

Abstract
Generative artificial intelligence offers a fresh view on molecular design. We present the first‐time prospective application of a deep learning model for designing new druglike compounds with desired activities. For this purpose, we trained a recurrent neural network to capture the constitution of a large set of known bioactive compounds represented as SMILES strings. By transfer learning, this general model was fine‐tuned on recognizing retinoid X and peroxisome proliferator‐activated receptor agonists. We synthesized five top‐ranking compounds designed by the generative model. Four of the compounds revealed nanomolar to low‐micromolar receptor modulatory activity in cell‐based assays. Apparently, the computational model intrinsically captured relevant chemical and biological knowledge without the need for explicit rules. The results of this study advocate generative artificial intelligence for prospective de novo molecular design, and demonstrate the potential of these methods for future medicinal chemistry.
Keywords: Automation, drug discovery, machine learning, medicinal chemistry, nuclear receptor
Computational de novo design aims to generate new chemical entities with desired properties.1 There are several such methodologies, largely differing in the process of chemical structure generation and the scoring methods employed.2,3 Recently, an innovative concept of de novo molecular design has been proposed that relies on generative artificial intelligence (AI). It bears promise as a way of learning from known bioactive compounds and autonomously designs novel compounds with inherited bioactivity and synthesizability (Figure 1).4,5 Importantly, these generative methods are expected to produce chemically correct structures without the need for explicitly including building block libraries or rules for their fusion and chemical transformation. However, until now, generative AI has only been applied to retrospective de novo design by reproducing known bioactive ligands or generating predicted actives. In this first prospective study, we apply generative AI to see if this approach lives up to its promise to deliver actually synthesizable bioactive de novo designs.
Figure 1.
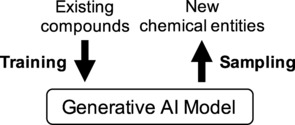
Concept of generative artificial intelligence (AI). A model of the training data (e. g., molecular structures) is obtained that can be used to emit new instances (new chemical entities) within the training domain by sampling.
The computational approach consisted of two basic steps. First, we developed a generic model that learned the constitution of druglike molecules from a large unfocussed compound set. In a second step, we fine‐tuned this generic model on more specific molecular features from a small target‐focused library of actives. For the generic model, we utilized a recently published deep recurrent neural network (RNN) with long short‐term memory (LSTM) cells,6 which had been trained on SMILES representations of 541,555 bioactive compounds (K D, K i, IC/EC50 values <1 μM) extracted from the ChEMBL227 compound database.5 Then, we fine‐tuned the model by transfer learning to enable the de novo generation of target‐specific ligands. For this purpose, we used 25 fatty acid mimetics8 with known agonistic activity on retinoid X receptors (RXR)9 and/or peroxisome proliferator‐activated receptors (PPAR).10 From the resulting fine‐tuned AI model, we sampled 1000 SMILES strings, applying fragment growing from the minimalist start fragment “−COOH”.
The generated set included 93 % valid and 90 % unique SMILES entries, all of which contained a carboxylic acid function by default. None of the computer‐generated chemical structures was identical to compounds from the training sets. Importantly, the newly generated molecules populate the chemical space of the training data, residing within the RXR/PPAR region of the fine‐tuning set (Figure 2). These observations corroborate the ability of the generative AI model to produce novel chemical entities within the training data domain.
Figure 2.
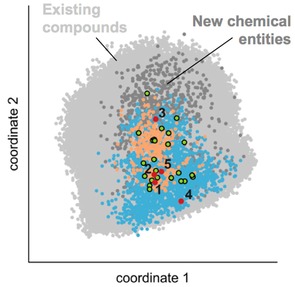
Chemical space analysis by multi‐dimensional scaling. Compounds were represented by Morgan substructure fingerprints (radius=0–4 bonds, length=1024 bit), and similarity was defined by the Jaccard‐Tanimoto index. Colored dots represent the training data (light grey), fine‐tuning set (green), known RXR (orange) and PPAR (blue) agonists, sampled molecules (dark grey), and the selected de novo designs 1–5 (red). Compounds 1, 2, 3 and 5 populate the same area as the known RXR and PPAR agonists, while 4 is similar to PPAR agonist but remote from known RXR actives.
