

Abstract
Background and purpose
To better characterize the effects of tafamidis in non‐Val30Met patients with transthyretin familial amyloid polyneuropathy, this post hoc analysis compared the neurological results from a 12‐month, open‐label study of non‐Val30Met versus Val30Met patients at month 12 from the 18‐month, double‐blind, placebo‐controlled registration study. A baseline covariate adjusted analysis was used to control for differences in baseline neurological severity.
Methods
Neurological function was assessed using the Neuropathy Impairment Score – Lower Limbs (NIS‐LL) in three cohorts: Val30Met tafamidis (n = 64), Val30Met placebo (n = 61) and non‐Val30Met tafamidis (n = 21). The change in NIS‐LL from baseline to month 12 for Val30Met and non‐Val30Met tafamidis‐treated patients was compared with the change from baseline at month 12 for Val30Met placebo‐treated patients using a mixed‐effects model for repeated measures (MMRM).
Results
The baseline adjusted mean (standard error) change in NIS‐LL values at month 12 was similar for Val30Met [1.60 (0.78)] and non‐Val30Met [1.62 (1.43)] tafamidis‐treated patients and less than that observed in the Val30Met placebo‐treated group [4.72 (0.77); P = 0.0055 for Val30Met and P = 0.0592 for non‐Val30Met]. Based on the MMRM, the magnitude of change in both tafamidis‐treated cohorts was similar across the range of observed baseline NIS‐LL values, and was consistently less than that observed in the Val30Met placebo‐treated group at month 12.
Conclusions
This baseline‐adjusted analysis demonstrated that tafamidis treatment delayed neurological progression comparably in Val30Met and non‐Val30Met patients across a range of baseline NIS‐LL values. Neurological progression in these two genotype groups may be more similar than previously considered.
Keywords: amyloidosis, baseline severity, familial amyloid polyneuropathy, neurological progression, non‐Val30Met, tafamidis, transthyretin, Val30Met
Background
Transthyretin familial amyloid polyneuropathy (TTR‐FAP) is a rare, life‐threatening, genetic disorder caused by TTR gene mutations that result in the formation of insoluble amyloid fibrils in peripheral nerves and organs 1. Progressive tissue damage and the severely debilitating symptoms of peripheral and autonomic neuropathy, along with potential complications due to organ involvement, can significantly shorten life expectancy 2, 3. Clinical presentation can vary widely amongst patients depending on a number of factors, including age of onset and TTR gene mutation 3, 4. Of the TTR mutations, the Val30Met genotype is believed to be the most common and widely studied mutation and is associated with a predominantly polyneuropathy phenotype 4, 5; non‐Val30Met mutations, although less well‐characterized, are generally associated with poorer prognosis, but considerable heterogeneity exists 6. Whether non‐Val30Met patients actually have a poorer prognosis continues to be an important question.
Tafamidis, a highly specific TTR stabilizer, is the only approved medicine to delay neurological progression of TTR‐FAP in Europe and parts of Asia and Latin America, and it has emerged as the new standard of care 7. Tafamidis has undergone extensive clinical evaluation, particularly amongst patients with the TTR mutation Val30Met. Clinical studies, including a multicentre, 18‐month, randomized, placebo‐controlled, double‐blind phase 3 study, along with its long‐term open‐label extensions, have demonstrated the safety and efficacy of tafamidis in delaying disease progression 8, 9, 10, 11, 12, 13.
Tafamidis has also been evaluated in a multicentre, open‐label, 12‐month study to assess the safety and efficacy in 21 patients with TTR‐FAP due to eight non‐Val30Met mutations 14. Following 12 months of treatment, all patients remaining in the study had achieved TTR stabilization, with some worsening of neurological function (the predominant symptom in these patients at baseline) but nutritional status (modified body mass index) and quality of life remained stable 14. Despite cardiac symptom involvement in these non‐Val30Met TTR‐FAP patients, little worsening in cardiac morbidity was seen during treatment 14. Interpretation of these results is limited due to the open‐label design and lack of a control group, as well as the limited information on the natural history of the expected progression of patients with non‐Val30Met mutations 6.
