

Abstract
Microvillus inclusion disease (MVID) is a rare but fatal autosomal recessive congenital diarrheal disorder caused by MYO5B mutations. In 2013, we launched an open‐access registry for MVID patients and their MYO5B mutations (www.mvid-central.org). Since then, additional unique MYO5B mutations have been identified in MVID patients, but also in non‐MVID patients. Animal models have been generated that formally prove the causality between MYO5B and MVID. Importantly, mutations in two other genes, STXBP2 and STX3, have since been associated with variants of MVID, shedding new light on the pathogenesis of this congenital diarrheal disorder. Here, we review these additional genes and their mutations. Furthermore, we discuss recent data from cell studies that indicate that the three genes are functionally linked and, therefore, may constitute a common disease mechanism that unifies a subset of phenotypically linked congenital diarrheal disorders. We present new data based on patient material to support this. To congregate existing and future information on MVID geno‐/phenotypes, we have updated and expanded the MVID registry to include all currently known MVID‐associated gene mutations, their demonstrated or predicted functional consequences, and associated clinical information.
Keywords: congenital diarrheal diseases, enteropathy, microvillus inclusion disease, munc18‐2, MYO5B, myosin VB, STX3, STXBP2, syntaxin‐3
1. MICROVILLUS INCLUSION DISEASE: AN EXPANDING SPECTRUM OF GENES AND PHENOTYPES
In 1978, Davidson and coworkers reported five infants that presented with an apparent congenital enteropathy characterized by persistent diarrhea from birth, the inability to absorb nutrients, failure to thrive, and leading to death (Davidson, Cutz, Hamilton, & Gall, 1978). At the cellular level, atrophy of the apical brush border membrane of the enterocytes, intracellular accumulation of brush border enzymes, and occasional microvilli‐lined inclusion bodies named microvillus inclusions were identified as characteristic features for this disease (Cutz et al., 1989; Davidson et al., 1978). This congenital enteropathy has been called Davidson Disease, intractable diarrhea of infancy, congenital familial protracted diarrhea with enterocyte brush border defects, congenital microvillus atrophy, or microvillus inclusion disease (MVID) (Cutz et al., 1989), and is listed in Online Mendelian Inheritance in Man (OMIM) as Diarrhea 2, with microvillus atrophy (DIAR2; #251850). To date, MVID is the most frequently used name for this disease in the scientific literature (source: Web of science).
The diagnosis of MVID is based on microscopical biopsy evaluation, and includes the detection of Periodic acid‐Shiff (PAS) staining in the apical cytoplasm, immunohistochemical detection of the brush border metallopeptidase CD10 in the apical cytoplasm, and the detection of microvillus inclusions in the enterocytes with electron microscopy. A focal appearance of MVID‐typical enterocyte defects has been reported, which seems associated with a late‐onset or milder clinical course of the disease (Perry et al., 2014). Notably, phenotypic variations have been reported in patients with clinical presentations that are entirely typical for MVID. These include for example microvilli at the lateral surface of MVID enterocytes (Croft et al., 2000; Morroni, Cangiotti, Guarino, & Cinti, 2006; Phillips & Schmitz, 1992; Wiegerinck et al., 2014) or aggregates of electron‐lucent membranous vesicles (Weeks, Zuppan, Malott, & Mierau, 2003). In some cases, microvillus inclusions could not be detected (Iancu, Mahajnah, Manov, & Shaoul, 2007; Mierau, Wills, Wyatt‐Ashmead, Hoffenberg, & Cutz, 2001) or only small ones could be detected after several attempts (Weeks et al., 2003). These phenotypic variations have led to suggest that MVID represents a heterogeneous disease (Iancu et al., 2007; Mierau et al., 2001; Weeks et al., 2003).
In 2008, mutations in the MYO5B gene (chromosome 18q21.1; MIM# 606540) were identified in MVID patients (Erickson, Larson‐Thomé, Valenzuela, Whitaker, & Shub, 2008; Müller et al., 2008), and confirmed MVID as an autosomal recessive disease. MYO5B encodes the myosin Vb protein, which belongs to the large myosin family of actin‐based molecular motor proteins and controls intracellular trafficking. RNAi‐mediated knockdown of myosin Vb in an intestinal cell line reproduced several of the disease phenotypes, supporting the causality between MYO5B and MVID (Ruemmele et al., 2010). Between 2008 and 2013, a total of 41 unique MYO5B mutations were identified, which were systematically analyzed, categorized, and collected in an online registry for MVID patients (van der Velde et al., 2013) (www.mvid-central.org). Since then, more unique homozygous and compound heterozygous MYO5B mutations have been identified in MVID patients. Furthermore, animal models for MVID have been developed, formally proving the connection between MYO5B and MVID. Notably, in some MVID patients, no MYO5B mutations, or only one heterozygous MYO5B mutation was detected (Müller et al., 2008; Perry et al., 2014; Szperl et al., 2011), and the possibility that other genes are involved was suggested.
Recently, mutations in two other genes have been associated with variant forms of MVID: STX3 (chromosome 11q12.1; MIM# 600876) (Wiegerinck et al., 2014) and STXBP2 (chromosome 19p13.2; MIM# 601717) (Stepensky et al., 2013; Vogel et al., 2017). STX3 encodes the syntaxin‐3 protein, which is a member of the Qa‐SNARE protein family that contributes a glutamine (Q) residue for the formation of the assembled core SNARE complex. STXBP2 encodes the syntaxin‐binding protein‐2 also called the mammalian uncoupled munc18‐2 protein, which belongs to the sec1/munc18‐like protein family. Both syntaxin‐3 and munc18‐2 play a role in membrane fusion. STX3 mutations were identified by whole exome sequencing in two patients diagnosed with MVID based on clinical symptoms but without MYO5B mutations (Wiegerinck et al., 2014). Immunohistochemical analyses of intestinal biopsies revealed intra‐cytoplasmic PAS staining, variable microvillus atrophy, microvillus inclusions and, unlike “classical” MVID, the appearance of microvilli at the basolateral plasma membrane (Wiegerinck et al., 2014). STXBP2 mutations were identified in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5, OMIM 613101). Although FHL5 is primarily regarded as a hyper‐inflammatory immune disorder, ∼40% of FHL5 patients show severe chronic diarrhea starting shortly after birth without signs of infection and often preceding the diagnosis of FHL5. Most of these patients require long‐term total parenteral nutrition (TPN) for survival. Therapies targeted against FHL5 failed to resolve the diarrhea which persisted even after full hematopoietic stem cell transplantation (Pagel et al., 2012), indicating that the intestinal symptoms are independent of immune cell defects. Immunohistochemical analyses of intestinal biopsies of these patients revealed the intracellular retention of apical brush border proteins such as CD10 and PAS‐positive material, variable microvillus atrophy and microvillus inclusions (Stepensky et al., 2013; Vogel et al., 2017). Thus, these patients show all intestine‐related clinical and cellular hallmarks of MVID (Stepensky et al., 2013; Vogel et al., 2017).
2. MVID‐ASSOCIATED GENES ARE FUNCTIONALLY LINKED
The striking overlap in intestinal symptoms and cellular phenotypes between patients carrying either MYO5B, STX3, or STXBP2 mutations and the previously reported roles of their encoded proteins in apical membrane trafficking in epithelial cells led to suggest that these three genes and their encoded proteins represent a common disease mechanism that unifies a subset of phenotypically linked congenital diarrheal disorders (Posovszky, 2016; Stepensky et al., 2013; Vogel et al., 2017; Wiegerinck et al., 2014). MYO5B, STX3, and STXBP2 regulate protein trafficking to the apical brush border. Myosin Vb is an actin‐based molecular motor protein that controls the trafficking of endosomes/transport vesicles to the apical brush border. Syntaxin‐3 and munc18‐2 are part of a protein complex that controls the membrane fusion of transport vesicles with the apical brush border. Indeed, the trafficking of proteins to the apical brush border membrane of intestinal epithelial Caco‐2 cells is inhibited upon loss‐of‐function of either myosin Vb (Knowles et al., 2014; Kravtsov et al., 2014, 2016; Ruemmele et al., 2010; Vogel et al., 2015), syntaxin‐3 (Breuza, Fransen, & Le Bivic, 2000; Collaco, Marathe, Kohnke, Kravstov, & Ameen, 2010; Riento, Kauppi, Keranen, & Olkkonen, 2000; Vogel et al., 2015; Wiegerinck et al., 2014), or munc18‐2 (Riento et al., 2000; Vogel et al., 2015, 2017). Also, apical microvillus atrophy was observed in Caco‐2 cells upon loss‐of‐function of either myosin Vb (Dhekne et al., 2014; Knowles et al., 2014; Ruemmele et al., 2010; Vogel et al., 2015) or syntaxin‐3 (Vogel et al., 2015; Wiegerinck et al., 2014). Further, loss of myosin Vb function in enterocytes of MVID patients with MYO5B mutations (Dhekne et al., 2014; Szperl et al., 2011) and in enterocytes of Myo5B knockout mice (Weis et al., 2016) resulted in the mislocalization of the myosin Vb‐binding protein rab11a (Szperl et al., 2011), and the loss of either myosin Vb or rab11a in murine enterocytes caused the mislocalization of syntaxin‐3 (Knowles et al., 2015; Weis et al., 2016). Moreover, myosin Vb—when bound to rab11a—was found to interact with syntaxin‐3 in intestinal epithelial cells, and the knockdown of myosin Vb protein expression resulted in a strongly reduced interaction between syntaxin‐3 and munc18‐2 (Vogel et al., 2015).
To provide further support that myosin Vb, syntaxin‐3 and munc18‐2 are also linked in patient tissue, we have analyzed the subcellular distribution of syntaxin‐3 and munc18‐2 in enterocytes of intestinal biopsies from patients with MYO5B or STXBP2 mutations. We found that munc18‐2 and syntaxin‐3 (and two additional members of the apical membrane fusion machinery, SNAP23 and cellubrevin; Galli, Brenna, Camilli de, & Meldolesi, 1976; Low et al., 1998; Riento et al., 2000)) all localized to the apical brush border plasma membrane in control enterocytes, but accumulated in intracellular puncta in the enterocytes of two MVID patients (Szperl et al., 2011), one with a homozygous c.4366C > T (p.Gln1456*) MYO5B mutation (Figure 1A–D) and one with compound heterozygous c.1540T > C (p.Cys514Arg) and IVS33+3753G > T MYO5B mutations (Supp. Figure S1A). Furthermore, in a biopsy of an FLH5 patient with a variant MVID phenotype and a homozygous c.693_695delGAT (p.Ile232del) mutation in STXBP2 (Stepensky et al., 2013), we found that syntaxin‐3 was mislocalized from the enterocyte brush border membrane to intracellular puncta (Figure 1E). This is in agreement with a previous study that demonstrated a function for munc18‐2 in the subcellular distribution of syntaxin‐3 in a different cell type (Hackmann et al., 2013). Investigation of the subcellular distribution of myosin Vb was precluded by the lack of good quality antibodies for use on paraffin‐embedded material.
Figure 1.
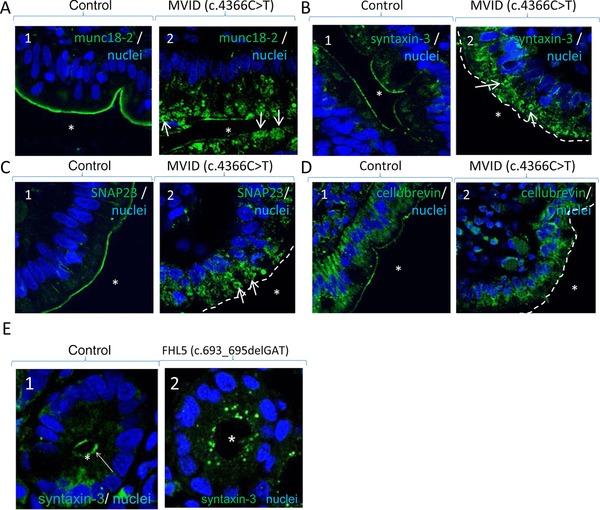
The apical SNARE machinery in duodenal enterocytes of patients with MYO5B or STXBP2 mutations. A: Labeling of munc18‐2 (green) and nuclei (blue) in control (1) and MVID (2) enterocytes. B: Labeling of syntaxin‐3 (green) and nuclei (blue) in control (1) and MVID (2) enterocytes. C: Labeling of SNAP23 (green) and DNA (blue) in control (1) and MVID (2) enterocytes. D: Labeling of cellubrevin (green) and DNA (blue) in control (1) and MVID (2) enterocytes. E: Labeling of syntaxin‐3 (green) and DNA (blue) in control (1) and FHL5 (2) enterocytes. Asterisks indicate position of the lumen. Arrows in A2, B2, and C2 point to globular structures in the apical cytoplasm
Together, the published in vitro data and the patient‐based data as shown in this study provide compelling evidence that myosin Vb, syntaxin‐3, and munc18‐2 are functionally linked in human enterocytes, and likely comprise a common molecular pathway and disease mechanism that unifies a subgroup of congenital diarrheal disorders centered around MVID (Overeem, Bryant, & van IJzendoorn, 2015; Posovszky, 2016; Vogel et al., 2017).
Although most of the cell line‐based studies involve the general knockdown of myosin Vb, syntaxin‐3, or munc18‐2, patients typically carry one or more of specific MYO5B, STX3, or STXBP2 mutations (van der Velde et al., 2013). These mutations are likely to result in the expression of mutated proteins, however direct experimental evidence of the mutations functional consequence is missing. Such data are eagerly awaited in order to understand and describe how the phenotype, impact, or onset of the disease might differ depending on the mutations. This is further emphasized by the fact that MYO5B, STX3, and STXBP2 mutations have also been identified in patients without intestinal symptoms (Chograni et al., 2015; Gonzales et al., 2017; Pagel et al., 2012; Qiu et al., 2017) (see below). Therefore, it is important to understand how the different MYO5B, STX3, and STXBP2 mutations and the affected residues may impact the function of the encoded proteins.
3. MYO5B GENE MUTATIONS
In 2013, we reviewed and categorized 41 reported MVID‐associated MYO5B mutations (van der Velde et al., 2013). Here we have retrieved from the literature all additional MVID patients reported between 2013 and 2017. Throughout this article, we have used the DNA and protein variant numbering system following the guidelines of the journal and Human Genome Variation Society (HGVS). Twenty of these additional patients were demonstrated to carry one or more MYO5B mutations, expanding the number of unique MVID‐associated MYO5B mutations to 62 (Figure 2A and B and Supp. Table S1A). Myosin Vb generates force via its motor domain upon conformational rearrangements driven by F‐actin binding coordinated with ATP hydrolysis. The motor domain is composed of four main subdomains: the N‐terminal, upper 50 kDa (U50), lower 50 kDa (L50) subdomains, and the converter (Figure 2A and C). The myosin V elongated lever arm (which contains the converter and the 6 IQ‐motifs bound to 6 light chains), swings and produces large displacements by amplifying small movements within the catalytic motor domain during the chemo‐mechanical acto‐myosin cycle (Houdusse & Sweeney, 2016). Finally, the myosin V C‐terminal tail binds several partners/cargoes and is composed of a long coiled‐coil region allowing motor dimerization and a terminal globular tail domain (Trybus, 2008). The localization of the consequential amino acid substitutions in a homology model of the 3D structure of the myosin Vb motor domain (Figure 2C) is depicted in Figure 2D. For MYO5B mutation analyses, reference sequence NM_001080467.2 was used.
Figure 2.