Following this preliminary analysis, we computationally ranked the de novo designs according to their potential modulatory effects on RXRs and PPARs. For this purpose, we employed a target prediction method (SPiDER),11 and molecular shape and partial charge descriptors to determine the similarity of the designed compounds to known bioactive ligands. The individual screening lists were merged, obtaining a final set of 49 high‐scoring designs (Supplementary Information). For proof‐of‐concept, we selected five compounds (1–5, Scheme 1) from this list for synthesis, taking into account their individual in silico ranks and building block availability. These five chemical entities were not present in the ChEMBL,7 PubChem,12 SureChEMBL,13 Reaxys14 and SciFinder15 databases, indicating their novelty.
Scheme 1.
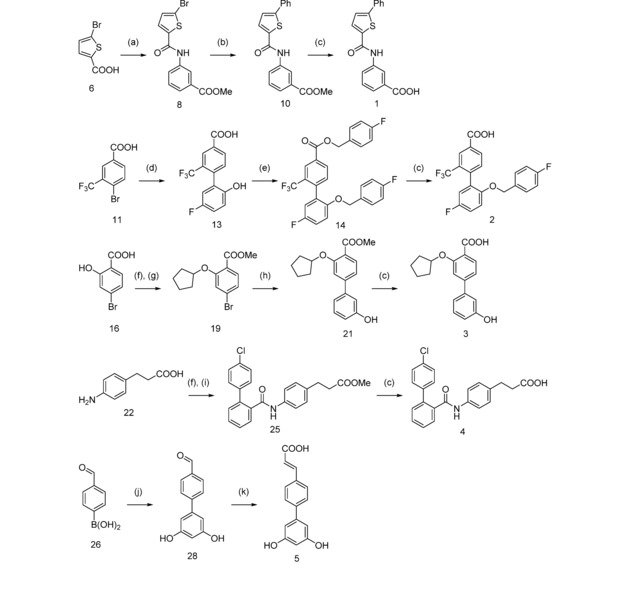
Synthesis of designs 1–5. Reagents & conditions: (a) H2N−C6H4−COOH (7), EDC, 4‐DMAP, THF, reflux, 4 h; (b) C6H5−B(OH)2 (9), Pd(PPh3)4, Cs2CO3, dioxane, 100 °C, 16 h; (c) KOH, MeOH/THF/H2O, μw, 70 °C, 30 min; (d) HO‐C6H3F−B(OH)2 (12), Pd(PPh3)4, Cs2CO3, toluene/EtOH, 100 °C, 20 h; (e) F‐C6H4‐CH2‐Br (15), K2CO3, DMF, μw, 100 °C, 120 min; (f) MeOH, H2SO4cc, reflux, 4 h; (g) C5H9Br (18), K2CO3, DMF, μw, 100 °C, 6 h; (h) HO‐C6H4‐B(OH)2 (20), Pd(PPh3)4, Cs2CO3, toluene/EtOH, 100 °C, 16 h; (i) C6H4Cl‐C6H4‐COOH (24), EDC, 4‐DMAP, CHCl3, relux, 12 h; (j) C6H3Br(OH)2 (27), Pd(PPh3)4, Cs2CO3, dioxane/DMF, reflux, 4 h; (k) malonic acid, pyridine/piperidine, μw, 100 °C, 30 min.
Compounds 1–5 were prepared over two to four steps (Scheme 1). Amide coupling of 5‐bromothiophene‐2‐carboxylic acid (6) and methyl 3‐aminobenzoate (7), using EDC/4‐DMAP to 8, followed by Suzuki reaction with benzeneboronic acid (9) to 10 and alkaline ester hydrolysis afforded compound 1. Compound 2 was available from 4‐bromo‐3‐trifluoromethylbenzoic acid (11) and 2‐hydroxy‐5‐fluorobenzeneboronic acid (12), forming 13 in a Suzuki reaction followed by Williamson ether synthesis to 14 with excess 4‐fluorobenzyl bromide (15) and subsequent hydrolysis of the resulting ester. For the preparation of compound 3, 4‐bromosalicylic acid (16) was esterified (17) with methanol and reacted with bromocyclopentane (18) to form ether 19. Suzuki reaction of 19 with 3‐hydroxybenzeneboronic acid (20) to 21 and alkaline ester hydrolysis yielded 3. Compound 4 was obtained from 3‐(4‐aminophenyl)propionic acid (22) by esterification (23), amide coupling with 2‐(4‐chlorophenyl)benzoic acid (24) to 25 using EDC/4‐DMAP, and alkaline ester hydrolysis. Suzuki reaction of 4‐formylphenylboronic acid (26) and 5‐bromoresorcinol (27) to 28 followed by Knoevenagel condensation in Doebner modification with malonic acid afforded compound 5.
