M, Alter HJ, Purcell RH. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci USA. 1996;93:15394–15399. doi: 10.1073/pnas.93.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu YK, Igarashi H, Kiyohara T, Cabezon T, Farci P, Purcell RH, Yoshikura H. A hyperimmune serum against a synthetic peptide corresponding to the hypervariable region 1 of hepatitis C virus can prevent viral infection in cell cultures. Virology. 1996;223:409–412. doi: 10.1006/viro.1996.0497. [DOI] [PubMed] [Google Scholar]
- 40.Pantua H, Diao J, Ultsch M, Hazen M, Mathieu M, McCutcheon K, Takeda K, Date S, Cheung TK, Phung Q, et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J Mol Biol. 2013;425:1899–1914. doi: 10.1016/j.jmb.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 41.Voisset C, Op de Beeck A, Horellou P, Dreux M, Gustot T, Duverlie G, Cosset FL, Vu-Dac N, Dubuisson J. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J Gen Virol. 2006;87:2577–2581. doi: 10.1099/vir.0.81932-0. [DOI] [PubMed] [Google Scholar]
- 42.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol. 2010;84:11905–11915. doi: 10.1128/JVI.01548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carrère-Kremer S, Montpellier C, Lorenzo L, Brulin B, Cocquerel L, Belouzard S, Penin F, Dubuisson J. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J Biol Chem. 2004;279:41384–41392. doi: 10.1074/jbc.M406315200. [DOI] [PubMed] [Google Scholar]
- 44.Lin C, Lindenbach BD, Prágai BM, McCourt DW, Rice CM. Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. J Virol. 1994;68:5063–5073. doi: 10.1128/jvi.68.8.5063-5073.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003;535:34–38. doi: 10.1016/s0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- 46.Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 2010;6:e1001087. doi: 10.1371/journal.ppat.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci USA. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Griffin S. Inhibition of HCV p7 as a therapeutic target. Curr Opin Investig Drugs. 2010;11:175–181. [PubMed] [Google Scholar]
- 49.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol. 2007;81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welbourn S, Pause A. The hepatitis C virus NS2/3 protease. Curr Issues Mol Biol. 2007;9:63–69. [PubMed] [Google Scholar]
- 51.Schregel V, Jacobi S, Penin F, Tautz N. Hepatitis C virus NS2 is a protease stimulated by cofactor domains in NS3. Proc Natl Acad Sci USA. 2009;106:5342–5347. doi: 10.1073/pnas.0810950106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dimitrova M, Imbert I, Kieny MP, Schuster C. Protein-protein interactions between hepatitis C virus nonstructural proteins. J Virol. 2003;77:5401–5414. doi: 10.1128/JVI.77.9.5401-5414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oem JK, Jackel-Cram C, Li YP, Zhou Y, Zhong J, Shimano H, Babiuk LA, Liu Q. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J Gen Virol. 2008;89:1225–1230. doi: 10.1099/vir.0.83491-0. [DOI] [PubMed] [Google Scholar]
- 54.Yi M, Ma Y, Yates J, Lemon SM. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog. 2009;5:e1000403. doi: 10.1371/journal.ppat.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gallinari P, Brennan D, Nardi C, Brunetti M, Tomei L, Steinkühler C, De Francesco R. Multiple enzymatic activities associated with recombinant NS3 protein of hepatitis C virus. J Virol. 1998;72:6758–6769. doi: 10.1128/jvi.72.8.6758-6769.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin L, Peterson DL. Expression, isolation, and characterization of the hepatitis C virus ATPase/RNA helicase. Arch Biochem Biophys. 1995;323:47–53. doi: 10.1006/abbi.1995.0008. [DOI] [PubMed] [Google Scholar]
- 57.Kim DW, Gwack Y, Han JH, Choe J. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem Biophys Res Commun. 1995;215:160–166. doi: 10.1006/bbrc.1995.2447. [DOI] [PubMed] [Google Scholar]
- 58.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J Virol. 1993;67:3835–3844. doi: 10.1128/jvi.67.7.3835-3844.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jennings TA, Chen Y, Sikora D, Harrison MK, Sikora B, Huang L, Jankowsky E, Fairman ME, Cameron CE, Raney KD. RNA unwinding activity of the hepatitis C virus NS3 helicase is modulated by the NS5B polymerase. Biochemistry. 2008;47:1126–1135. doi: 10.1021/bi701048a. [DOI] [PubMed] [Google Scholar]
- 60.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, Hart A, Hesk D, et al. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob Agents Chemother. 2006;50:1013–1020. doi: 10.1128/AAC.50.3.1013-1020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, et al. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob Agents Chemother. 2006;50:899–909. doi: 10.1128/AAC.50.3.899-909.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mauss S, Berg T, Rockstroh J, Sarrazin C, Wedemeyer H. Hepatology - Clinical textbook. 8th Edition. Hamburg: Medizin Fokus Verlag; 2017. Available from: www.hepatologytextbook.com/download/hepatology2017.pdf. [Google Scholar]
- 64.Matsuura Y, Tani H, Suzuki K, Kimura-Someya T, Suzuki R, Aizaki H, Ishii K, Moriishi K, Robison CS, Whitt MA, et al. Characterization of pseudotype VSV possessing HCV envelope proteins. Virology. 2001;286:263–275. doi: 10.1006/viro.2001.0971. [DOI] [PubMed] [Google Scholar]
- 65.Selimović D, Hassan M. Inhibition of hepatitis C virus (HCV) core protein- induced cell growth by non-structural protein 4A (NS4A) is mediated by mitochondrial dysregulation. Bosn J Basic Med Sci. 2008;8:4–11. doi: 10.17305/bjbms.2008.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nomura-Takigawa Y, Nagano-Fujii M, Deng L, Kitazawa S, Ishido S, Sada K, Hotta H. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J Gen Virol. 2006;87:1935–1945. doi: 10.1099/vir.0.81701-0. [DOI] [PubMed] [Google Scholar]
- 67.Welsch C, Albrecht M, Maydt J, Herrmann E, Welker MW, Sarrazin C, Scheidig A, Lengauer T, Zeuzem S. Structural and functional comparison of the non-structural protein 4B in flaviviridae. J Mol Graph Model. 2007;26:546–557. doi: 10.1016/j.jmgm.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 68.Egger D, Wölk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol. 2002;76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones DM, Patel AH, Targett-Adams P, McLauchlan J. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J Virol. 2009;83:2163–2177. doi: 10.1128/JVI.01885-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macdonald A, Crowder K, Street A, McCormick C, Harris M. The hepatitis C virus NS5A protein binds to members of the Src family of tyrosine kinases and regulates kinase activity. J Gen Virol. 2004;85:721–729. doi: 10.1099/vir.0.19691-0. [DOI] [PubMed] [Google Scholar]