The objective of the present analysis was to more fully characterize the results of the non‐Val30Met study 14 and to better understand the effects of tafamidis in non‐Val30Met patients by comparing the neurological data to those of Val30Met patients of the registration study 8. Importantly, because a much higher level of baseline disease burden (potentially due to the longer duration of disease in the non‐Val30Met study population) was observed in non‐Val30Met patients than in Val30Met patients, the present baseline covariate‐adjusted analysis addresses the difference in baseline disease severity. The importance of controlling for baseline disease severity was recently demonstrated in a post hoc analysis of Val30Met patients treated with tafamidis 12. By controlling for baseline disease severity, the current analysis helps to answer the question of whether patients with non‐Val30Met mutations have more rapid disease progression than those with a Val30Met mutation.
Methods
Study design and participants
The combined intent‐to‐treat (ITT) data from the tafamidis 18‐month, placebo‐controlled, double‐blind registration study in Val30Met patients (ClinicalTrials.gov identifier NCT00409175) and the 12‐month open‐label study in non‐Val30Met patients (NCT00630864) were analysed based on treatment assignment in the original studies: Val30Met treated with tafamidis (n = 64), Val30Met treated with placebo (n = 61) and non‐Val30Met treated with tafamidis (n = 21; Asp38Ala, n = 1; Gly47Ala, n = 3; Leu58His, n = 4; Thr60Ala, n = 4; Phe64Leu, n = 4; Ser77Phe, n = 1; Ser77Tyr, n = 2; Ile107Val, n = 2) 8, 14. The ITT population for the Val30Met study was defined as all patients who received at least one dose of oral study medication (placebo or tafamidis meglumine 20 mg once daily) and had at least one post baseline assessment for both the Neuropathy Impairment Score – Lower Limbs (NIS‐LL) and the Norfolk Quality of Life – Diabetic Neuropathy questionnaire, or discontinued the study due to death or liver transplant. For the non‐Val30Met study, the ITT population included all patients who received at least one dose of oral tafamidis meglumine 20 mg 14. Additional details on trial design and study participants are available in the primary publications of the registration study for Val30Met patients 8 and the open‐label study for non‐Val30Met patients 14. Both studies received approval of local institutional review boards or independent ethics committees (Box S1) and were conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization Guideline for Good Clinical Practice and local regulatory requirements. All study participants provided written informed consent.
Outcome measure
Neurological function was assessed using the NIS‐LL [scores range from 0 (normal) to 88 (total impairment)], a validated and sensitive measure of neurological impairment in patients with TTR‐FAP 15.
Statistical analysis
The change in NIS‐LL from baseline to month 12 for Val30Met and non‐Val30Met tafamidis‐treated patients was compared with the change from baseline to month 12 for Val30Met placebo patients using a mixed‐effects model for repeated measures (MMRM), with change from baseline in NIS‐LL as the dependent variable, treatment, visit and treatment‐by‐visit as fixed effects, baseline NIS‐LL as a covariate, and subject as a random effect in the model, using an unstructured covariance matrix. MMRM is an analysis method commonly used to assess effects in data collected from patients at multiple visits over time. The method is able to address the correlated observations that result from the repeated measurements of a patient progressing through a study over time. Partial data (e.g. from patients who do not fully complete the study) can also be included in the analysis.
Results
The baseline demographic and clinical characteristics of the Val30Met (placebo and tafamidis) and non‐Val30Met (tafamidis) groups have been published previously 8, 14. The Val30Met and non‐Val30Met patients were mostly from clinical sites in Portugal and the USA, respectively. All groups were predominantly Caucasian (~90%), comprising both male and female patients (Val30Met, 46.4% male; non‐Val30Met, 61.9% male). Non‐Val30Met tafamidis versus Val30Met tafamidis and placebo patients, respectively, were older [mean (SD) 63.1 (9.9) vs. 39.8 (12.7) and 38.4 (12.9) years] and had longer symptom duration [mean (SD) 64.7 (60.8) vs. 47.0 (48.4) and 34.7 (32.9) months] and more advanced neurological impairment [mean (SD) NIS‐LL score 27.6 (24.7) vs. 8.4 (11.4) and 11.4 (13.5)] 8, 14.
Baseline covariate adjusted change in NIS‐LL scores from baseline to month 12 was analysed to assess treatment effect and account for differences in baseline disease severity amongst treatment groups. The baseline adjusted mean change scores in NIS‐LL at month 12 were similar in Val30Met and non‐Val30Met tafamidis‐treated patients (1.60 vs. 1.62, respectively) and less than that observed in the Val30Met placebo group (4.72) (Table 1). The magnitude of separation from the Val30Met placebo group was statistically significant in the Val30Met tafamidis group (P = 0.0055) but not in the non‐Val30Met tafamidis group (P = 0.0592), possibly due to the lower number of patients in the non‐Val30Met group (Table 1).
Table 1.
Change from baseline in NIS‐LL to month 12 for Val30Met and non‐Val30Met groups
| Tafamidis Val30Met | Placebo Val30Met | Tafamidis Val30Met versus placebo Val30Met | Tafamidis non‐Val30Met | Tafamidis non‐Val30Met versus placebo Val30Met | |
|---|---|---|---|---|---|
| Baseline | |||||
| n | 64 | 61 | – | 21 | – |
| Mean (SD) | 8.4 (11.4) | 11.4 (13.5) | – | 27.6 (24.7) | – |
| Month 12 change from baseline | |||||
| n | 49 | 50 | – | 18 | – |
| LS mean (SE) | 1.60 (0.78) | 4.72 (0.77) | –3.11 (1.10) | 1.62 (1.43) | –3.10 (1.63) |
| 95% CI | 0.05, 3.15 | 3.19, 6.25 | –5.30, –0.93 | –1.21, 4.45 | –6.31, 0.12 |
| P value | – | – | 0.0055 | – | 0.0592 |
CI, confidence interval; LS mean, least squares mean; NIS‐LL, Neuropathy Impairment Score – Lower Limbs.
Figure 1 shows the change from baseline across a range of baseline NIS‐LL scores based on predicted values from the MMRM analysis. The magnitude of change in both the Val30Met and non‐Val30Met tafamidis‐treated patients was remarkably similar across the range of observed baseline NIS‐LL values, and was consistently less than that observed in the Val30Met placebo group. The data also illustrate the impact of baseline NIS‐LL on projected level of neurological progression: as baseline NIS‐LL values increased, the predicted level of disease progression as measured by the change in NIS‐LL from baseline to month 12 also increased. This relationship was evident regardless of treatment or genotype.
Figure 1.
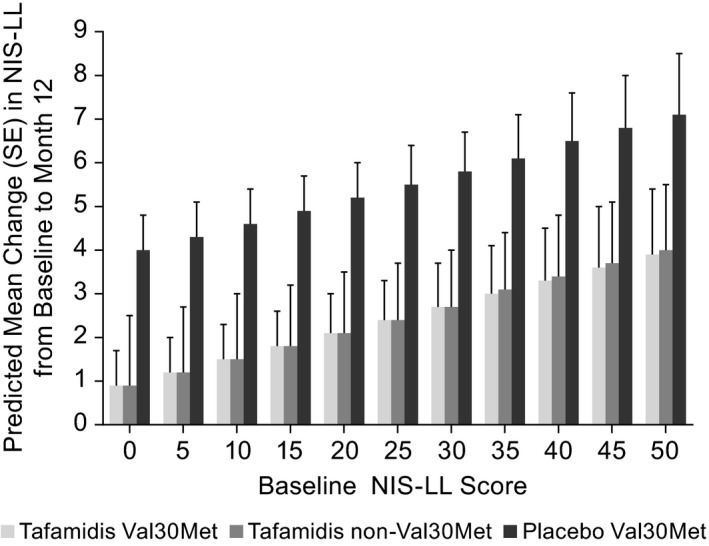
Predicted LS mean NIS‐LL change from baseline to month 12 across a range of baseline NIS‐LL values in the intent‐to‐treat placebo and tafamidis cohorts. NIS‐LL, Neuropathy Impairment Score – Lower Limbs.
Discussion
This use of combined data from two clinical trials, whilst controlling for baseline disease severity, demonstrated that tafamidis delayed neurological progression comparably in non‐Val30Met and Val30Met patients. The magnitude of neurological change from baseline to month 12 (based on predicted values from the MMRM analysis) in both Val30Met and non‐Val30Met tafamidis‐treated patients was remarkably similar across a range of baseline NIS‐LL values and, in all cases, less than the change from baseline observed in the Val30Met placebo group. These comparable trajectories suggest that patients with non‐Val30Met mutations may not progress faster than those with a Val30Met mutation. Differences in disease progression observed to date may be related to the fact that non‐Val30Met patients are more probably sporadic cases and from non‐endemic areas, where time to diagnosis is delayed due to a lack of recognition of the disease 16 and not related to an inherent worse severity or progression of disease associated with non‐Val30Met genotypes.
This analysis provides further support for the benefit of tafamidis treatment in delaying disease progression in patients with TTR‐FAP and has particular relevance for non‐Val30Met tafamidis‐treated patients where the original single‐arm clinical study revealed some worsening of neurological function that was difficult to interpret in the absence of a control group 14. The present post hoc analysis provided context by comparing the non‐Val30Met data with a placebo group of another study (whilst adjusting for baseline disease severity) and, although not statistically significant, it demonstrated a slower neurological progression in non‐Val30Met tafamidis‐treated patients compared with the Val30Met placebo group, suggesting a beneficial effect of tafamidis.
The predicted values from the MMRM analysis demonstrate the beneficial effect of tafamidis across a range of baseline NIS‐LL values, and also highlight the importance of baseline disease severity in predicting natural disease progression and treatment response. As baseline disease burden increased, the rate of neurological progression in the subsequent 12 months also increased. This relationship held true regardless of genotype or treatment and was also demonstrated in another study based on longer‐term data (the tafamidis registration study and the subsequent, open‐label, extension studies; L. Amass, H. Li, B.K. Gundapaneni, J.H. Schwartz, D.J. Keohane, submitted).
The similar trajectories of neurological progression across tafamidis‐treated Val30Met and non‐Val30Met patients when controlling for baseline disease burden suggest that these two genotype groups are more similar than previously thought 3, 4, 5, 17. Despite the considerable phenotypic heterogeneity observed across non‐Val30Met patients with TTR‐FAP, the similarity between genotype groups is noteworthy, especially considering that the two clinical trials were conducted at different sites and under different protocols. Identifying and understanding the importance of key demographic and/or clinical characteristics (e.g. baseline disease severity) may ultimately reveal more similarities than differences between genotypes and become important considerations when studying disease progression and the impact of treatment.
Limitations
These results are limited by their post hoc nature and the combining of non‐contemporaneous open‐label, non‐Val30Met study data with double‐blind, placebo‐controlled Val30Met data. They are also limited by the relatively small number of patients (comprising eight genotypes) in the non‐Val30Met group.
Conclusions
This post hoc analysis of combined clinical data, whilst controlling for baseline disease severity, demonstrated similar trajectories in the Val30Met and non‐Val30Met patients, suggesting that neurological progression in these two genotype groups may be more similar than previously considered. Further, the predicted values from the MMRM analysis illustrated the beneficial effects of tafamidis across a range of baseline NIS‐LL values, and also highlighted the importance of baseline disease severity when evaluating the natural history of disease progression and the impact of treatment. This analysis demonstrated that treatment with tafamidis delayed neurological progression comparably in non‐Val30Met and Val30Met patients.
Disclosure of conflicts of interest
MB Sultan, DJ Keohane and JH Schwartz are employees of Pfizer and hold stock and/or stock options. BK Gundapaneni, an employee of inVentiv Health, was a paid contractor to Pfizer in providing statistical support for this analysis and the development of this paper.
Supporting information
Box S1. List of independent ethics committees and institutional review boards.
Acknowledgements
Medical writing support was provided by Diane Hoffman, PhD, of Engage Scientific Solutions and was funded by Pfizer. This analysis was sponsored by Pfizer.
Trial registration: ClinicalTrials.gov NCT00409175, NCT00630864.
References
- 1. Plante‐Bordeneuve V. Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol 2014; 261: 1227–1233. [DOI] [PubMed] [Google Scholar]
- 2. Coelho T, Inês M, Concieção I, Saramago P, de Carvalho M, Costa J. Temporal trends in transthyretin familial amyloid polyneuropathy survival over a century. Int J Clin Neurosci Ment Health 2016; 3: P130. [Google Scholar]
- 3. Mariani LL, Lozeron P, Theaudin M, et al Genotype−phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol 2015; 78: 901–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coelho T, Maurer MS, Suhr OB. THAOS − the Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild‐type transthyretin amyloidosis. Curr Med Res Opin 2013; 29: 63–76. [DOI] [PubMed] [Google Scholar]
- 5. Sekijima Y, Yoshida K, Tokuda T, Ikeda S. of website. http://www.ncbi.nlm.nih.gov/books/NBK1194/ (accessed 13 March 2017) [Google Scholar]
- 6. Coelho T, Waddington Cruz M, Fallet S, Carlsson M, Ong M‐L. The natural history of transthyretin familial amyloidosis polyneuropathy: an analysis from the Transthyretin Amyloidosis Outcomes Survey. Presented at the XV International Symposium on Amyloidosis; Uppsala: Sweden, 3–7 July 2016.
- 7. Coelho T, Merlini G, Bulawa CE, et al Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther 2016; 5: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coelho T, Maia LF, Martins da Silva A, et al Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79: 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coelho T, Maia LF, Martins da Silva A, et al Long‐term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol 2013; 260: 2802–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barroso FA, Judge DP, Ebede B, et al Long‐term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid 2017; 24: 194–204. [DOI] [PubMed] [Google Scholar]
- 11. Ando Y, Sekijima Y, Obayashi K, et al Effects of tafamidis treatment on transthyretin (TTR) stabilization, efficacy, and safety in Japanese patients with familial amyloid polyneuropathy (TTR‐FAP) with Val30Met and non‐Val30Met: a phase III, open‐label study. J Neurol Sci 2016; 362: 266–271. [DOI] [PubMed] [Google Scholar]
- 12. Waddington Cruz M, Amass L, Keohane D, Schwartz J, Li H, Gundapaneni B. Early intervention with tafamidis provides long term (5.5 year) delay of neurologic progression in transthyretin familial amyloid polyneuropathy. Amyloid 2016; 23: 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keohane D, Schwartz J, Gundapaneni B, Stewart M, Amass L. Tafamidis delays disease progression in patients with early stage transthyretin familial amyloid polyneuropathy: additional supportive analyses from the pivotal trial. Amyloid 2017; 24: 30–36. [DOI] [PubMed] [Google Scholar]
- 14. Merlini G, Plante‐Bordeneuve V, Judge DP, et al Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non‐Val30Met transthyretin amyloidosis. Cardiovasc Transl Res 2013; 6: 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coelho T, Vinik A, Vinik EJ, Tripp T, Packman J, Grogan DR. Clinical measures in transthyretin familial amyloid polyneuropathy. Muscle Nerve 2017; 55: 323–332. [DOI] [PubMed] [Google Scholar]
- 16. Parman Y, Adams D, Obici L, et al Sixty years of transthyretin familial amyloid polyneuropathy (TTR‐FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR‐FAP. Curr Opin Neurol 2016; 29(Suppl. 1): S3–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suhr OB, Larsson M, Ericzon BG, Wilczek HE. Survival after transplantation in patients with mutations other than Val30Met: extracts from the FAP World Transplant Registry. Transplantation 2016; 100: 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Box S1. List of independent ethics committees and institutional review boards.