We then characterized designs 1–5 in hybrid reporter gene assays for their agonistic effects on nuclear receptors RXRα/β/γ and PPARα/γ/δ in HEK293T cells.16 These in vitro tests involved constitutively expressed hybrid receptors composed of the ligand binding domain of the respective human nuclear receptor and the DNA‐binding domain of the nuclear receptor Gal4 from yeast. Gal4 responsive firefly luciferase served as reporter gene, and constitutively expressed Renilla luciferase was used for normalization of transfection efficiency and toxicity control.
The in vitro characterization of 1–5 revealed agonistic activity on PPAR and RXR subtypes (Table 1). Four of the compounds were active, and for each receptor studied, we identified at least one agonist. Designs 1 and 2 turned out as dual agonists of RXRs and PPARγ, whereas 3 and 4 each activated two PPAR subtypes but were inactive on RXRs. Only design 5 showed neither RXR nor PPAR transactivation activity. EC50 values of 1–4 ranged between double‐digit nanomolar for RXR agonist 1, despite moderate transactivation efficacy, and double‐digit micromolar for design 4 on PPARδ. Design 2 revealed micromolar potency on RXRs but markedly higher transactivation efficacy than 1. With regard to PPARγ, design 2 showed micromolar agonistic activity with equivalent efficacy as the reference agonist pioglitazone. Design 3 behaved as a micromolar superagonist on PPARγ, with about 2.5‐fold greater transactivation efficacy than pioglitazone. 4 turned out as the least potent design and showed partial agonistic activity on both PPARγ and PPARδ.
Table 1.
In vitro activity of designs 1–5 on RXRs and PPARs (EC50 values ± SEM [μM]; n=2 (when inactive) or 4 (when active) independent experiments in duplicates; inactive, no statistically significant reporter transactivation at a compound concentration of 30 μM).
| Compound no. | RXRα | RXRβ | RXRγ | PPARα | PPARγ | PPARδ |
|---|---|---|---|---|---|---|
| 1 | 0.13±0.01 | 1.1±0.3 | 0.06±0.02 | inactive | 2.3±0.2 | inactive |
| 2 | 13.0±0.1 | 9±2 | 8.0±0.7 | inactive | 2.8±0.3 | inactive |
| 3 | inactive | inactive | inactive | 4.0±1.0 | 10.1±0.3 | inactive |
| 4 | inactive | inactive | inactive | inactive | 9±3 | 14±2 |
| 5 | inactive | inactive | inactive | inactive | inactive | inactive |
| reference agonistsa) | 0.033±0.002 | 0.024±0.004 | 0.025±0.002 | 0.006±0.002 | 0.6±0.1 | 0.5±0.1 |
To exclude unspecific effects, we repeated the in vitro assays in the absence of a hybrid receptor for every active molecule, using a concentration at or above its EC80 value. This time, only the reporter gene and control luciferase, but no hybrid receptor, were transfected. Designs 1–4 caused no observable reporter transactivation without a hybrid receptor, confirming that their activity was actually mediated via RXRs and PPARs, respectively (Supplementary Information).
These results experimentally validate the applicability of generative AI to prospective de novo molecule design. The computational approach led to the discovery of new agonists of therapeutically relevant nuclear receptors. The bioactive designs 1–4 possess considerable potency, as well as diverse selectivity profiles on RXRs and PPARs, and may serve as starting points for hit‐to‐lead expansion. All of the selected compounds were easily prepared from commercially available building blocks, suggesting that their chemical synthesizability was intrinsically learned by the computer model. The results also suggest that a proper choice of compound libraries for model fine‐tuning by transfer learning enables application‐tailored AI support for de novo design. This particular concept might even be suitable for concerted multi‐target drug design. By providing rapid knowledge‐driven access to innovative small molecules, generative AI bears potential for medicinal chemistry and chemical biology.
Conflict of Interest
G. S. declares a potential financial conflict of interest in his role as life‐science industry consultant and cofounder of inSili.com GmbH, Zurich.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank P. Schneider for compiling the subsets of the ChEMBL database and A. T. Müller for technical support. This research was financially supported by the Swiss National Science Foundation (grant no. IZSEZ0_177477). D. M. was supported by an ETH Zurich Postdoctoral Fellowship (grant no. 16‐2 FEL‐07).