- 71.Reyes GR. The nonstructural NS5A protein of hepatitis C virus: an expanding, multifunctional role in enhancing hepatitis C virus pathogenesis. J Biomed Sci. 2002;9:187–197. doi: 10.1007/BF02256065. [DOI] [PubMed] [Google Scholar]
- 72.Singh P, Schnitzlein WM, Tripathy DN. Reticuloendotheliosis virus sequences within the genomes of field strains of fowlpox virus display variability. J Virol. 2003;77:5855–5862. doi: 10.1128/JVI.77.10.5855-5862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 74.Moradpour D, Brass V, Penin F. Function follows form: the structure of the N-terminal domain of HCV NS5A. Hepatology. 2005;42:732–735. doi: 10.1002/hep.20851. [DOI] [PubMed] [Google Scholar]
- 75.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Izumi N, Marumo F, Sato C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J Clin Invest. 1995;96:224–230. doi: 10.1172/JCI118025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gale MJ Jr, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 77.Tamura R, Kanda T, Imazeki F, Wu S, Nakamoto S, Tanaka T, Arai M, Fujiwara K, Saito K, Roger T, et al. Hepatitis C Virus nonstructural 5A protein inhibits lipopolysaccharide-mediated apoptosis of hepatocytes by decreasing expression of Toll-like receptor 4. J Infect Dis. 2011;204:793–801. doi: 10.1093/infdis/jir381. [DOI] [PubMed] [Google Scholar]
- 78.Carranza-Rosales P, Santiago-Mauricio MG, Guzmán-Delgado NE, Vargas-Villarreal J, Lozano-Garza G, Ventura-Juárez J, Balderas-Rentería I, Morán-Martínez J, Gandolfi AJ. Precision-cut hamster liver slices as an ex vivo model to study amoebic liver abscess. Exp Parasitol. 2010;126:117–125. doi: 10.1016/j.exppara.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 79.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 80.De Francesco R, Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 81.Walewski JL, Keller TR, Stump DD, Branch AD. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA. 2001;7:710–721. doi: 10.1017/s1355838201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu Z, Choi J, Yen TS, Lu W, Strohecker A, Govindarajan S, Chien D, Selby MJ, Ou J. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001;20:3840–3848. doi: 10.1093/emboj/20.14.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Varaklioti A, Vassilaki N, Georgopoulou U, Mavromara P. Alternate translation occurs within the core coding region of the hepatitis C viral genome. J Biol Chem. 2002;277:17713–17721. doi: 10.1074/jbc.M201722200. [DOI] [PubMed] [Google Scholar]
- 84.Roussel J, Pillez A, Montpellier C, Duverlie G, Cahour A, Dubuisson J, Wychowski C. Characterization of the expression of the hepatitis C virus F protein. J Gen Virol. 2003;84:1751–1759. doi: 10.1099/vir.0.19065-0. [DOI] [PubMed] [Google Scholar]
- 85.Xu Z, Choi J, Lu W, Ou JH. Hepatitis C virus f protein is a short-lived protein associated with the endoplasmic reticulum. J Virol. 2003;77:1578–1583. doi: 10.1128/JVI.77.2.1578-1583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pisani F, Fazio A, Artesi C, Russo M, Trio R, Oteri G, Perucca E, Di Perri R. Elevation of plasma phenytoin by viloxazine in epileptic patients: a clinically significant drug interaction. J Neurol Neurosurg Psychiatry. 1992;55:126–127. doi: 10.1136/jnnp.55.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Samrat SK, Li W, Singh S, Kumar R, Agrawal B. Alternate reading frame protein (F protein) of hepatitis C virus: paradoxical effects of activation and apoptosis on human dendritic cells lead to stimulation of T cells. PLoS One. 2014;9:e86567. doi: 10.1371/journal.pone.0086567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pomper GJ, Wu Y, Snyder EL. Risks of transfusion-transmitted infections: 2003. Curr Opin Hematol. 2003;10:412–418. doi: 10.1097/00062752-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 89.Koziol DE, Holland PV, Alling DW, Melpolder JC, Solomon RE, Purcell RH, Hudson LM, Shoup FJ, Krakauer H, Alter HJ. Antibody to hepatitis B core antigen as a paradoxical marker for non-A, non-B hepatitis agents in donated blood. Ann Intern Med. 1986;104:488–495. doi: 10.7326/0003-4819-104-4-488. [DOI] [PubMed] [Google Scholar]
- 90.Donahue JG, Muñoz A, Ness PM, Brown DE Jr, Yawn DH, McAllister HA Jr, Reitz BA, Nelson KE. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992;327:369–373. doi: 10.1056/NEJM199208063270601. [DOI] [PubMed] [Google Scholar]
- 91.Mauser-Bunschoten EP, Bresters D, van Drimmelen AA, Roosendaal G, Cuypers HT, Reesink HW, van der Poel CL, van den Berg HM, Lelie PN. Hepatitis C infection and viremia in Dutch hemophilia patients. J Med Virol. 1995;45:241–246. doi: 10.1002/jmv.1890450302. [DOI] [PubMed] [Google Scholar]
- 92.Chiaramonte M, Stroffolini T, Lorenzoni U, Minniti F, Conti S, Floreani A, Ntakirutimana E, Vian A, Ngatchu T, Naccarato R. Risk factors in community-acquired chronic hepatitis C virus infection: a case-control study in Italy. J Hepatol. 1996;24:129–134. doi: 10.1016/s0168-8278(96)80020-1. [DOI] [PubMed] [Google Scholar]
- 93.Frank C, Mohamed MK, Strickland GT, Lavanchy D, Arthur RR, Magder LS, El Khoby T, Abdel-Wahab Y, Aly Ohn ES, Anwar W, et al. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet. 2000;355:887–891. doi: 10.1016/s0140-6736(99)06527-7. [DOI] [PubMed] [Google Scholar]
- 94.Centers for Disease Control and Prevention (CDC) Acute hepatitis C virus infections attributed to unsafe injection practices at an endoscopy clinic--Nevada, 2007. MMWR Morb Mortal Wkly Rep. 2008;57:513–517. [PubMed] [Google Scholar]
- 95.Gutelius B, Perz JF, Parker MM, Hallack R, Stricof R, Clement EJ, Lin Y, Xia GL, Punsalang A, Eramo A, et al. Multiple clusters of hepatitis virus infections associated with anesthesia for outpatient endoscopy procedures. Gastroenterology. 2010;139:163–170. doi: 10.1053/j.gastro.2010.03.053. [DOI] [PubMed] [Google Scholar]
- 96.Terrault NA. Sexual activity as a risk factor for hepatitis C. Hepatology. 2002;36:S99–105. doi: 10.1053/jhep.2002.36797. [DOI] [PubMed] [Google Scholar]
- 97.Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. Centers for Disease Control and Prevention. MMWR Recomm Rep. 1998;47:1–39. [PubMed] [Google Scholar]
- 98.Thomas DL, Zenilman JM, Alter HJ, Shih JW, Galai N, Carella AV, Quinn TC. Sexual transmission of hepatitis C virus among patients attending sexually transmitted diseases clinics in Baltimore--an analysis of 309 sex partnerships. J Infect Dis. 1995;171:768–775. doi: 10.1093/infdis/171.4.768. [DOI] [PubMed] [Google Scholar]
- 99.Keiserman DR, Both CT, Mattos AA, Remiao J, Alexandre CO, Sherman KE. Intrafamilial transmission of hepatitis C virus in patients with hepatitis C and human immunodeficiency virus coinfection. Am J Gastroenterol. 2003;98:878–883. doi: 10.1111/j.1572-0241.2003.07340.x. [DOI] [PubMed] [Google Scholar]
- 100.Wandeler G, Gsponer T, Bregenzer A, Günthard HF, Clerc O, Calmy A, Stöckle M, Bernasconi E, Furrer H, Rauch A; Swiss HIV Cohort Study. Hepatitis C virus infections in the Swiss HIV Cohort Study: a rapidly evolving epidemic. Clin Infect Dis. 2012;55:1408–1416. doi: 10.1093/cid/cis694. [DOI] [PubMed] [Google Scholar]
- 101.American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C. Available from: http://hcvguidelines.org/full-report-view. Accessed February 18, 2015. [DOI] [PMC free article] [PubMed]
- 102.Yeung LT, King SM, Roberts EA. Mother-to-infant transmission of hepatitis C virus. Hepatology. 2001;34:223–229. doi: 10.1053/jhep.2001.25885. [DOI] [PubMed] [Google Scholar]
- 103.Polis CB, Shah SN, Johnson KE, Gupta A. Impact of maternal HIV coinfection on the vertical transmission of hepatitis C virus: a meta-analysis. Clin Infect Dis. 2007;44:1123–1131. doi: 10.1086/512815. [DOI] [PubMed] [Google Scholar]
- 104.Ruiz-Extremera A, Muñoz-Gámez JA, Salmerón-Ruiz MA, de Rueda PM, Quiles-Pérez R, Gila-Medina A, Casado J, Belén Martín A, Sanjuan-Nuñez L, Carazo A, et al. Genetic variation in interleukin 28B with respect to vertical transmission of hepatitis C virus and spontaneous clearance in HCV-infected children. Hepatology. 2011;53:1830–1838. doi: 10.1002/hep.24298. [DOI] [PubMed] [Google Scholar]
- 105.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yeung LT, To T, King SM, Roberts EA. Spontaneous clearance of childhood hepatitis C virus infection. J Viral Hepat. 2007;14:797–805. doi: 10.1111/j.1365-2893.2007.00873.x. [DOI] [PubMed] [Google Scholar]
- 107.Mast EE. Mother-to-infant hepatitis C virus transmission and breastfeeding. Adv Exp Med Biol. 2004;554:211–216. doi: 10.1007/978-1-4757-4242-8_18. [DOI] [PubMed] [Google Scholar]
- 108.Pondé RA. Hidden hazards of HCV transmission. Med Microbiol Immunol. 2011;200:7–11. doi: 10.1007/s00430-010-0159-9. [DOI] [PubMed] [Google Scholar]
- 109.Polywka S, Pembrey L, Tovo PA, Newell ML. Accuracy of HCV-RNA PCR tests for diagnosis or exclusion of vertically acquired HCV infection. J Med Virol. 2006;78:305–310. doi: 10.1002/jmv.20540. [DOI] [PubMed] [Google Scholar]
- 110.European Paediatric Hepatitis C Virus Network. A significant sex--but not elective cesarean section--effect on mother-to-child transmission of hepatitis C virus infection. J Infect Dis. 2005;192:1872–1879. doi: 10.1086/497695. [DOI] [PubMed] [Google Scholar]
- 111.Prüss-Ustün A, Rapiti E, Hutin Y. Estimation of the global burden of disease attributable to contaminated sharps injuries among health-care workers. Am J Ind Med. 2005;48:482–490. doi: 10.1002/ajim.20230. [DOI] [PubMed] [Google Scholar]
- 112.Makary MA, Al-Attar A, Holzmueller CG, Sexton JB, Syin D, Gilson MM, Sulkowski MS, Pronovost PJ. Needlestick injuries among surgeons in training. N Engl J Med. 2007;356:2693–2699. doi: 10.1056/NEJMoa070378. [DOI] [PubMed] [Google Scholar]
- 113.Kubitschke A, Bader C, Tillmann HL, Manns MP, Kuhn S, Wedemeyer H. [Injuries from needles contaminated with hepatitis C virus: how high is the risk of seroconversion for medical personnel really?] Internist (Berl) 2007;48:1165–1172. doi: 10.1007/s00108-007-1912-z. [DOI] [PubMed] [Google Scholar]
- 114.Wilkins T, Malcolm JK, Raina D, Schade RR. Hepatitis C: diagnosis and treatment. Am Fam Physician. 2010;81:1351–1357. [PubMed] [Google Scholar]
- 115.Centers for Disease Control and Prevention. Healthcare-associated hepatitis B and C outbreaks reported to the Centers for Disease Control and Prevention (CDC) in 2008-2012. Available from: http://www.cdc.gov/hepatitis/Statistics/HealthcareOutbreakTable.htm.Accessed February 18, 2015.
- 116.Younai FS, Murphy DC, Kotelchuck D. Occupational exposures to blood in a dental teaching environment: results of a ten-year surveillance study. J Dent Educ. 2001;65:436–448. [PubMed] [Google Scholar]
- 117.Marasco S, Woods S. The risk of eye splash injuries in surgery. Aust N Z J Surg. 1998;68:785–787. doi: 10.1111/j.1445-2197.1998.tb04677.x. [DOI] [PubMed] [Google Scholar]
- 118.Mele A, Corona R, Tosti ME, Palumbo F, Moiraghi A, Novaco F, Galanti C, Bernacchia R, Ferraro P. Beauty treatments and risk of parenterally transmitted hepatitis: results from the hepatitis surveillance system in Italy. Scand J Infect Dis. 1995;27:441–444. doi: 10.3109/00365549509047042. [DOI] [PubMed] [Google Scholar]
- 119.Sun DX, Zhang FG, Geng YQ, Xi DS. Hepatitis C transmission by cosmetic tattooing in women. Lancet. 1996;347:541. doi: 10.1016/s0140-6736(96)91172-1. [DOI] [PubMed] [Google Scholar]
- 120.Hwang LY, Kramer JR, Troisi C, Bull L, Grimes CZ, Lyerla R, Alter MJ. Relationship of cosmetic procedures and drug use to hepatitis C and hepatitis B virus infections in a low-risk population. Hepatology. 2006;44:341–351. doi: 10.1002/hep.21252. [DOI] [PubMed] [Google Scholar]
- 121.Hurley SF, Jolley DJ, Kaldor JM. Effectiveness of needle-exchange programmes for prevention of HIV infection. Lancet. 1997;349:1797–1800. doi: 10.1016/S0140-6736(96)11380-5. [DOI] [PubMed] [Google Scholar]
- 122.Briggs ME, Baker C, Hall R, Gaziano JM, Gagnon D, Bzowej N, Wright TL. Prevalence and risk factors for hepatitis C virus infection at an urban Veterans Administration medical center. Hepatology. 2001;34:1200–1205. doi: 10.1053/jhep.2001.29303. [DOI] [PubMed] [Google Scholar]
- 123.Balasekaran R, Bulterys M, Jamal MM, Quinn PG, Johnston DE, Skipper B, Chaturvedi S, Arora S. A case-control study of risk factors for sporadic hepatitis C virus infection in the southwestern United States. Am J Gastroenterol. 1999;94:1341–1346. doi: 10.1111/j.1572-0241.1999.01084.x. [DOI] [PubMed] [Google Scholar]
- 124.Karmochkine M, Carrat F, Dos Santos O, Cacoub P, Raguin G. A case-control study of risk factors for hepatitis C infection in patients with unexplained routes of infection. J Viral Hepat. 2006;13:775–782. doi: 10.1111/j.1365-2893.2006.00742.x. [DOI] [PubMed] [Google Scholar]
- 125.Ernst E, Sherman KJ. Is acupuncture a risk factor for hepatitis? Systematic review of epidemiological studies. J Gastroenterol Hepatol. 2003;18:1231–1236. doi: 10.1046/j.1440-1746.2003.03135.x. [DOI] [PubMed] [Google Scholar]
- 126.Davies D, King SM, Parekh RS, D’Angelo G. Psoas abscess caused by Haemophilus influenzae type B. Pediatr Infect Dis J. 1991;10:411–412. [PubMed] [Google Scholar]
- 127.Centers for Disease Control and Prevention CDC. Hepatitis C: general information. Available from: http://www.cdc.gov/correctionalhealth/docs/CDCHep_HepC.pdf. Accessed February 18, 2015.
- 128.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pawlotsky JM, Tsakiris L, Roudot-Thoraval F, Pellet C, Stuyver L, Duval J, Dhumeaux D. Relationship between hepatitis C virus genotypes and sources of infection in patients with chronic hepatitis C. J Infect Dis. 1995;171:1607–1610. doi: 10.1093/infdis/171.6.1607. [DOI] [PubMed] [Google Scholar]
- 130.McOmish F, Chan SW, Dow BC, Gillon J, Frame WD, Crawford RJ, Yap PL, Follett EA, Simmonds P. Detection of three types of hepatitis C virus in blood donors: investigation of type-specific differences in serologic reactivity and rate of alanine aminotransferase abnormalities. Transfusion. 1993;33:7–13. doi: 10.1046/j.1537-2995.1993.33193142314.x. [DOI] [PubMed] [Google Scholar]
- 131.Driesel G, Wirth D, Stark K, Baumgarten R, Sucker U, Schreier E. Hepatitis C virus (HCV) genotype distribution in German isolates: studies on the sequence variability in the E2 and NS5 region. Arch Virol. 1994;139:379–388. doi: 10.1007/BF01310799. [DOI] [PubMed] [Google Scholar]
- 132.Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens G, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 133.Zein NN. Clinical significance of hepatitis C virus genotypes. Clin Microbiol Rev. 2000;13:223–235. doi: 10.1128/cmr.13.2.223-235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paintsil E, Binka M, Patel A, Lindenbach BD, Heimer R. Hepatitis C virus maintains infectivity for weeks after drying on inanimate surfaces at room temperature: implications for risks of transmission. J Infect Dis. 2014;209:1205–1211. doi: 10.1093/infdis/jit648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zeisel MB, Fofana I, Fafi-Kremer S, Baumert TF. Hepatitis C virus entry into hepatocytes: molecular mechanisms and targets for antiviral therapies. J Hepatol. 2011;54:566–576. doi: 10.1016/j.jhep.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 137.International symposium on the first steps towards an international harmonization of veterinary biologicals: 1993 and free circulation of vaccines within the E. E.C. Ploufragan, France, 14-16 January 1992. Proceedings. Dev Biol Stand. 1992;79:1–224. [PubMed] [Google Scholar]
- 138.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol. 2012;86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Albecka A, Belouzard S, Op de Beeck A, Descamps V, Goueslain L, Bertrand-Michel J, Tercé F, Duverlie G, Rouillé Y, Dubuisson J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology. 2012;55:998–1007. doi: 10.1002/hep.25501. [DOI] [PubMed] [Google Scholar]
- 140.Bocchetta S, Maillard P, Yamamoto M, Gondeau C, Douam F, Lebreton S, Lagaye S, Pol S, Helle F, Plengpanich W, et al. Up-regulation of the ATP-binding cassette transporter A1 inhibits hepatitis C virus infection. PLoS One. 2014;9:e92140. doi: 10.1371/journal.pone.0092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Belouzard S, Cocquerel L, Dubuisson J. Hepatitis C virus entry into the hepatocyte. Cent Eur J Biol. 2011;6:933–945. doi: 10.2478/s11535-011-0076-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, et al. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol. 2010;84:34–43. doi: 10.1128/JVI.02199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, Crotta S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J Virol. 2008;82:8316–8329. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sharma NR, Mateu G, Dreux M, Grakoui A, Cosset FL, Melikyan GB. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J Biol Chem. 2011;286:30361–30376. doi: 10.1074/jbc.M111.263350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Harris HJ, Farquhar MJ, Mee CJ, Davis C, Reynolds GM, Jennings A, Hu K, Yuan F, Deng H, Hubscher SG, et al. CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol. 2008;82:5007–5020. doi: 10.1128/JVI.02286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Coller KE, Berger KL, Heaton NS, Cooper JD, Yoon R, Randall G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009;5:e1000702. doi: 10.1371/journal.ppat.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology. 2008;47:17–24. doi: 10.1002/hep.21959. [DOI] [PubMed] [Google Scholar]
- 150.Ciczora Y, Callens N, Penin F, Pécheur EI, Dubuisson J. Transmembrane domains of hepatitis C virus envelope glycoproteins: residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J Virol. 2007;81:2372–2381. doi: 10.1128/JVI.02198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Albecka A, Montserret R, Krey T, Tarr AW, Diesis E, Ball JK, Descamps V, Duverlie G, Rey F, Penin F, et al. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J Virol. 2011;85:1777–1792. doi: 10.1128/JVI.02170-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wahid A, Helle F, Descamps V, Duverlie G, Penin F, Dubuisson J. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J Virol. 2013;87:1605–1617. doi: 10.1128/JVI.02659-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vieyres G, Thomas X, Descamps V, Duverlie G, Patel AH, Dubuisson J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J Virol. 2010;84:10159–10168. doi: 10.1128/JVI.01180-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Helle F, Goffard A, Morel V, Duverlie G, McKeating J, Keck ZY, Foung S, Penin F, Dubuisson J, Voisset C. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J Virol. 2007;81:8101–8111. doi: 10.1128/JVI.00127-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rocha-Perugini V, Montpellier C, Delgrange D, Wychowski C, Helle F, Pillez A, Drobecq H, Le Naour F, Charrin S, Levy S, et al. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One. 2008;3:e1866. doi: 10.1371/journal.pone.0001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Montpellier C, Tews BA, Poitrimole J, Rocha-Perugini V, D’Arienzo V, Potel J, Zhang XA, Rubinstein E, Dubuisson J, Cocquerel L. Interacting regions of CD81 and two of its partners, EWI-2 and EWI-2wint, and their effect on hepatitis C virus infection. J Biol Chem. 2011;286:13954–13965. doi: 10.1074/jbc.M111.220103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Potel J, Rassam P, Montpellier C, Kaestner L, Werkmeister E, Tews BA, Couturier C, Popescu CI, Baumert TF, Rubinstein E, et al. EWI-2wint promotes CD81 clustering that abrogates Hepatitis C Virus entry. Cell Microbiol. 2013;15:1234–1252. doi: 10.1111/cmi.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rocha-Perugini V, Lavie M, Delgrange D, Canton J, Pillez A, Potel J, Lecoeur C, Rubinstein E, Dubuisson J, Wychowski C, et al. The association of CD81 with tetraspanin-enriched microdomains is not essential for Hepatitis C virus entry. BMC Microbiol. 2009;9:111. doi: 10.1186/1471-2180-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Voisset C, Lavie M, Helle F, Op De Beeck A, Bilheu A, Bertrand-Michel J, Tercé F, Cocquerel L, Wychowski C, Vu-Dac N, et al. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol. 2008;10:606–617. doi: 10.1111/j.1462-5822.2007.01070.x. [DOI] [PubMed] [Google Scholar]
- 160.Blanchard E, Brand D, Trassard S, Goudeau A, Roingeard P. Hepatitis C virus-like particle morphogenesis. J Virol. 2002;76:4073–4079. doi: 10.1128/JVI.76.8.4073-4079.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouillé Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Meertens L, Bertaux C, Dragic T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol. 2006;80:11571–11578. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci USA. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dreux M, Chisari FV. Impact of the autophagy machinery on hepatitis C virus infection. Viruses. 2011;3:1342–1357. doi: 10.3390/v3081342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mohl BP, Tedbury PR, Griffin S, Harris M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J Virol. 2012;86:10724–10732. doi: 10.1128/JVI.01667-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol. 2012;86:8705–8712. doi: 10.1128/JVI.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Huang H, Kang R, Wang J, Luo G, Yang W, Zhao Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy. 2013;9:175–195. doi: 10.4161/auto.22791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, Machida K, Ou JH. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem. 2012;287:18036–18043. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lindenbach BD, Rice CM. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol. 2013;11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rice CM. New insights into HCV replication: potential antiviral targets. Top Antivir Med. 2011;19:117–120. [PMC free article] [PubMed] [Google Scholar]
- 172.Murakami K, Ishii K, Ishihara Y, Yoshizaki S, Tanaka K, Gotoh Y, Aizaki H, Kohara M, Yoshioka H, Mori Y, et al. Production of infectious hepatitis C virus particles in three-dimensional cultures of the cell line carrying the genome-length dicistronic viral RNA of genotype 1b. Virology. 2006;351:381–392. doi: 10.1016/j.virol.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 173.Ezelle HJ, Markovic D, Barber GN. Generation of hepatitis C virus-like particles by use of a recombinant vesicular stomatitis virus vector. J Virol. 2002;76:12325–12334. doi: 10.1128/JVI.76.23.12325-12334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J Virol. 2008;82:7964–7976. doi: 10.1128/JVI.00826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hansen MD, Johnsen IB, Stiberg KA, Sherstova T, Wakita T, Richard GM, Kandasamy RK, Meurs EF, Anthonsen MW. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc Natl Acad Sci USA. 2017;114:E3462–E3471. doi: 10.1073/pnas.1616683114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Calattini S, Fusil F, Mancip J, Dao Thi VL, Granier C, Gadot N, Scoazec JY, Zeisel MB, Baumert TF, Lavillette D, et al. Functional and Biochemical Characterization of Hepatitis C Virus (HCV) Particles Produced in a Humanized Liver Mouse Model. J Biol Chem. 2015;290:23173–23187. doi: 10.1074/jbc.M115.662999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dao Thi VL, Granier C, Zeisel MB, Guérin M, Mancip J, Granio O, Penin F, Lavillette D, Bartenschlager R, Baumert TF, et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J Biol Chem. 2012;287:31242–31257. doi: 10.1074/jbc.M112.365924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bartenschlager R, Penin F, Lohmann V, André P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 181.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 182.Sumpter R Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McLauchlan J, Lemberg MK, Hope G, Martoglio B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002;21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ettelaie C, Collier ME, Maraveyas A, Ettelaie R. Characterization of physical properties of tissue factor-containing microvesicles and a comparison of ultracentrifuge-based recovery procedures. J Extracell Vesicles. 2014:3. doi: 10.3402/jev.v3.23592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Conde-Vancells J, Rodriguez-Suarez E, Embade N, Gil D, Matthiesen R, Valle M, Elortza F, Lu SC, Mato JM, Falcon-Perez JM. Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J Proteome Res. 2008;7:5157–5166. doi: 10.1021/pr8004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cannito S, Morello E, Bocca C, Foglia B, Benetti E, Novo E, Chiazza F, Rogazzo M, Fantozzi R, Povero D, et al. Microvesicles released from fat-laden cells promote activation of hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS One. 2017;12:e0172575. doi: 10.1371/journal.pone.0172575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lagaye S, Shen H, Rousseau G, Pol S. Major models of Hepatitis C virus infection. Future Virol. 2013;8:493–506. [Google Scholar]
- 189.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 190.Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 191.Yanagi M, Purcell RH, Emerson SU, Bukh J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122:352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 194.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Friebe P, Lohmann V, Krieger N, Bartenschlager R. Sequences in the 5’ nontranslated region of hepatitis C virus required for RNA replication. J Virol. 2001;75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Guo JT, Bichko VV, Seeger C. Effect of alpha interferon on the hepatitis C virus replicon. J Virol. 2001;75:8516–8523. doi: 10.1128/JVI.75.18.8516-8523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Krieger N, Lohmann V, Bartenschlager R. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol. 2001;75:4614–4624. doi: 10.1128/JVI.75.10.4614-4624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lohmann V, Körner F, Dobierzewska A, Bartenschlager R. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J Virol. 2001;75:1437–1449. doi: 10.1128/JVI.75.3.1437-1449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 200.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Murray EM, Grobler JA, Markel EJ, Pagnoni MF, Paonessa G, Simon AJ, Flores OA. Persistent replication of hepatitis C virus replicons expressing the beta-lactamase reporter in subpopulations of highly permissive Huh7 cells. J Virol. 2003;77:2928–2935. doi: 10.1128/JVI.77.5.2928-2935.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yi M, Bodola F, Lemon SM. Subgenomic hepatitis C virus replicons inducing expression of a secreted enzymatic reporter protein. Virology. 2002;304:197–210. doi: 10.1006/viro.2002.1652. [DOI] [PubMed] [Google Scholar]
- 203.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset FL. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem. 2003;278:41624–41630. doi: 10.1074/jbc.M305289200. [DOI] [PubMed] [Google Scholar]
- 205.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhang J, Randall G, Higginbottom A, Monk P, Rice CM, McKeating JA. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J Virol. 2004;78:1448–1455. doi: 10.1128/JVI.78.3.1448-1455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Matsumura T, Hu Z, Kato T, Dreux M, Zhang YY, Imamura M, Hiraga N, Juteau JM, Cosset FL, Chayama K, et al. Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology. 2009;137:673–681. doi: 10.1053/j.gastro.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Date T, Kato T, Miyamoto M, Zhao Z, Yasui K, Mizokami M, Wakita T. Genotype 2a hepatitis C virus subgenomic replicon can replicate in HepG2 and IMY-N9 cells. J Biol Chem. 2004;279:22371–22376. doi: 10.1074/jbc.M311120200. [DOI] [PubMed] [Google Scholar]
- 209.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 210.Jensen TB, Gottwein JM, Scheel TK, Hoegh AM, Eugen-Olsen J, Bukh J. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J Infect Dis. 2008;198:1756–1765. doi: 10.1086/593021. [DOI] [PubMed] [Google Scholar]
- 211.Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci USA. 2008;105:997–1002. doi: 10.1073/pnas.0711044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Manickam C, Reeves RK. Modeling HCV disease in animals: virology, immunology and pathogenesis of HCV and GBV-B infections. Front Microbiol. 2014;5:690. doi: 10.3389/fmicb.2014.00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Carpentier A, Tesfaye A, Chu V, Nimgaonkar I, Zhang F, Lee SB, Thorgeirsson SS, Feinstone SM, Liang TJ. Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J Clin Invest. 2014;124:4953–4964. doi: 10.1172/JCI75456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bissig KD, Le TT, Woods NB, Verma IM. Repopulation of adult and neonatal mice with human hepatocytes: a chimeric animal model. Proc Natl Acad Sci USA. 2007;104:20507–20511. doi: 10.1073/pnas.0710528105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gaska JM, Ploss A. Study of viral pathogenesis in humanized mice. Curr Opin Virol. 2015;11:14–20. doi: 10.1016/j.coviro.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.von Schaewen M, Ding Q, Ploss A. Visualizing hepatitis C virus infection in humanized mice. J Immunol Methods. 2014;410:50–59. doi: 10.1016/j.jim.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ploss A. Mouse models for human infectious diseases. J Immunol Methods. 2014;410:1–2. doi: 10.1016/j.jim.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 219.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature. 2013;501:237–241. doi: 10.1038/nature12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Billerbeck E, de Jong Y, Dorner M, de la Fuente C, Ploss A. Animal models for hepatitis C. Curr Top Microbiol Immunol. 2013;369:49–86. doi: 10.1007/978-3-642-27340-7_3. [DOI] [PubMed] [Google Scholar]
- 221.von Schaewen M, Ploss A. Murine models of hepatitis C: what can we look forward to? Antiviral Res. 2014;104:15–22. doi: 10.1016/j.antiviral.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gondeau C, Pichard-Garcia L, Maurel P. Cellular models for the screening and development of anti-hepatitis C virus agents. Pharmacol Ther. 2009;124:1–22. doi: 10.1016/j.pharmthera.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 223.Funakoshi N, Duret C, Pascussi JM, Blanc P, Maurel P, Daujat-Chavanieu M, Gerbal-Chaloin S. Comparison of hepatic-like cell production from human embryonic stem cells and adult liver progenitor cells: CAR transduction activates a battery of detoxification genes. Stem Cell Rev. 2011;7:518–531. doi: 10.1007/s12015-010-9225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Schwartz RE, Trehan K, Andrus L, Sheahan TP, Ploss A, Duncan SA, Rice CM, Bhatia SN. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109:2544–2548. doi: 10.1073/pnas.1121400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lagaye S, Shen H, Saunier B, Nascimbeni M, Gaston J, Bourdoncle P, Hannoun L, Massault PP, Vallet-Pichard A, Mallet V, et al. Efficient replication of primary or culture hepatitis C virus isolates in human liver slices: a relevant ex vivo model of liver infection. Hepatology. 2012;56:861–872. doi: 10.1002/hep.25738. [DOI] [PubMed] [Google Scholar]
- 227.Lagaye S, Brun S, Gaston J, Shen H, Stranska R, Camus C, Dubray C, Rousseau G, Massault PP, Courcambeck J, et al. Anti-hepatitis C virus potency of a new autophagy inhibitor using human liver slices model. World J Hepatol. 2016;8:902–914. doi: 10.4254/wjh.v8.i21.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.CDC. Hepatitis C FAQs for the public. Available from: http://www.cdc.gov/hepatitis/c/cfaq.htm. Accessed February 18, 2015.
- 229.McGuinness PH, Bishop GA, Painter DM, Chan R, McCaughan GW. Intrahepatic hepatitis C RNA levels do not correlate with degree of liver injury in patients with chronic hepatitis C. Hepatology. 1996;23:676–687. doi: 10.1002/hep.510230404. [DOI] [PubMed] [Google Scholar]
- 230.Demetris AJ. Evolution of hepatitis C virus in liver allografts. Liver Transpl. 2009;15 Suppl 2:S35–S41. doi: 10.1002/lt.21890. [DOI] [PubMed] [Google Scholar]
- 231.Spengler U, Nattermann J. Immunopathogenesis in hepatitis C virus cirrhosis. Clin Sci (Lond) 2007;112:141–155. doi: 10.1042/CS20060171. [DOI] [PubMed] [Google Scholar]
- 232.Koziel MJ, Dudley D, Wong JT, Dienstag J, Houghton M, Ralston R, Walker BD. Intrahepatic cytotoxic T lymphocytes specific for hepatitis C virus in persons with chronic hepatitis. J Immunol. 1992;149:3339–3344. [PubMed] [Google Scholar]
- 233.Schirren CA, Jung MC, Gerlach JT, Worzfeld T, Baretton G, Mamin M, Hubert Gruener N, Houghton M, Pape GR. Liver-derived hepatitis C virus (HCV)-specific CD4(+) T cells recognize multiple HCV epitopes and produce interferon gamma. Hepatology. 2000;32:597–603. doi: 10.1053/jhep.2000.9635. [DOI] [PubMed] [Google Scholar]
- 234.Alatrakchi N, Koziel M. Regulatory T cells and viral liver disease. J Viral Hepat. 2009;16:223–229. doi: 10.1111/j.1365-2893.2009.01081.x. [DOI] [PubMed] [Google Scholar]
- 235.Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64:830–841. doi: 10.1136/gutjnl-2014-306842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Franceschini D, Paroli M, Francavilla V, Videtta M, Morrone S, Labbadia G, Cerino A, Mondelli MU, Barnaba V. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. J Clin Invest. 2009;119:551–564. doi: 10.1172/JCI36604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Heydtmann M, Adams DH. Chemokines in the immunopathogenesis of hepatitis C infection. Hepatology. 2009;49:676–688. doi: 10.1002/hep.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zeremski M, Petrovic LM, Chiriboga L, Brown QB, Yee HT, Kinkhabwala M, Jacobson IM, Dimova R, Markatou M, Talal AH. Intrahepatic levels of CXCR3-associated chemokines correlate with liver inflammation and fibrosis in chronic hepatitis C. Hepatology. 2008;48:1440–1450. doi: 10.1002/hep.22500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ming-Ju H, Yih-Shou H, Tzy-Yen C, Hui-Ling C. Hepatitis C virus E2 protein induce reactive oxygen species (ROS)-related fibrogenesis in the HSC-T6 hepatic stellate cell line. J Cell Biochem. 2011;112:233–243. doi: 10.1002/jcb.22926. [DOI] [PubMed] [Google Scholar]
- 242.Clément S, Pascarella S, Conzelmann S, Gonelle-Gispert C, Guilloux K, Negro F. The hepatitis C virus core protein indirectly induces alpha-smooth muscle actin expression in hepatic stellate cells via interleukin-8. J Hepatol. 2010;52:635–643. doi: 10.1016/j.jhep.2009.10.035. [DOI] [PubMed] [Google Scholar]
- 243.Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, Washington MK, Germanidis G, Flaherty JF, Aguilar Schall R, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381:468–475. doi: 10.1016/S0140-6736(12)61425-1. [DOI] [PubMed] [Google Scholar]
- 244.D’Ambrosio R, Aghemo A, Rumi MG, Ronchi G, Donato MF, Paradis V, Colombo M, Bedossa P. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology. 2012;56:532–543. doi: 10.1002/hep.25606. [DOI] [PubMed] [Google Scholar]
- 245.Lee YA, Friedman SL. Reversal, maintenance or progression: what happens to the liver after a virologic cure of hepatitis C? Antiviral Res. 2014;107:23–30. doi: 10.1016/j.antiviral.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.van der Meer AJ, Veldt BJ, Feld JJ, Wedemeyer H, Dufour JF, Lammert F, Duarte-Rojo A, Heathcote EJ, Manns MP, Kuske L, et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA. 2012;308:2584–2593. doi: 10.1001/jama.2012.144878. [DOI] [PubMed] [Google Scholar]
- 247.Mehal WZ, Iredale J, Friedman SL. Scraping fibrosis: expressway to the core of fibrosis. Nat Med. 2011;17:552–553. doi: 10.1038/nm0511-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Friedman SL. Liver fibrosis in 2012: Convergent pathways that cause hepatic fibrosis in NASH. Nat Rev Gastroenterol Hepatol. 2013;10:71–72. doi: 10.1038/nrgastro.2012.256. [DOI] [PubMed] [Google Scholar]
- 249.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 250.Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147:577–594.e1. doi: 10.1053/j.gastro.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 251.Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta. 2013;1832:1061–1069. doi: 10.1016/j.bbadis.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 253.Poynard T, Ratziu V, McHutchison J, Manns M, Goodman Z, Zeuzem S, Younossi Z, Albrecht J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology. 2003;38:75–85. doi: 10.1053/jhep.2003.50267. [DOI] [PubMed] [Google Scholar]
- 254.Cross TJ, Quaglia A, Hughes S, Joshi D, Harrison PM. The impact of hepatic steatosis on the natural history of chronic hepatitis C infection. J Viral Hepat. 2009;16:492–499. doi: 10.1111/j.1365-2893.2009.01098.x. [DOI] [PubMed] [Google Scholar]
- 255.Abenavoli L, Masarone M, Peta V, Milic N, Kobyliak N, Rouabhia S, Persico M. Insulin resistance and liver steatosis in chronic hepatitis C infection genotype 3. World J Gastroenterol. 2014;20:15233–15240. doi: 10.3748/wjg.v20.i41.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Goossens N, Negro F. Is genotype 3 of the hepatitis C virus the new villain? Hepatology. 2014;59:2403–2412. doi: 10.1002/hep.26905. [DOI] [PubMed] [Google Scholar]
- 257.Roque-Afonso AM, Ducoulombier D, Di Liberto G, Kara R, Gigou M, Dussaix E, Samuel D, Féray C. Compartmentalization of hepatitis C virus genotypes between plasma and peripheral blood mononuclear cells. J Virol. 2005;79:6349–6357. doi: 10.1128/JVI.79.10.6349-6357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zehender G, De Maddalena C, Bernini F, Ebranati E, Monti G, Pioltelli P, Galli M. Compartmentalization of hepatitis C virus quasispecies in blood mononuclear cells of patients with mixed cryoglobulinemic syndrome. J Virol. 2005;79:9145–9156. doi: 10.1128/JVI.79.14.9145-9156.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sung VM, Shimodaira S, Doughty AL, Picchio GR, Can H, Yen TS, Lindsay KL, Levine AM, Lai MM. Establishment of B-cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: the apoptotic effects of virus infection. J Virol. 2003;77:2134–2146. doi: 10.1128/JVI.77.3.2134-2146.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis. 2007;16:65–73. [PubMed] [Google Scholar]
- 261.Grignoli R, Goossens N, Negro F. Extrahepatic manifestations of HCV. Minerva Gastroenterol Dietol. 2015;61:31–38. [PubMed] [Google Scholar]
- 262.Sène D, Limal N, Cacoub P. Hepatitis C virus-associated extrahepatic manifestations: a review. Metab Brain Dis. 2004;19:357–381. doi: 10.1023/b:mebr.0000043982.17294.9b. [DOI] [PubMed] [Google Scholar]
- 263.Saadoun D, Delluc A, Piette JC, Cacoub P. Treatment of hepatitis C-associated mixed cryoglobulinemia vasculitis. Curr Opin Rheumatol. 2008;20:23–28. doi: 10.1097/BOR.0b013e3282f1330c. [DOI] [PubMed] [Google Scholar]
- 264.Himoto T, Masaki T. Extrahepatic manifestations and autoantibodies in patients with hepatitis C virus infection. Clin Dev Immunol. 2012;2012:871401. doi: 10.1155/2012/871401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.White DL, Ratziu V, El-Serag HB. Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. J Hepatol. 2008;49:831–844. doi: 10.1016/j.jhep.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Modi AA, Liang TJ. Hepatitis C: a clinical review. Oral Dis. 2008;14:10–14. doi: 10.1111/j.1601-0825.2007.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tarr AW, Urbanowicz RA, Ball JK. The role of humoral innate immunity in hepatitis C virus infection. Viruses. 2012;4:1–27. doi: 10.3390/v4010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Maasoumy B, Wedemeyer H. Natural history of acute and chronic hepatitis C. Best Pract Res Clin Gastroenterol. 2012;26:401–412. doi: 10.1016/j.bpg.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 269.Thomas DL, Seeff LB. Natural history of hepatitis C. Clin Liver Dis. 2005;9:383–398, vi. doi: 10.1016/j.cld.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 270.Scott JD, Gretch DR. Molecular diagnostics of hepatitis C virus infection: a systematic review. JAMA. 2007;297:724–732. doi: 10.1001/jama.297.7.724. [DOI] [PubMed] [Google Scholar]
- 271.Mederacke I, Wedemeyer H, Ciesek S, Steinmann E, Raupach R, Wursthorn K, Manns MP, Tillmann HL. Performance and clinical utility of a novel fully automated quantitative HCV-core antigen assay. J Clin Virol. 2009;46:210–215. doi: 10.1016/j.jcv.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 272.Medici MC, Furlini G, Rodella A, Fuertes A, Monachetti A, Calderaro A, Galli S, Terlenghi L, Olivares M, Bagnarelli P, et al. Hepatitis C virus core antigen: analytical performances, correlation with viremia and potential applications of a quantitative, automated immunoassay. J Clin Virol. 2011;51:264–269. doi: 10.1016/j.jcv.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 273.Vermehren J, Susser S, Berger A, Perner D, Peiffer KH, Allwinn R, Zeuzem S, Sarrazin C. Clinical utility of the ARCHITECT HCV Ag assay for early treatment monitoring in patients with chronic hepatitis C genotype 1 infection. J Clin Virol. 2012;55:17–22. doi: 10.1016/j.jcv.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 274.Sarrazin C, Berg T, Ross RS, Schirmacher P, Wedemeyer H, Neumann U, Schmidt HH, Spengler U, Wirth S, Kessler HH, et al. [Prophylaxis, diagnosis and therapy of hepatitis C virus (HCV) infection: the German guidelines on the management of HCV infection] Z Gastroenterol. 2010;48:289–351. doi: 10.1055/s-0028-1110008. [DOI] [PubMed] [Google Scholar]
- 275.Yoshida EM, Sulkowski MS, Gane EJ, Herring RW Jr, Ratziu V, Ding X, Wang J, Chuang SM, Ma J, McNally J, Stamm LM, Brainard DM, Symonds WT, McHutchison JG, Beavers KL, Jacobson IM, Reddy KR, Lawitz E. Concordance of sustained virological response 4, 12, and 24 weeks post-treatment with sofosbuvir-containing regimens for hepatitis C virus. Hepatology. 2015;61:41–45. doi: 10.1002/hep.27366. [DOI] [PubMed] [Google Scholar]
- 276.Lange CM, Jacobson IM, Rice CM, Zeuzem S. Emerging therapies for the treatment of hepatitis C. EMBO Mol Med. 2014;6:4–15. doi: 10.1002/emmm.201303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Bowden DS, Berzsenyi MD. Chronic hepatitis C virus infection: genotyping and its clinical role. Future Microbiol. 2006;1:103–112. doi: 10.2217/17460913.1.1.103. [DOI] [PubMed] [Google Scholar]
- 278.Mauss S, Berg T, Rockstroh J, Sarrazin C, Wedemeyer H. Hepatology - a Clinical Textbook. 6th ed. Düsseldorf: Flying Publisher, 2015 [Google Scholar]
- 279.World Health Organization. The Alcohol, Smoking and Substance nvolvement Screening Test (ASSIST) Available from: http://whqlibdoc.who.int/publications/2010/9789241599382_eng.pdf.
- 280.Martin NK, Hickman M, Hutchinson SJ, Goldberg DJ, Vickerman P. Combination interventions to prevent HCV transmission among people who inject drugs: modeling the impact of antiviral treatment, needle and syringe programs, and opiate substitution therapy. Clin Infect Dis. 2013;57 Suppl 2:S39–S45. doi: 10.1093/cid/cit296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Shivkumar S, Peeling R, Jafari Y, Joseph L, Pant Pai N. Accuracy of rapid and point-of-care screening tests for hepatitis C: a systematic review and meta-analysis. Ann Intern Med. 2012;157:558–566. doi: 10.7326/0003-4819-157-8-201210160-00006. [DOI] [PubMed] [Google Scholar]
- 282.Aspinall EJ, Corson S, Doyle JS, Grebely J, Hutchinson SJ, Dore GJ, Goldberg DJ, Hellard ME. Treatment of hepatitis C virus infection among people who are actively injecting drugs: a systematic review and meta-analysis. Clin Infect Dis. 2013;57 Suppl 2:S80–S89. doi: 10.1093/cid/cit306. [DOI] [PubMed] [Google Scholar]
- 283.World Health Organization. Guidelines for the screening, care and treatment of persons with hepatitis C infection. Available from: http://apps.who.int/iris/bitstream/10665/111747/1/9789241548755_eng.pdf. [PubMed]
- 284.Chen TY, Ding EL, Seage Iii GR, Kim AY. Meta-analysis: increased mortality associated with hepatitis C in HIV-infected persons is unrelated to HIV disease progression. Clin Infect Dis. 2009;49:1605–1615. doi: 10.1086/644771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Thein HH, Yi Q, Dore GJ, Krahn MD. Natural history of hepatitis C virus infection in HIV-infected individuals and the impact of HIV in the era of highly active antiretroviral therapy: a meta-analysis. AIDS. 2008;22:1979–1991. doi: 10.1097/QAD.0b013e32830e6d51. [DOI] [PubMed] [Google Scholar]
- 286.Chou R, Wasson N. Blood tests to diagnose fibrosis or cirrhosis in patients with chronic hepatitis C virus infection: a systematic review. Ann Intern Med. 2013;158:807–820. doi: 10.7326/0003-4819-158-11-201306040-00005. [DOI] [PubMed] [Google Scholar]
- 287.Suthar AB, Harries AD. A public health approach to hepatitis C control in low- and middle-income countries. PLoS Med. 2015;12:e1001795. doi: 10.1371/journal.pmed.1001795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. The French METAVIR Cooperative Study Group. Hepatology. 1994;20:15–20. [PubMed] [Google Scholar]
- 289.Vallet-Pichard A, Mallet V, Nalpas B, Verkarre V, Nalpas A, Dhalluin-Venier V, Fontaine H, Pol S. FIB-4: an inexpensive and accurate marker of fibrosis in HCV infection. Comparison with liver biopsy and fibrotest. Hepatology. 2007;46:32–36. doi: 10.1002/hep.21669. [DOI] [PubMed] [Google Scholar]
- 290.Pol S, Corouge M. Treatment of hepatitis C: perspectives. Med Mal Infect. 2014;44:449–454. doi: 10.1016/j.medmal.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 291.European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of hepatitis C virus infection. J Hepatol. 2011;55:245–264. doi: 10.1016/j.jhep.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 292.Feeney ER, Chung RT. Antiviral treatment of hepatitis C. BMJ. 2014;348:g3308. doi: 10.1136/bmj.g3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med. 2013;368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.van de Ven N, Fortunak J, Simmons B, Ford N, Cooke GS, Khoo S, Hill A. Minimum target prices for production of direct-acting antivirals and associated diagnostics to combat hepatitis C virus. Hepatology. 2015;61:1174–1182. doi: 10.1002/hep.27641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Schneider MD, Sarrazin C. Antiviral therapy of hepatitis C in 2014: do we need resistance testing? Antiviral Res. 2014;105:64–71. doi: 10.1016/j.antiviral.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 296.Wyles DL, Gutierrez JA. Importance of HCV genotype 1 subtypes for drug resistance and response to therapy. J Viral Hepat. 2014;21:229–240. doi: 10.1111/jvh.12230. [DOI] [PubMed] [Google Scholar]
- 297.Tornai I. [Significance of hepatitis C virus baseline polymorphism during the antiviral therapy] Orv Hetil. 2015;156:849–854. doi: 10.1556/650.2015.30180. [DOI] [PubMed] [Google Scholar]
- 298.Sun D, Dai M, Shen S, Li C, Yan X. Analysis of naturally occurring resistance-associated variants to NS3/4A protein inhibitors, NS5A protein inhibitors and NS5B polymerase inhibitors in patients with chronic hepatitis C. Gene Expr. 2017 doi: 10.3727/105221617X15100607143377. [DOI] [PMC free article] [PubMed] [Google Scholar]