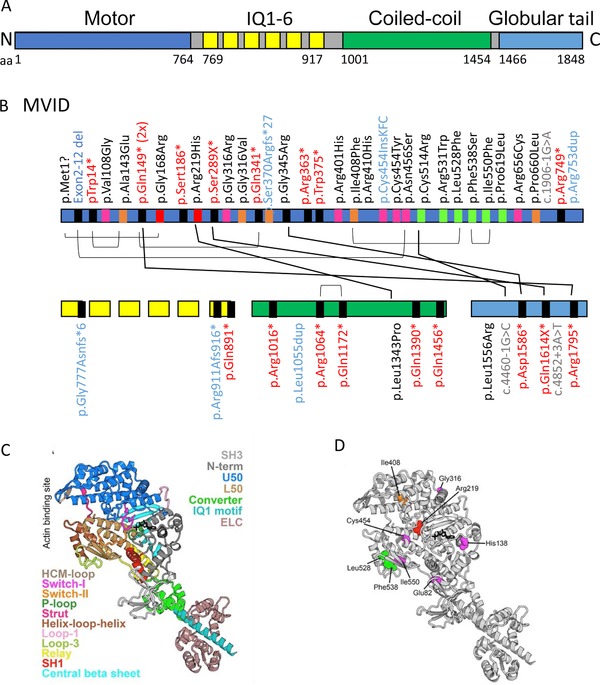
Schematic representation of the myosin Vb protein. A: A schematic overview of the myosin Vb protein and its functional domains. Protein data are deduced from Genbank RefSeq‐file accession number NG_012925.1 for the human MYO5B gene. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to the journal guidelines (www.hgvs.org/mutnomen). The initiation codon is 1. B: Overviews of the MVID‐associated MYO5B mutations in the different domains of the myosin Vb protein. IQ1‐6 refers to the six calmodulin‐binding IQ domains that conform to the consensus sequence [I,L,V]QxxxRGxxx[R,K]. Mutations indicated in black, red, blue, and gray letter color represent missense, frameshift/nonsense, deletions/insertions, and splicing mutations, respectively. The differently colored blocks associated with each mutation in the protein domains represent the predicted consequences for the protein (black: premature termination, magenta: mutations in regions important for the allosteric rearrangements of the myosin motor head during the kinetic cycle, orange: mutations that may lead to protein misfolding, red: mutations in the ATP‐binding site, green: mutations in regions that are important for actin interactions). Black lines between individual mutations indicate their combined presence in a single patient. C: Model of the myosin Vb motor domain. A. Homology model of myosin Vb based on myosin Va post‐rigor structure with ATP nucleotide bound (PDB ID: 1W7J) is shown. The motor domain contains four subdomains (the N‐terminal: gray, including the SH3: light gray, the U50: marine blue, the L50: sand, and the converter: green). Conformational changes in the motor domain are amplified by the lever arm (converter, green; IQ motif 1, pale cyan helix associated with a calmodulin (light pink); the 5 other IQ motifs of the lever arm are not shown). These conformational changes are coordinated within the motor by elements of the transducer (loop 1: light purple, central beta sheet: cyan) as well as the connectors (Relay: yellow, SH1 helix: red, Strut: hot pink), which are linkers between subdomains. The nucleotide‐binding elements (P‐loop: pale green, Switch I: magenta, and switch II: orange) surrounding the nucleotide (represented in black sticks) and the actin‐binding elements (helix‐loop‐helix: brown, HCM loop: wheat) are also depicted. D: The MVID‐associated missense mutations found in the motor domain of myosin Vb are depicted with the following color code: (green: mutations in regions that are important for actin interactions, red: mutations in the ATP‐binding site, Magenta: mutations in regions of importance for allosteric rearrangements of the myosin head during the motor cycle, orange: mutations that may lead to protein misfolding)
Of the 25 MYO5B mutations found in these additional patients, four compound heterozygous MYO5B mutations and one homozygous MYO5B mutation present in five patients of different families were previously reported in other MVID patients of unrelated families (this study). These patients were indeed found to carry either a heterozygous (c.3163‐3165dupCTC (p.Leu1055dup) and c.445C > T (p.Gln149*)), compound heterozygous (c.3163‐3165dupCTC (p.Leu1055dup)), or homozygous (c.656G > A (p.Arg219His), c.5383A > T (p.1795Leu) and c.1323‐2A > G (IVS10‐2A > G)) mutations (van der Velde et al., 2013). The homozygous patient with the c.656G > A (p.Arg219His) mutation displayed early onset MVID, whereas the patient who was compound heterozygous for this mutation displayed late‐onset MVID. Furthermore, heterozygous c.656G > A (p.Arg219His) mutations have been associated with colon carcinoma in the COSMIC database (cancer.sanger.ac.uk). The compound heterozygous c.3163‐3165dupCTC mutation (p.Leu1055dup) currently has been reported in three unrelated families of different ethnic origin, also including patients with early‐onset and patients with late‐onset MVID. The c.3163‐3165dupCTC (p.Leu1055dup) mutation (rs10625857)) is not likely to be pathogenic as such as it has been reported at least 41,262 times of which 7,704 in a homozygous manner, and has the highest reported allele frequency (0.3420) in the ExAC database (exac.broadinstitute.org). One heterozygous patient carrying this c.3163‐3165dupCTC (p.Leu1055dup) mutation and a c.1347delC (p.Phe450leufs*30) mutation displayed late onset MVID and could be weaned of TPN and moved to normal enteral feeding (Perry et al., 2014). Given the autosomal recessive inheritance pattern of MVID, this suggests that the c.3163‐3165dupCTC (p.Leu1055dup) mutation may contribute to the onset of clinical symptoms.
The 21 new MVID‐associated MYO5B mutations include seven nonsense, eight missense, and five splicing mutations and one small deletion. These mutations are found in both the N‐terminal motor domain and in the C‐terminal tail domain of the myosin Vb protein (Figure 2A and B). We have examined these mutations in the light of current understanding of how myosin motors produce force, as previously reviewed (Houdusse & Sweeney, 2016) as well as based on the current structural data available for the myosin V tail (Nascimento et al., 2013; Pylypenko et al., 2013; Velvarska & Niessing, 2013). The results of these examinations are summarized in Supp. Table S1A. Interestingly, the three newly reported amino acid substitutions in the motor domain p.Leu528Phe, p.Phe538Ser, p.Ile550Phe, resulting from the c.1582C > T, c.1316T > C, and c. 1648A > T MYO5B mutations, respectively, affect residues that interact with each other, forming a buried hydrophobic core of the lower part of the L50 subdomain (see model in Figure 2C and D). All three substitutions will have destabilizing effect in the folding of this region of the L50 subdomain. The mutant side chains are also close to the activation loop and loop3 that are both involved in actin binding; thus, these mutations likely affect filamentous actin‐binding and motility properties. Specifically, Phe538 is a part of loop‐3 that is involved in F‐actin binding; the bulky hydrophobic side chain determines the relative orientation of the helix‐loop‐helix and loop‐3. The Phe538 substitution to the small polar serine side chain will impact the conformational stability of the actin‐binding elements and will thus likely affect the force produced by the motor on actin. Leu528 belongs to the second helix of the helix‐loop‐helix motif that plays an essential role in F‐actin binding, while Ile550 is located on a beta strand preceding loop‐3. Both aliphatic side chains are tightly packed in the L50 subdomain hydrophobic core and their substitution to a bulky phenylalanine aromatic side chain may change the relative orientation of the secondary structure elements of this actin‐binding unit. In conclusion, these substitutions can be classified as both affecting the stability of the L50 fold and actin‐binding properties of myosin Vb and thus the force produced by this myosin motor.
3.1. MYO5B mutations that are not associated with MVID
Some but not all MVID patients have been reported to develop intrahepatic cholestasis that results in high levels of plasma bile acids. It was suggested that the MYO5B mutations and consequential loss of myosin Vb protein function may lead to defective trafficking of the bile acid transporter ABCB11/BSEP to the apical bile canalicular surface of hepatocytes in the liver, thereby blocking the enterohepatic circulation of bile acids and leading to elevated plasma bile acid levels (Girard et al., 2014; Halac et al., 2011). Recently, 26 MYO5B mutations were reported in 17 patients with intrahepatic cholestasis but without symptoms of MVID (Gonzales et al., 2017; Qiu et al., 2017) (Supp. Table S1B). Interestingly, two MYO5B missense mutations which affect the same arginine residue at position 401 were identified in two unrelated patients presenting with either intrahepatic cholestasis without MVID (c.1201C > T, p.Arg401Cys), and with MVID (c.1202G > A, p.Arg401His), respectively (Qiu et al., 2017; Ruemmele et al., 2010; van der Velde et al., 2013) (Supp. Table S1B). The previously described p.Arg401His substitution likely affects the motor U50 subdomain fold and thus the allosteric motor activity (van der Velde et al., 2013), whereas the p.Arg401Cys substitution is compatible with the motor structure in different states and might be less damaging (Supp. Table S1B and Figure 3A and B). Another MYO5B nonsense mutation (c.1021C > T (p.Gln341*)) was identified in a heterozygous patient with isolated intrahepatic cholestasis without MVID and also in an unrelated homozygous patient with MVID (Müller et al., 2008; Qiu et al., 2017) (Supp. Table S1B). Additionally, the monoallelic c.1136G > C mutation (Qiu et al., 2017) leads to a p.Arg379Pro substitution at the beginning of the actin‐binding HCM loop in the U50 subdomain (the name of this loop “hypertrophic cardiomyopathy (HCM)” is linked to the deadly disease caused by the Arg403Gln substitution in beta‐cardiac myosin (Nag et al., 2015)). While a proline is a naturally occurring residue at this position for some myosin II, whether the p.Arg379Pro substitution can alter the performance of myosin V and its processivity requires further investigations. Further, we found that the heterozygous p.Gln1079His substitution in the coiled‐coil region of the myosin Vb protein, resulting from the heterozygous c.3237G > C mutation (Qiu et al., 2017), naturally occurs in the myosin Vb sequence of the platypus (Ornithorhynchus anatinus), and is thus unlikely to have much influence on motor function. Patients with isolated cholestasis and MYO5B mutations are more often heterozygous (82% (14 out of 17)) when compared to patients with MVID and MYO5B mutations (67% heterozygosity (35 out of 52)), although this is not statistically significant (Fisher Exact test P = 0.35). The myosin VB homology protein structure‐based analyses (summarized in Supp. Table S1B, Figures 2A and 3) show that the compound heterozygous patients with isolated cholestasis and MYO5B mutations often carry in one allele a mutation that corresponds to more peripheral residues of the motor domain that are predicted to be less damaging for motor function. It is possible that this results in sufficient activity for at least one of the mutated myosin Vb proteins. Qiu and colleagues noted that biallelic severe MYO5B mutations (that is, truncations and splicing mutations) and biallelic MYO5B mutations that were predicted to affect the myosin Vb‐rab11a interaction domains were less frequent in patients with isolated cholestasis when compared to patients with MVID (Figures 2B and 3A). This led to suggest that the severity of the (combined) MYO5B mutations contributes to the clinical phenotype (that is, isolated MVID, MVID with cholestasis or isolated cholestasis). However, as siblings with the same mutations were reported to show differential age‐of‐onset and course of liver symptoms a modifying role of other genes, epigenetics or environmental factors should also be considered (Qiu et al., 2017).
Figure 3.
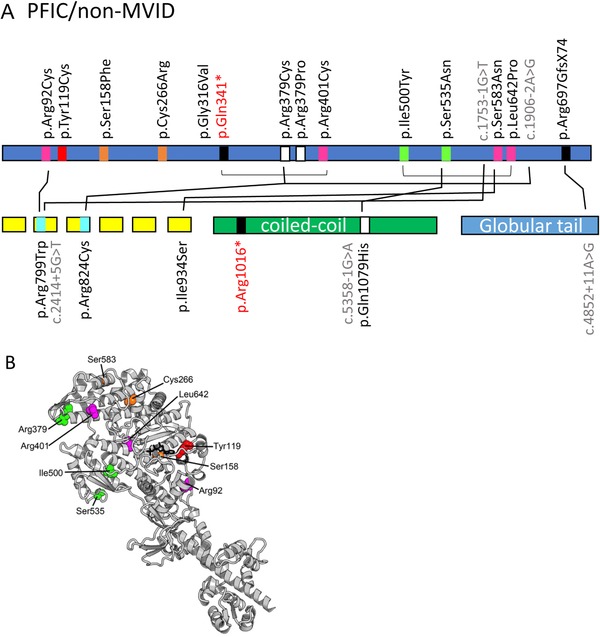
A and B: Overview of the non‐MVID/intrahepatic cholestasis‐associated MYO5B mutations in the different domains of the myosin Vb protein. Mutations indicated in black, red, blue, and gray letter color represent missense, frameshift/nonsense, deletions/insertions, and splicing mutations, respectively. The differently colored blocks associated with each mutation in the protein domains represent the predicted consequences for the protein (black: premature termination, magenta: mutations in regions important for the allosteric rearrangements of the myosin motor head during the kinetic cycle, orange: mutations that may lead to protein misfolding, red: mutations in the ATP‐binding site, green: mutations in regions that are important for actin interactions. Black lines between individual mutations indicate their combined presence in a single patient. B: The missense mutations found in the motor domain of myosin Vb associated with non‐MVID patients with intrahepatic cholestasis are depicted with the following color code: green: mutations in regions that are important for actin interactions, red: mutations in the ATP‐binding site, magenta: mutations in regions of importance for allosteric rearrangements of the myosin head during the motor cycle, orange: mutations that may lead to protein misfolding)
How can mild MYO5B mutations lead to isolated cholestasis without intestinal symptoms while severe MYO5B mutations can lead to intestinal symptoms without cholestasis? As a possible explanation we propose that severe MYO5B mutations prevent the reabsorption of bile acids from the intestine via the apical sodium‐dependent bile acid transporter ASBT/SLC10A2. Given that in a normal situation ∼95% of the circulating bile acids are reabsorbed in the intestine and transported back to the liver via the portal vein, loss of ASBT/SLC10A2‐mediated reabsorption would drastically limit the bile acid load on the liver and thereby the cholestasis phenotype. Importantly, such a scenario implies that physiological parameters co‐determine the clinical outcome in patients with (a) given MYO5B mutation(s).
4. STX3 AND STXBP2 MUTATIONS
We have retrieved three unique STX3 and 51 unique STXBP disease‐relevant mutations from the scientific literature. At least two and six of these STX3 and STXBP2 mutations, respectively, have been reported to be associated with MVID‐resembling congenital diarrheal disorder (based on immunohistological biopsy inspection) (Stepensky et al., 2013; Wiegerinck et al., 2014) (Supp. Tables S2A and S3A), and nine more STXBP2 mutations have been associated with FLH5‐independent chronic diarrhea (Supp. Table S3A).
The STX3 encoded syntaxin‐3 is a member of the SNARE protein family. STX3 contains a transmembrane C‐terminal anchor (TMD) and cytoplasmic SNARE motif (also referred to as H3 domain), a three α‐helix bundle (Habc domain), and a short N‐terminal extension, known as N‐peptide (Figure 4A). The SNARE motifs from different SNARE proteins assemble to form a parallel four‐helix bundle that bridge membranes and mediate their fusion (Rizo & Südhof, 2012). The STXBP2 encoded munc18‐2 belongs to the sec1/munc18‐like protein family. Munc18‐2 is composed of three domains (Figure 4B): the N‐terminal domain 1 together with domain 2 forms one‐half of the protein arch‐shaped structure, whereas the other half of the arch is predominantly formed by domain 3 (Hackmann et al., 2013). Munc18‐2 forms a complex with syntaxin‐3 and other SNARE proteins, thereby regulating the specificity and productivity of SNARE‐mediated membrane fusion at the brush border membrane (Rizo & Südhof, 2012).
Figure 4.
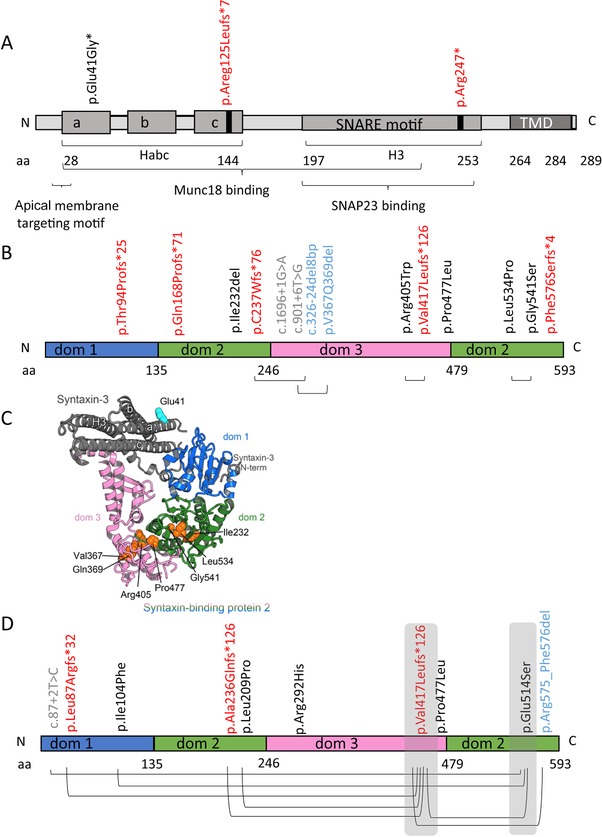
Overview of mutations in STX3 and STXBP2. A: The syntaxin‐3 protein with known domains and locations of STX3 mutations. B: Overview of the munc18‐2 protein with known domains and locations of STXBP2 mutations associated with chronic diarrhea in patients. C: A homology model of syntaxin‐3‐munc18‐2 complex based on syntaxin‐1/munc18‐1 and munc18‐2 crystal structures (PDB ID 4JEU, 4CCA). In closed conformation of syntaxin‐3 regulatory Habc domain (composed of three helixes a, b, c) interacts with H3 domain containing a SNARE motif. Munc18‐2 domains 1 (dom 1) and 3 create a main syntaxin‐3‐binding surface. Syntaxin‐3 N‐terminal peptide binds to the opposite surface of the domain 1. Residues affected by MVID related mutations are shown in spheres and labeled. D: Overview of the munc18‐2 protein with known domains and locations of STXBP2 mutations associated with absence of chronic diarrhea in patients. Mutations indicted in black, red, gray, and blue represent missense, frameshift/nonsense, splicing, and deletions, respectively
The munc18‐2 structure is known (Hackmann et al., 2013), but there is no structural information available for syntaxin‐3. However, the structure of a homologous syntaxin‐1A and its complex with syntaxin‐binding protein 1 (STXBP1 or munc18‐1) has been extensively characterized (Burkhardt, Hattendorf, Weis, & Fasshauer, 2008; Colbert et al., 2013; Lerman, Robblee, Fairman, & Hughson, 2000). In the syntaxin‐1A/munc18‐1 crystal structure (Burkhardt et al., 2008), syntaxin‐1A bound to munc18‐1 adopts a closed conformation in which a regulatory Habc domain three helix bundle (Fernandez et al., 1998), interacts with H3 (SNARE motif) domain, and masks the SNARE motif required for SNARE complex assembly (Figure 4C). The closed syntaxin‐1A binds within the munc18‐1 cleft that is formed by two of the three domains (domain‐1 and domain‐3) (Figure 4D). The N‐terminal region of syntaxin‐1A also interacts with domain‐1 and this serves as a second binding site in the syntaxin‐1A:munc18‐1 complex (Burkhardt et al., 2008). Munc13‐1 MUN domain binding to the syntaxin‐1A linker between Habc and H3 domains initiates its conformational change favoring a transition from the munc18‐1/syntaxin‐1 complex to the SNARE complex (Wang et al., 2017). Syntaxin‐3 shares 63% sequence identity and 80% sequence homology with syntaxin‐1A. Munc18‐2 shares 62% sequence identity and 80% sequence homology with syntaxin‐1A and has a similar structure (Hackmann et al., 2013). This allowed us to model a putative syntaxin‐3:munc18‐2 complex structure (Figure 4C) in order to address the possible functional consequences of identified STX3 and STXBP2 mutations.
4.1. STX3
For STX3 mutation analyses, Genbank sequence (reference AJ002076.1) was used. The two homozygous STX3 mutations, which cosegregated with the disease in the families, included a nonsense mutation c.739C > T (p.Arg247*) in exon 9 and a frame‐shifting insertion c.372_373dup (p.Arg125Leufs*7) in exon 6 (Wiegerinck et al., 2014) (Supp. Table S2A). The c.372_373dup (p.Arg125Leufs*7) mutation causes the introduction of a stop codon in the N‐terminal syntaxin domain (HC) (Figure 4A) and resulted in the loss of syntaxin‐3 protein expression, as evidenced by Western blot analysis (Wiegerinck et al., 2014). The c.739C > T (p.Arg247*) STX3 mutation introduces a stop codon in the central SNARE domain (Figure 4A) and resulted in a truncated protein lacking part of the SNARE‐motif and the entire C‐terminal transmembrane domain (TMD) and extracellular part of the protein, rendering the protein cytosolic (Wiegerinck et al., 2014).
Notably, a homozygous missense STX3 mutation was also identified in a patient with autosomal recessive congenital cataract and intellectual disability phenotype without reported intestinal symptoms (Chograni et al., 2015). This c.122A > G mutation is predicted to lead to the substitution of a glutamic acid with a glycine at position 41 which is on the surface of the first helix of the Habc regulatory domain of syntaxin‐3 (Figure 4A and C). The residue is not involved in munc18‐2 binding. Substitution of the residue to a glycine introduces flexibility in the protein main chain and may destabilize the helix. The Glu41 residue contributes to a negatively charged cluster on the surface of helix‐a of syntaxin that overlaps with the apical membrane targeting motif (aa 31–36) at the beginning of the helix‐a of syntaxin‐3 (Sharma, Low, Misra, Pallavi, & Weimbs, 2006). This suggests a potential contribution of the Glu41 residue to the correct membrane localization of syntaxin‐3 (Supp. Table S2B). While the retina and brain (both affected in this patient) differentially express the alternatively spliced syntaxin‐3B and ‐3A variants, respectively (Curtis et al., 2008), the Glu41 residue is conserved in all syntaxin‐3 splice variants, which explains why both organs can be affected. However, the absence of intestinal symptoms in this patient is not easily explained and will require further investigations to reveal whether the mutation indeed affects the stability of the protein and whether in the case of intestinal cells, the syntaxin‐3 function is compensated by other proteins. The identification of disease‐causing STX3 mutations in patients with completely different clinical features highlights the need for functional genotype–phenotype correlation studies.
4.2. STXBP2
For STXBP2 mutation analyses, nucleotide/protein sequence from Genbank (U63533.1) was used. In the munc18‐2 protein architecture, domain‐2 supports the relative orientation of the domains ‐1 and ‐3, forming a syntaxin‐binding surface (Figure 4C). All the substituted residues resulting from the missense mutations (Supp. Table S3A) as well as the deletion mutations (p.Ile232del, p.Val367_Gln369del) in STXBP2 are buried in the protein core and would destabilize the folding of this protein and therefore perturb the integrity of the munc18‐2 structure and its functions (Supp. Table S3A and Figure 4C). The substitutions in domain‐2 include p.Pro477Leu (found in a buried environment within the protein core that cannot accommodate a bigger side chain), p.Leu534Pro (a critical residue in the central strand of the domain‐2 beta sheet; its mutation to a proline would drastically destabilize the fold), and p.Gly541Ser (the absence of a side‐chain for this residue is critical for the fold of subdomain‐2) (Hackmann et al., 2013). In addition, the p.Arg405Trp substitution is localized in the interface between domain‐2 and ‐3. The Arg405 residue maintains a salt bridge that stabilizes the arched domain‐3 conformation (Hackmann et al., 2013). Its substitution by a bulky tryptophan would destabilize the relative orientation of domain‐2 and ‐3 and therefore also the binding site for syntaxin that requires a precise relative positioning of subdomain ‐1 and ‐3.
In FHL5 families with more than one child affected, intestinal symptoms were either present or absent in all affected siblings, suggesting that the presence or absence of intestinal symptoms in FHL5 patients is related to family‐specific STXBP2 mutations. An important question therefore is whether and, if so how, specific STXBP2 mutations can be correlated to intestinal symptoms. Pagel and colleagues noted that most FHL5 patients that carried the exon 15‐skipping frameshift mutation p.Val417Leufs*126 in STXBP2 (Figure 4D) on at least one allele appeared protected from intestinal symptoms (Pagel et al., 2012) (Supp. Table S3B, 11 out of 13 patients). Nevertheless, two FHL5 patients, one heterozygous and one homozygous for this p.Val417Leufs*126 mutation, were reported positive for intestinal symptoms (Figure 4B) (Supp. Table S3A). In the collected patient data, we also identified a male FHL5 patient with a homozygous p.Pro774Leu missense mutation in STXBP2 with reported intestinal symptoms (Figure 4B), whereas two male siblings with the same homozygous mutation were reported without intestinal symptoms (Figure 4D and Supp. Table S3A and B). Further, a heterozygous p.Gly541Ser substitution in STXBP2 resulting from a c.1621G > A missense mutation was predicted in a male FHL5 patient with intestinal symptoms (Figure 4B) and in two female FHL5 patients without intestinal symptoms (Figure 4D and Supp. Table S3A and B). Substitution of the Gly541 residue with a glutamate in munc18‐2 inhibited its capacity to bind syntaxin‐3 by ∼60% (Riento et al., 2000). Thus, at least for these three STXBP2 mutations, there is as yet no clear genotype–phenotype correlation with regard to the occurrence of intestinal symptoms in FHL5 patients.
Taken together, we conclude that in vitro and patient tissue‐based data clearly indicate that myosin Vb, syntaxin‐3, and munc18‐2 are functionally linked in the process of brush border protein trafficking and brush border development, but that the precise disease mechanism associated with specific mutations in the individual proteins requires further study. Clearly, our structure–function analyses only provide predictions and supporting biochemical analyses are eagerly awaited. Our analyses may help to delineate which mutations would be most useful to study, for example MYO5B mutations that based on structural prediction should strongly impair the protein's motor function but do not have much or highly variable consequences for the disease in terms of clinical symptoms.
5. AN EXPANDED MVID PATIENT REGISTRY AND DATABASE OF MVID‐ASSOCIATED GENE MUTATIONS
We have updated the dataset containing all known MYO5B mutations including information on gender, ancestry, consanguinity, the intron/exon involved and predicted consequence for the protein. In addition, we have expanded the registry with a dataset that includes patients with (variant) MVID and STX3 mutations and a dataset that includes FLH‐5 patients with (variant) MVID and their STXBP2 mutations, together with clinical information with regard to their intestinal symptoms. The registry now includes 188 patients with (variant) MVID and 106 mutations. The inclusion of patients with STX3 or STXBP2 mutations in the registry allows the future correlation and comparison of the clinical course of the disease and the genes involved.
6. ANIMAL MODELS FOR MVID: VERTEBRATES ONLY?
Animal models have been developed which demonstrated that the germline or conditional deletion of the Myo5b gene in mice or its ortholog in zebrafish causes the clinical and cellular hallmarks of MVID (Cartón‐García et al., 2015; Schneeberger et al., 2015; Sidhaye et al., 2016; Weis et al., 2016), thereby formally proving the causality between loss of myosin Vb function and MVID (Cartón‐García et al., 2015). There are no reported mouse models in which Stxbp2 or Stx3 is deleted.
In addition to Myo5b, mice in which Rab8a, Rab11a, or Cdc42 was deleted in the intestine also showed typical hallmarks of MVID. No mutations in RAB8, RAB11A, or CDC42 have thus far been reported in MVID patients, although RAB11A single nucleotide polymorphisms have been detected in at least two MVID patients (Szperl et al., 2011). Interestingly, rab8a expression was shown to be downregulated at the protein level in intestinal mucosa of at least one MVID patient (Sato et al., 2007), and rab11a was shown to be mislocalized in the enterocytes of MVID patient small intestinal biopsies (Dhekne et al., 2014; Golachowska et al., 2012; Szperl et al., 2011). Likewise, cdc42 has been reported to be mislocalized in the enterocytes of MVID patient small intestine biopsies (Michaux et al., 2015). These and other data from cell lines (Knowles et al., 2014) suggest the functional involvement of rab8a, rab11a and cdc42 with myosin Vb in the pathogenesis of MVID. The animal models will be of great value for further elucidation of the cellular and molecular mechanisms that underlie congenital diarrheal disorders centered around MVID, and for the preclinical testing of therapeutic strategies. The zebrafish model for MVID may be particularly useful for high‐throughput testing of potential therapeutic compounds.
Notably, in contrast to vertebrate mice and zebrafish, the invertebrate Drosophila melanogaster (fruit fly) in which the myosin V ortholog didum was deleted did not show an intestinal phenotype, despite the abundant expression level of myosin V in the wild‐type digestive system and its distribution to punctate subapical vesicles (Mermall et al., 2005), which is very similar to its distribution to human epithelial cells (Dhekne et al., 2014; Goldenring et al., 1996). Similarly, the deletion of the MYO5B ortholog Hum‐2 in Caenorhabditis elegans (nematode) did not show an intestinal phenotype (Winter et al., 2012). In contrast to humans, mice, and zebrafish, which have three MYO5 family members (A–C), the invertebrate D. melanogaster and C. elegans have only a single MYO5 gene. Thus, it appears that the critical role of myosin V in the differentiation and function of enterocytes is restricted to vertebrates and evolved with the appearance of multiple myosin V isoforms. There are no reports with regard to the function of syntaxin‐3 or munc18‐2 in the invertebrate digestive system.
7. FUTURE PERSPECTIVES
Clearly, MVID presents as a heterogeneous disease with an expanding spectrum of genotypes and phenotypes. Diagnosis of MVID must include, besides the clinical symptoms, immunohistochemistry analyses of CD10 and PAS staining ad electron microscopy in duodenal biopsies followed by mutation analyses of the MYO5B, STX3, and STXBP2 genes.
Supporting information
Supplementary Figure S1
Supp. Table S1A
ACKNOWLEDGMENTS
We are grateful to Rodrigo Perez‐Ortega, Nick Schubert, and Fabian Peeks for excellent assistance with some of the experiments and Max den Uijl for assistance with the literature analyses.
DISCLOSURE STATEMENT
The authors declare no conflict of interest.
Dhekne HS, Pylypenko O, Overeem AW, et al. MYO5B, STX3 and STXBP2 mutations reveal a common disease mechanism that unifies a subset of congenital diarrheal disorders: a mutation update. Human Mutation. 2018;39:333–344. https://doi.org/10.1002/humu.23386
Funding information Contract Grant Sponsors: Daniel Courtney Trust Foundation; University Medical Center Groningen; Centre National de la Recherche Scientifique (ANR‐13‐BSV8‐0019‐01); Association pour la Recherche sur le Cancer (SFI20121205398).
Communicated by Jürgen Horst
REFERENCES
- Breuza, L. , Fransen, J. , & Le Bivic, A. (2000). Transport and function of syntaxin 3 in human epithelial intestinal cells. American Journal of Physiology. Cell Physiology, 279(4), C1239–1248. [DOI] [PubMed] [Google Scholar]
- Burkhardt, P. , Hattendorf, D. A. , Weis, W. I. , & Fasshauer, D. (2008). Munc18a controls SNARE assembly through its interaction with the syntaxin N‐peptide. The EMBO Journal, 27(7), 923–933. https://doi.org/10.1038/emboj.2008.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartón‐García, F. , Overeem, A. W. , Nieto, R. , Bazzocco, S. , Dopeso, H. , Macaya, I. , … Arango, D. (2015). Myo5b knockout mice as a model of microvillus inclusion disease. Scientific Reports, 5, 12312 https://doi.org/10.1038/srep12312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chograni, M. , Alkuraya, F. S. , Ourteni, I. , Maazoul, F. , Lariani, I. , & Chaabouni, H. B. (2015). Autosomal recessive congenital cataract, intellectual disability phenotype linked to STX3 in a consanguineous Tunisian family. Clinical Genetics, 88(3), 283–287. https://doi.org/10.1111/cge.12489 [DOI] [PubMed] [Google Scholar]
- Colbert, K. N. , Hattendorf, D. A. , Weiss, T. M. , Burkhardt, P. , Fasshauer, D. , & Weis, W. I. (2013). Syntaxin1a variants lacking an N‐peptide or bearing the LE mutation bind to Munc18a in a closed conformation. Proceedings of the National Academy of Sciences of the United States of America, 110(31), 12637–12642. https://doi.org/10.1073/pnas.1303753110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaco, A. , Marathe, J. , Kohnke, H. , Kravstov, D. , & Ameen, N. (2010). Syntaxin 3 is necessary for cAMP‐ and cGMP‐regulated exocytosis of CFTR: Implications for enterotoxigenic diarrhea. American Journal of Physiology. Cell Physiology, 299(6), C1450–1460. https://doi.org/10.1152/ajpcell.00029.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft, N. M. , Howatson, A. G. , Ling, S. C. , Nairn, L. , Evans, T. J. , & Weaver, L. T. (2000). Microvillous inclusion disease: An evolving condition. Journal of Pediatric Gastroenterology and Nutrition, 31(2), 185–189. [DOI] [PubMed] [Google Scholar]
- Curtis, L. B. , Doneske, B. , Liu, X. , Thaller, C. , McNew, J. A. , & Janz, R. (2008). Syntaxin 3b is a t‐SNARE specific for ribbon synapses of the retina. The Journal of Comparative Neurology, 510(5), 550–559. https://doi.org/10.1002/cne.21806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutz, E. , Rhoads, J. M. , Drumm, B. , Sherman, P. M. , Durie, P. R. , & Forstner, G. G. (1989). Microvillus inclusion disease: An inherited defect of brush‐border assembly and differentiation. The New England Journal of Medicine, 320(10), 646–651. https://doi.org/10.1056/NEJM198903093201006 [DOI] [PubMed] [Google Scholar]
- Davidson, G. P. , Cutz, E. , Hamilton, J. R. , & Gall, D. G. (1978). Familial enteropathy: A syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology, 75(5), 783–790. [PubMed] [Google Scholar]
- Dhekne, H. S. , Hsiao, N.‐H. , Roelofs, P. , Kumari, M. , Slim, C. L. , Rings, E. H. H. M. , & van Ijzendoorn, S. C. D. (2014). Myosin Vb and Rab11a regulate phosphorylation of ezrin in enterocytes. Journal of Cell Science, 127(Pt 5), 1007–1017. https://doi.org/10.1242/jcs.137273 [DOI] [PubMed] [Google Scholar]
- Erickson, R. P. , Larson‐Thomé, K. , Valenzuela, R. K. , Whitaker, S. E. , & Shub, M. D. (2008). Navajo microvillous inclusion disease is due to a mutation in MYO5B. American Journal of Medical Genetics. Part A, 146A(24), 3117–3119. https://doi.org/10.1002/ajmg.a.32605 [DOI] [PubMed] [Google Scholar]
- Fernandez, I. , Ubach, J. , Dulubova, I. , Zhang, X. , Südhof, T. C. , & Rizo, J. (1998). Three‐dimensional structure of an evolutionarily conserved N‐terminal domain of syntaxin 1A. Cell, 94(6), 841–849. [DOI] [PubMed] [Google Scholar]
- Galli, P. , Brenna, A. , Camilli de, P. , & Meldolesi, J. (1976). Extracellular calcium and the organization of tight junctions in pancreatic acinar cells. Experimental Cell Research, 99(1), 178–183. [DOI] [PubMed] [Google Scholar]
- Girard, M. , Lacaille, F. , Verkarre, V. , Mategot, R. , Feldmann, G. , Grodet, A. , … Debray, D. (2014). MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology, 60(1), 301–310. https://doi.org/10.1002/hep.26974 [DOI] [PubMed] [Google Scholar]
- Golachowska, M. R. , van Dael, C. M. L. , Keuning, H. , Karrenbeld, A. , Hoekstra, D. , Gijsbers, C. F. M. , … van Ijzendoorn, S. C. D. (2012). MYO5B mutations in patients with microvillus inclusion disease presenting with transient renal Fanconi syndrome. Journal of Pediatric Gastroenterology and Nutrition, 54(4), 491–498. https://doi.org/10.1097/MPG.0b013e3182353773 [DOI] [PubMed] [Google Scholar]
- Goldenring, J. R. , Smith, J. , Vaughan, H. D. , Cameron, P. , Hawkins, W. , & Navarre, J. (1996). Rab11 is an apically located small GTP‐binding protein in epithelial tissues. The American Journal of Physiology, 270(3 Pt 1), G515–525. [DOI] [PubMed] [Google Scholar]
- Gonzales, E. , Taylor, S. A. , Davit‐Spraul, A. , Thébaut, A. , Thomassin, N. , Guettier, C. , … Jacquemin, E. (2017). MYO5B mutations cause cholestasis with normal serum gamma‐glutamyl transferase activity in children without microvillous inclusion disease. Hepatology, 65(1), 164–173. https://doi.org/10.1002/hep.28779 [DOI] [PubMed] [Google Scholar]
- Hackmann, Y. , Graham, S. C. , Ehl, S. , Höning, S. , Lehmberg, K. , Aricò, M. , … Griffiths, G. M. (2013). Syntaxin binding mechanism and disease‐causing mutations in Munc18‐2. Proceedings of the National Academy of Sciences of the United States of America, 110(47), E4482–4491. https://doi.org/10.1073/pnas.1313474110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halac, U. , Lacaille, F. , Joly, F. , Hugot, J.‐P. , Talbotec, C. , Colomb, V. , … Goulet, O. (2011). Microvillous inclusion disease: How to improve the prognosis of a severe congenital enterocyte disorder. Journal of Pediatric Gastroenterology and Nutrition, 52(4), 460–465. https://doi.org/10.1097/MPG.0b013e3181fb4559 [DOI] [PubMed] [Google Scholar]
- Houdusse, A. , & Sweeney, H. L. (2016). How myosin generates force on actin filaments. Trends in Biochemical Sciences, 41(12), 989–997. https://doi.org/10.1016/j.tibs.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu, T. C. , Mahajnah, M. , Manov, I. , & Shaoul, R. (2007). Microvillous inclusion disease: Ultrastructural variability. Ultrastructural Pathology, 31(3), 173–188. https://doi.org/10.1080/01913120701350712 [DOI] [PubMed] [Google Scholar]
- Knowles, B. C. , Roland, J. T. , Krishnan, M. , Tyska, M. J. , Lapierre, L. A. , Dickman, P. S. , … Shub, M. D. (2014). Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. The Journal of Clinical Investigation, 124(7), 2947–2962. https://doi.org/10.1172/JCI71651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, B. C. , Weis, V. G. , Yu, S. , Roland, J. T. , Williams, J. A. , Alvarado, G. S. , … Goldenring, J. R. (2015). Rab11a regulates Syntaxin 3 localization and microvillus assembly in enterocytes. Journal of Cell Science, https://doi.org/10.1242/jcs.163303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravtsov, D. , Mashukova, A. , Forteza, R. , Rodriguez, M. M. , Ameen, N. A. , & Salas, P. J. (2014). Myosin 5b loss of function leads to defects in polarized signaling: Implication for microvillus inclusion disease pathogenesis and treatment. American Journal of Physiology. Gastrointestinal and Liver Physiology, 307(10), G992–G1001. https://doi.org/10.1152/ajpgi.00180.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravtsov, D. V. , Ahsan, M. K. , Kumari, V. , van Ijzendoorn, S. C. D. , Reyes‐Mugica, M. , Kumar, A. , … Ameen, N. A. (2016). Identification of intestinal ion transport defects in microvillus inclusion disease. American Journal of Physiology. Gastrointestinal and Liver Physiology, 311(1), G142–155. https://doi.org/10.1152/ajpgi.00041.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman, J. C. , Robblee, J. , Fairman, R. , & Hughson, F. M. (2000). Structural analysis of the neuronal SNARE protein syntaxin‐1A. Biochemistry, 39(29), 8470–8479. [DOI] [PubMed] [Google Scholar]
- Low, S. H. , Chapin, S. J. , Wimmer, C. , Whiteheart, S. W. , Kömüves, L. G. , Mostov, K. E. , & Weimbs, T. (1998). The SNARE machinery is involved in apical plasma membrane trafficking in MDCK cells. The Journal of Cell Biology, 141(7), 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermall, V. , Bonafé, N. , Jones, L. , Sellers, J. R. , Cooley, L. , & Mooseker, M. S. (2005). Drosophila myosin V is required for larval development and spermatid individualization. Developmental Biology, 286(1), 238–255. https://doi.org/10.1016/j.ydbio.2005.07.028 [DOI] [PubMed] [Google Scholar]
- Michaux, G. , Massey‐Harroche, D. , Nicolle, O. , Rabant, M. , Brousse, N. , Goulet, O. , … Ruemmele, F. M. (2015). The localisation of the apical Par/Cdc42 polarity module is specifically affected in microvillus inclusion disease. Biology of the Cell, 108(1), 19–28. https://doi.org/10.1111/boc.201500034 [DOI] [PubMed] [Google Scholar]
- Mierau, G. W. , Wills, E. J. , Wyatt‐Ashmead, J. , Hoffenberg, E. J. , & Cutz, E. (2001). Microvillous inclusion disease: Report of a case with atypical features. Ultrastructural Pathology, 25(6), 517–521. [DOI] [PubMed] [Google Scholar]
- Morroni, M. , Cangiotti, A. M. , Guarino, A. , & Cinti, S. (2006). Unusual ultrastructural features in microvillous inclusion disease: A report of two cases. Virchows Archiv, 448(6), 805–810. https://doi.org/10.1007/s00428-006-0180-y [DOI] [PubMed] [Google Scholar]
- Müller, T. , Hess, M. W. , Schiefermeier, N. , Pfaller, K. , Ebner, H. L. , Heinz‐Erian, P. , … Janecke, A. R. (2008). MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nature Genetics, 40(10), 1163–1165. https://doi.org/10.1038/ng.225 [DOI] [PubMed] [Google Scholar]
- Nag, S. , Sommese, R. F. , Ujfalusi, Z. , Combs, A. , Langer, S. , Sutton, S. , … Spudich, J. A. (2015). Contractility parameters of human β‐cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Science Advances, 1(9), e1500511 https://doi.org/10.1126/sciadv.1500511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento, A. F. Z. , Trindade, D. M. , Tonoli, C. C. C. , de Giuseppe, P. O. , Assis, L. H. P. , Honorato, R. V. , … Murakami, M. T. (2013). Structural insights into functional overlapping and differentiation among myosin V motors. The Journal of Biological Chemistry, 288(47), 34131–34145. https://doi.org/10.1074/jbc.M113.507202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overeem, A. W. , Bryant, D. M. , & van IJzendoorn, S. C. D. (2015). Mechanisms of apical‐basal axis orientation and epithelial lumen positioning. Trends in Cell Biology, 25(8), 476–485. https://doi.org/10.1016/j.tcb.2015.04.002 [DOI] [PubMed] [Google Scholar]
- Pagel, J. , Beutel, K. , Lehmberg, K. , Koch, F. , Maul‐Pavicic, A. , Rohlfs, A.‐K. , … Janka, G. (2012). Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood, 119(25), 6016–6024. https://doi.org/10.1182/blood-2011-12-398958 [DOI] [PubMed] [Google Scholar]
- Perry, A. , Bensallah, H. , Martinez‐Vinson, C. , Berrebi, D. , Arbeille, B. , Salomon, J. , … Hugot, J.‐P. (2014). Microvillous atrophy: Atypical presentations. Journal of Pediatric Gastroenterology and Nutrition, 59(6), 779–785. https://doi.org/10.1097/MPG.0000000000000526 [DOI] [PubMed] [Google Scholar]
- Phillips, A. D. , & Schmitz, J. (1992). Familial microvillous atrophy: A clinicopathological survey of 23 cases. Journal of Pediatric Gastroenterology and Nutrition, 14(4), 380–396. [PubMed] [Google Scholar]
- Posovszky, C. (2016). Congenital intestinal diarrhoeal diseases: A diagnostic and therapeutic challenge. Best Practice & Research. Clinical Gastroenterology, 30(2), 187–211. https://doi.org/10.1016/j.bpg.2016.03.004 [DOI] [PubMed] [Google Scholar]
- Pylypenko, O. , Attanda, W. , Gauquelin, C. , Lahmani, M. , Coulibaly, D. , Baron, B. , … Houdusse, A. M. (2013). Structural basis of myosin V Rab GTPase‐dependent cargo recognition. Proceedings of the National Academy of Sciences of the United States of America, 110(51), 20443–20448. https://doi.org/10.1073/pnas.1314329110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, Y.‐L. , Gong, J.‐Y. , Feng, J.‐Y. , Wang, R.‐X. , Han, J. , Liu, T. , … Wang, J.‐S. (2017). Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ‐glutamyltransferase cholestasis. Hepatology, 65(5), 1655–1669. https://doi.org/10.1002/hep.29020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento, K. , Kauppi, M. , Keranen, S. , & Olkkonen, V. M. (2000). Munc18‐2, a functional partner of syntaxin 3, controls apical membrane trafficking in epithelial cells. The Journal of Biological Chemistry, 275(18), 13476–13483. [DOI] [PubMed] [Google Scholar]
- Rizo, J. , & Südhof, T. C. (2012). The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annual Review of Cell and Developmental Biology, 28, 279–308. https://doi.org/10.1146/annurev-cellbio-101011-155818 [DOI] [PubMed] [Google Scholar]
- Ruemmele, F. M. , Müller, T. , Schiefermeier, N. , Ebner, H. L. , Lechner, S. , Pfaller, K. , … Huber, L. A. (2010). Loss‐of‐function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo‐2 RNAi cell model. Human Mutation, 31(5), 544–551. https://doi.org/10.1002/humu.21224 [DOI] [PubMed] [Google Scholar]
- Sato, T. , Mushiake, S. , Kato, Y. , Sato, K. , Sato, M. , Takeda, N. , … Harada, A. (2007). The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature, 448(7151), 366–369. https://doi.org/10.1038/nature05929 [DOI] [PubMed] [Google Scholar]
- Schneeberger, K. , Vogel, G. F. , Teunissen, H. , van Ommen, D. D. , Begthel, H. , El Bouazzaoui, L. , … Middendorp, S. (2015). An inducible mouse model for microvillus inclusion disease reveals a role for myosin Vb in apical and basolateral trafficking. Proceedings of the National Academy of Sciences of the United States of America, 112(40), 12408–12413. https://doi.org/10.1073/pnas.1516672112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, N. , Low, S. H. , Misra, S. , Pallavi, B. , & Weimbs, T. (2006). Apical targeting of syntaxin 3 is essential for epithelial cell polarity. The Journal of Cell Biology, 173(6), 937–948. https://doi.org/10.1083/jcb.200603132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhaye, J. , Pinto, C. S. , Dharap, S. , Jacob, T. , Bhargava, S. , & Sonawane, M. (2016). The zebrafish goosepimples/myosin Vb mutant exhibits cellular attributes of human microvillus inclusion disease. Mechanisms of Development, 142, 62–74. https://doi.org/10.1016/j.mod.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepensky, P. , Bartram, J. , Barth, T. F. , Lehmberg, K. , Walther, P. , Amann, K. , … Posovszky, C. (2013). Persistent defective membrane trafficking in epithelial cells of patients with familial hemophagocytic lymphohistiocytosis type 5 due to STXBP2/MUNC18‐2 mutations. Pediatric Blood & Cancer, 60(7), 1215–1222. https://doi.org/10.1002/pbc.24475 [DOI] [PubMed] [Google Scholar]
- Szperl, A. M. , Golachowska, M. R. , Bruinenberg, M. , Prekeris, R. , Thunnissen, A.‐M. W. H. , Karrenbeld, A. , … van IJzendoorn, S. C. D. (2011). Functional characterization of mutations in the myosin Vb gene associated with microvillus inclusion disease. Journal of Pediatric Gastroenterology and Nutrition, 52(3), 307–313. https://doi.org/10.1097/MPG.0b013e3181eea177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trybus, K. M. (2008). Myosin V from head to tail. Cellular and Molecular Life Sciences, 65(9), 1378–1389. https://doi.org/10.1007/s00018-008-7507-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velde, K. J. , Dhekne, H. S. , Swertz, M. A. , Sirigu, S. , Ropars, V. , Vinke, P. C. , … van Ijzendoorn, S. C. D. (2013). An overview and online registry of microvillus inclusion disease patients and their MYO5B mutations. Human Mutation, 34(12), 1597–1605. https://doi.org/10.1002/humu.22440 [DOI] [PubMed] [Google Scholar]
- Velvarska, H. , & Niessing, D. (2013). Structural insights into the globular tails of the human type v myosins Myo5a, Myo5b, And Myo5c. PloS One, 8(12), e82065 https://doi.org/10.1371/journal.pone.0082065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, G. F. , Klee, K. M. C. , Janecke, A. R. , Müller, T. , Hess, M. W. , & Huber, L. A. (2015). Cargo‐selective apical exocytosis in epithelial cells is conducted by Myo5B, Slp4a, Vamp7, and Syntaxin 3. The Journal of Cell Biology, 211(3), 587–604. https://doi.org/10.1083/jcb.201506112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, G. F. , van Rijn, J. M. , Krainer, I. M. , Janecke, A. R. , Posovzsky, C. , Cohen, M. , … Huber, L. A. (2017). Disrupted apical exocytosis of cargo vesicles causes enteropathy in FHL5 patients with Munc18‐2 mutations. JCI Insight, 2(14). https://doi.org/10.1172/jci.insight.94564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Choi, U. B. , Gong, J. , Yang, X. , Li, Y. , Wang, A. L. , … Ma, C. (2017). Conformational change of syntaxin linker region induced by Munc13s initiates SNARE complex formation in synaptic exocytosis. The EMBO Journal, 36(6), 816–829. https://doi.org/10.15252/embj.201695775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks, D. A. , Zuppan, C. W. , Malott, R. L. , & Mierau, G. W. (2003). Microvillous inclusion disease with abundant vermiform, electron‐lucent vesicles. Ultrastructural Pathology, 27(5), 337–340. [PubMed] [Google Scholar]
- Weis, G. V. , Knowles, B. C. , Choi, E. , Goldstein, A. E. , Williams, J. A. , Manning, E. H. , … Goldenring, J. R. (2016). Loss of MYO5B in mice recapitulates Microvillus Inclusion Disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cellular and Molecular Gastroenterology and Hepatology, 2(2), 131–157. https://doi.org/10.1016/j.jcmgh.2015.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegerinck, C. L. , Janecke, A. R. , Schneeberger, K. , Vogel, G. F. , van Haaften‐Visser, D. Y. , Escher, J. C. , … Middendorp, S. (2014). Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology, 147(1), 65–68.e10. https://doi.org/10.1053/j.gastro.2014.04.002 [DOI] [PubMed] [Google Scholar]
- Winter, J. F. , Höpfner, S. , Korn, K. , Farnung, B. O. , Bradshaw, C. R. , Marsico, G. , … Zerial, M. (2012). Caenorhabditis elegans screen reveals role of PAR‐5 in RAB‐11‐recycling endosome positioning and apicobasal cell polarity. Nature Cell Biology, 14(7), 666–676. https://doi.org/10.1038/ncb2508 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supp. Table S1A