D. Merk, L. Friedrich, F. Grisoni, G. Schneider, Mol. Inf. 2018, 37, 1700153.
References
- 1.G. Schneider, Nat. Rev. Drug Discov 2018, doi: nrd.2017.232.
- 2. Schneider P., Schneider G., J. Med. Chem. 2016, 59, 4077–4086. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Schneider G., Fechner U., U., Nat. Rev. Drug Discov. 2005, 4, 649–663; [DOI] [PubMed] [Google Scholar]
- 3b. Hartenfeller M., Schneider G., Methods Mol. Biol., 2011, 672, 299–323. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Olivecrona M., Blaschke T., Engkvist O., Chen H., J. Cheminform. 2017, 9, 48; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Blaschke T., Olivecrona M., Engkvist O., Bajorath J., Chen H., Mol. Inf. 2018, 37, 1700123; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Miyao T., Funatsu K., Mol. Inf. 2017, 36, 1700030. [DOI] [PubMed] [Google Scholar]
- 5. Gupta A., Müller A. T., Huisman B. J. H., Fuchs J. A., Schneider P., Schneider G., Mol. Inf. 2018, 37, 1700111. [Google Scholar]
- 6. Hochreiter S., Schmidhuber J., Neural Comput. 1997, 9, 1735–1780. [DOI] [PubMed] [Google Scholar]
- 7. Bento A. P., Gaulton A., Hersey A., Bellis L. J., Chambers J., Davies M., Krüger F. A., Light Y., Mak L., McGlinchey S., Nowotka M., Papadatos G., Santos R., Overington J. P., Nucleic Acids Res. 2014, 42, 1083–1090; https://www.ebi.ac.uk/chembl/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Proschak E., Heitel P., Kalinowsky L., Merk D., J. Med. Chem. 2017, 60, 5235–5266. [DOI] [PubMed] [Google Scholar]
- 9. Germain P., Chambon P., Eichele G., Evans R. M., Lazar M. A., Leid M., De Lera A. R., Lotan R., Mangelsdorf D. J., Gronemeyer H., Pharmacol. Rev. 2006, 58, 760–772. [DOI] [PubMed] [Google Scholar]
- 10. Michalik L., Auwerx J., Berger J. P., Chatterjee V. K., Glass C. K., Gonzalez F. J., Grimaldi P. A., Kadowaki T., Lazar M. A., O'Rahilly S., Palmer C. N., Plutzky J., Reddy J. K., Spiegelman B. M., Staels B., Wahli W., Pharmacol. Rev. 2006, 58, 726–741. [DOI] [PubMed] [Google Scholar]
- 11. Reker D., Rodrigues T., Schneider P., Schneider G., Proc. Natl. Acad. Sci. USA 2014, 111, 4067–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim S., Thiessen P. A., Bolton E. E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B. A., Wang J., Yu B., Zhang J., Bryant S. H., Nucleic Acids Res. 2016, 44, D1202–D1213; https://pubchem.ncbi.nlm.nih.gov/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Papadatos G., Davies M., Dedman N., Chambers J., Gaulton A., Siddle J., Koks R., Irvine S. A., Pettersson J., Goncharoff N., Hersey A., Overington J. P., Nucleic Acids Res. 2016, 44, D1220–D1228; https://www.surechembl.org/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reaxys, Elsevier 2017; https://www.reaxys.com/.
- 15.SciFinder, Chemical Abstracts Service 2017; https://scifinder.cas.org/.
- 16. Schmidt J., Rotter M., Weiser T., Wittmann S., Weizel L., Kaiser A., Heering J., Goebel T., Angioni C., Wurglics M., Paulke A., Geisslinger G., Kahnt A., Steinhilber D., Proschak E., Merk D., J. Med. Chem. 2017, 60, 7703–7724. [DOI] [PubMed] [Google Scholar]
- 17. Boehm M., Zhang L., Badea B., White S., Mais D., Berger E., Suto C., Goldman M., Heyman R., J. Med. Chem. 1994, 37, 2930–2941. [DOI] [PubMed] [Google Scholar]
- 18. Brown P., Stuart L., Hurley K., Lewis M., Winegar D., Wilson J., Wilkison W., Ittoop O., Willson T., Bioorg. Med. Chem. Lett. 2001, 11, 1225–1227. [DOI] [PubMed] [Google Scholar]
- 19. Willson T., Brown P., Sternbach D., Henke B., J. Med. Chem. 2000, 43, 527–550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary