

Abstract
Background
The interleukin‐33 (IL‐33)/suppressor of tumorigenicity 2 (ST2) pathway is suggested to play an important role in fibrosis, remodelling and the progression of heart failure (HF). Increased soluble (sST2) levels are associated with adverse outcome in the average HF population. Less is known about sST2 levels in end‐stage HF. Therefore, we studied sST2 levels in end‐stage HF and the effect of unloading by left ventricular assist device (LVAD) support on sST2 levels.
Method and results
Serial plasma measurements of sST2 were performed pre‐implantation and 1, 3 and 6 months after (LVAD) implantation in 38 patients. sST2 levels were elevated in end‐stage HF just prior to LVAD implantation (74.2 ng/mL [IQR 54.7‐116.9]; normal <35 ng/mL) and decreased substantially during LVAD support, to 29.5 ng/mL [IQR 24.7‐46.6](P < .001). Patients with INTERMACS profile I had significantly higher sST2 levels compared to patients in profile II and profile III. A moderate correlation was found between sST2 and C‐reactive protein (r = .580, P < .010).
Conclusion
Levels of sST2 are elevated in end‐stage HF patients with variability that suggests multiple inputs to a pro‐inflammatory and pro‐fibrotic pathway. Cardiogenic shock and increased C‐reactive protein levels are associated with higher sST2 levels. LVAD support results in a significant drop in sST2 levels with normalization within 3 months postimplantation. This suggests that LVAD support leads to lessening of fibrosis and inflammation, which might eventually be used to target medical policy: explantation of the LVAD versus permanent use or cardiac transplantation.
Keywords: biomarker, heart failure, left ventricular assist device, mechanical circulatory support, soluble ST2
1. INTRODUCTION
The interleukin‐33 (IL‐33)/suppressor of tumorigenicity 2 (ST2) pathway is part of the IL‐1 family and was initially associated with inflammation and immunity. Recently, the IL‐33/ST2 pathway has also been suggested to play an important role in the progression of heart failure (HF).1 It is thought to be a cardioprotective signalling pathway that is activated by cell damage and mechanical stress, through the release of IL‐33 from cardiac cells (predominantly stromal). Activation of IL‐33 prevents myocardial hypertrophy and fibrosis through interaction of IL‐33 and transmembrane bound ST2L.2 The soluble form of ST2 (sST2) acts as a “decoy” receptor that results in attenuation of the beneficial (myocardial and vascular) effects of IL‐33. Increased sST2 levels are associated with an activation of a pro‐fibrotic pathway leading to adverse outcomes in the average HF population.1, 3 Soluble ST2 has an additive value to natriuretic peptides and superior prognostic performance compared with all other biomarkers (high sensitive troponin T, growth differentiation factor‐15, B‐type natriuretic peptide (BNP) and N‐terminal‐proBNP.4 Serial monitoring of sST2 in particular may benefit risk prediction and therapy guidance.5, 6 However, most studies are performed in NYHA class II‐III heart failure patients and little is known about sST2 in end‐stage HF patients (NYHA IV). Left ventricular assist device (LVAD) patients offer an opportunity to investigate sST2 concentrations both during severe end‐stage HF as well as during LV unloading. Although sST2 is suggested to be a marker of cardiac fibrosis and remodelling, the association with clinical factors in end‐stage heart failure patients has not yet been elucidated. Therefore, the aim of this study was to assess sST2 levels in end‐stage heart failure and assess the effect of unloading of the left ventricle by mechanical circulatory support on sST2 levels. Furthermore, we evaluated prespecified potential determinants of sST2 concentrations in end‐stage heart failure patients.
2. METHOD
2.1. Study population
In this retrospective study, 38 consecutive patients were included who received a continuous flow LVAD as bridge to heart transplantation with a support duration of more than 180 days. Exclusion criteria were severe kidney dysfunction, peripheral vasculopathy and also elderly patients. Inclusion was only carried out if plasma samples of all predefined time points were available. This study was approved by an ethics committee, and all patients agreed to collection and banking of blood samples for research purposes at the time of LVAD implant (number 12/387). Reporting of the study conforms to STROBE statement.7
2.2. sST2 measurements
Blood samples were collected within 24 hours prior to LVAD implantation and 1, 3 and 6 months after implantation through a peripheral vein into EDTA‐containing tubes. Blood was centrifuged within 6 hours after withdrawal, and plasma was collected in aliquots and stored at −80°C. Levels of sST2 were analysed by double measurements in one batch, using the Presage ST2 ELISA assay according to manufacturer's instructions (Critical diagnostics, San Diego, CA, USA). The assay has an intra‐ and interassay coefficient of variation of 5.1% and 5.2%, respectively, and a total coefficient of variation ranging from 4.2 to 12.0%.
2.3. Clinical parameters
Data regarding HF aetiology, INTERMACS profile, blood pressure (BP), HF duration and medication were gathered from the medical file. Diagnostic blood measurements were used for laboratory assessments of BNP (ng/L), C‐reactive protein (CRP) (mg/L) and kidney function (eGFR, mL min−1 m−2; estimated by MDRD calculation). Echocardiography was used to evaluate right ventricular function (RVF) and left ventricular end‐diastolic dimension (LVEDD). RVF was classified as poor, moderate, reasonable or good. Categories were paired for analysis: poor and moderate RVF as “<reasonable” and reasonable and good function as “≥reasonable”. LVEDD was measured in millimetres. Cardiac output (CO, L/min), right atrial pressure (RAp, mm Hg) and pulmonary capillary wedge pressure (PCWP, mm Hg) were measured with a Swan‐Ganz catheter shortly before LVAD implantation.
2.4. Statistical analysis
Continuous data are expressed as medians (interquartile range; IQR, with 25th‐75th percentile) and categorical variables as number (%). The overall effect of LVAD implantation on sST2 levels after 6 months was analysed with the Friedman test. Comparison at different time points was performed with Wilcoxon‐signed‐rank test, corrected for multiple testing with the Bonferroni method. The distribution of sST2 levels was analysed for several clinical factors using Mann‐Whitney U tests. Spearman's correlation was used to calculate correlations. A P‐value <.05 was considered statistically significant. All analyses were performed with SPSS 21.0 software (SPSS, Inc., Chicago, IL, USA).
3. RESULTS
3.1. Patient characteristics
Baseline patient characteristics are presented in Table 1. The mean age was 50 years (range 17‐68). Twenty‐four patients (63%) were male. The aetiology of HF was ischaemic in 10 patients (26%) and nonischaemic in 28 patients (74%), with a majority of idiopathic dilated cardiomyopathy. All patients had New York Heart Association (NYHA) class IV. Ten patients (26%) were in INTERMACS profile I (“crash and burn”) prior to LVAD implantation, while the majority was in INTERMACS profile II or III (dependent on inotropes with progressive decline resp. clinical stability). Most patients received a HeartMate II (HMII; Abbott, Illinois, United States) device (92%), AND three patients (8%) received a HVAD (HeartWare; Medtronic, Minneapolis, United States).
Table 1.
Baseline characteristics
| Baseline characteristics | n = 38 |
|---|---|
| Age (years) | 50 (range 17‐68) |
| Male/female | 24 (63%)/14 (37%) |
| NYHA classification IV | 38 (100%) |
| Heart failure duration (weeks) | 220 (range 1‐936) |
| CRT/ICD | 23 (61%) |
| Ischaemic/nonischaemic aetiology | 10 (26%)/28 (74%) |
| Idiopathic dilated | 13 (34%) |
| Hypertrophic | 3 (8%) |
| Other | 12 (32%) |
| Peripartum, myocarditis, tachy | 3 (8%) |
| PLN mutation, familial, noncompaction | 6 (16%) |
| Limb girdle, haemachromatosis, toxic | 3 (8%) |
| Inotropic medication | 30 (79%) |
| INTERMACS profile I/II‐III | 10 (26%)/28 (74%) |
| LVAD | 38 (100%) |
| HeartMate II | 35 (92%) |
| HeartWare | 3 (8%) |
NYHA, New York Heart Association; CRT, cardiac resynchronization therapy; ICD, implantable cardioverter defibrillator; PLN, phospholamban; INTERMACS, Interagency Registry for Mechanically Assisted Circulatory Support; LVAD, left ventricular assist device.
3.2. sST2 levels before and after LVAD implantation
Prior to LVAD implantation, sST2 was severely elevated (74.2 ng/mL [IQR 54.7‐116.9 ng/mL], normal <35 ng/mL1) in the majority of patients (95%). Levels significantly decreased during LVAD support (P < .001). In most patients, sST2 levels normalized after 6 months (29.5 ng/mL [IQR 24.7‐46.6 ng/mL]). A maximum drop in sST2 levels was seen after 3 months, without a significant decrease thereafter (Figure 1). The great variation of sST2 levels between individuals before LVAD implantation is striking and becomes less prominent during LVAD support.
Figure 1.
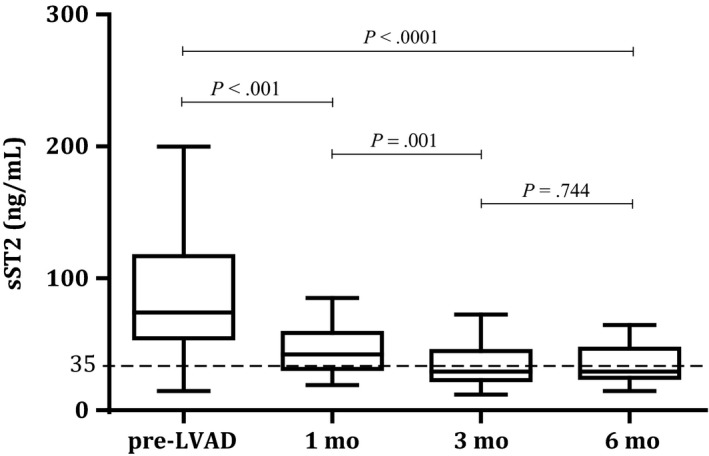
Median sST2 levels before and during LVAD support with IQR (25%‐75%). The dotted line represents the cut‐off value for normal sST2 levels (<35 ng/mL)
3.3. sST2 and clinical factors
To explore the variation of sST2 prior to LVAD implantation, various clinical factors were evaluated in relation to sST2 levels (Table 2). The distribution of sST2 with regard to patient profile at implant (INTERMACS) showed a significant difference. Patients with INTERMACS profile I had significantly higher sST2 levels compared to patients in profile II and profile III (P = .030; Figure 2A).
Table 2.
The relation of sST2 levels was analysed for several clinical factors using Mann‐Whitney U testsa [gender, heart failure aetiology (ischaemic vs nonischaemic), RV function (≥reasonable vs <reasonable), kidney function (normal vs abnormal), inotropic medication (yes vs no) and mineralocorticoid receptor antagonists (yes vs no)] and nonparametric (Spearman) correlationsb
| Clinical parameter | sST2 level (ng/mL) | P‐value n = 38 | |
|---|---|---|---|
| Gender | (male/female) | 77.1/65.8 | .78a |
| HF aetiology | (Ischaemic/Nonischaemic) | 74.2/73.5 | .91a |
| INTERMACS | (Profile I/Profile II‐III) | 120.2/65.7 | .03a |
| RV function | (≥Reasonable/<Reasonable) | 104.3/74.8 | .23a |
| Kidney function | (eGFR≥60/eGFR<60) | 64.9/74.8 | .19a |
| Inotropic medication | (Yes/No) | 87.7/60.8 | .16a |
| MRA | (Yes/No) | 74.8/64.9 | .74a |
| Correlation (r) | P‐value n = 38 | ||
|---|---|---|---|
| BNP | pmol/L | .19 | .29b |
| CRP | mg/L | .58 | <.01b |
| PCWP | mm Hg | .25 | .19b |
| RAp | mm Hg | .22 | .28b |
| LVEDD | mm | .07 | .72b |
| Pulse | beats/min | .02 | .90b |
| BP syst | mm Hg | .09 | .62b |
| BP diast | mm Hg | .18 | .32b |
| CO | (L/min) | −.07 | .74b |
| HF duration | wk | .14 | .41b |
HF, heart failure; INTERMACS, Interagency Registry for Mechanically Assisted Circulatory Support; RV, right ventricular; MRA, mineralocorticoid receptor antagonist; BNP, brain natriuretic peptide; CRP, C‐reactive protein; PCWP, pulmonary capillary wedge pressure; RAp, right atrial pressure; LVEDD, left ventricular end‐diastolic dimension; BP, blood pressure; CO, cardiac output.
Figure 2.
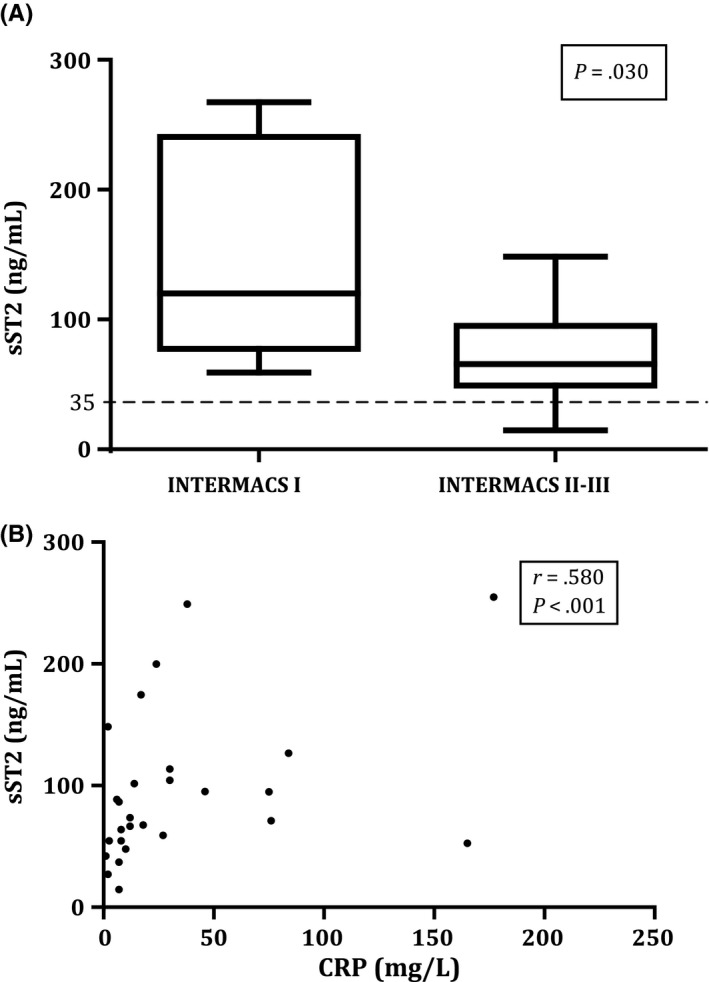
sST2 levels in relation to INTERMACS profile (A) and in correlation with C‐reactive protein (CRP) (B) The dotted line represents the cut‐off value for normal sST2 levels (<35 ng/mL)
C‐reactive protein and sST2 levels showed a moderate positive and significant correlation (r = .580, P < .010; Figure 2B), suggesting higher CRP blood levels in patients with higher sST2 values. No differences in sST2 levels were seen with regard to gender, heart failure aetiology (ischaemic vs nonischaemic), RV function (≥reasonable vs <reasonable), kidney function (normal vs abnormal), inotropic medication (yes vs no) and mineralocorticoid receptor antagonists (yes vs no) (Table 2).
No significant correlations were found between sST2 levels in end‐stage HF and other tested clinical parameters, that is heart failure duration, BNP levels and hemodynamic parameters (Table 2).
4. DISCUSSION
This study presents the first reported analysis of sST2 levels in patients with end‐stage heart failure before and after LVAD implantation. We demonstrated severely elevated sST2 levels in patients with end‐stage heart failure just before LVAD implantation, which decreased to near‐normal levels during LVAD support. Furthermore, an association between clinical patient profile before LVAD support and sST2 levels was found.
In extension to reported sST2 levels in the NYHA class II and III HF population,1 we found increased sST2 levels in almost all patients prior to LVAD implantation with a wide distribution of sST2 levels. Despite the diversity of the study population with regard to HF aetiology and duration, we found significantly higher sST2 levels in patients with INTERMACS profile I vs II‐III before LVAD support. This suggests that cardiogenic shock results in higher sST2 levels. As we could not correlate this in our study to hemodynamic data, the inflammatory state coinciding with cardiogenic shock might be responsible for this rise. Moreover, inflammation as an important determinant of sST2 levels corresponds with previous reports on the role of IL‐33/ST2 signalling8, 9 and with the moderate positive correlation between sST2 and CRP levels found in this study. The wide variation of sST2 before LVAD implant could not be explained by other clinical factors that have previously been described to affect sST2 levels, like gender, RV function and hemodynamic parameters.10, 11 It is not surprising that we could not find a “typical” patient‐specific characteristic for the elevated sST2 levels, because sST2 is not merely a reflection of ventricular wall stress, but also of inflammation, active fibrosis and even subendocardial ischaemia.12, 13 The effect of other related clinical factors on sST2 levels may have been diminished by the impact of cardiogenic shock.
After 6 months of LVAD support sST2 levels were significantly decreased to normal levels in most of the patients (and the distribution narrowed). The biggest drop was seen after 1 month and reached a maximum at 3 months. The decrease in sST2 levels after unloading of the heart suggests that the inflammatory and pro‐fibrotic state has dissipated, and in fact reverse remodelling may be occurring. The significant decrease in elevated sST2 levels after unloading of the left ventricle confirms the value of serial ST2 measurements as described previously14 and indicates a potential role for ST2 as biomarker to monitor therapy, not only in NYHA class II‐III patients, but also in patients with end‐stage heart failure. However, clinical benefits of sST2 measurements in patients with end‐stage heart failure before and after LVAD implantation remain unclear.
By identifying those factors that drive ST2, we may be able to use ST2 as a biomarker to stratify patients and assist in therapy guidance, perhaps even as a response predictor that will help clinicians in decision‐making. As LVADs become small and more portable, sST2 levels may be used to help profile those patients in whom LVAD discontinuation could be discussed.
5. LIMITATIONS
While there were relatively small numbers of patients in this study, the LVAD effect on sST2 levels was clear. Given the correlation with baseline CRP, sST2 after LVAD implantation may have been influenced by infectious postoperative complications that occurred in 7 patients. Another drawback is the variable timing of hemodynamic measurements prior to LVAD implantation. Unlike eGFR and CRP, measured maximum 48 hours before LVAD, the calculated correlation may not completely represent the hemodynamics on the day of LVAD implantation. BNP levels were also determined at different time frames, within 24 hours to 1 month before LVAD implantation.
The strength of our analysis, however, lies in the consecutive measurements of sST2 in the same patient, before and after LVAD implantation, eliminating in this way variable results due to variations in patient population.
6. CONCLUSION
We extend the reported elevated sST2 levels in class II‐III heart failure patients to patients with end‐stage heart failure, qualifying for mechanical circulatory support as a bridge to heart transplantation. High sST2 levels predict a worse outcome in patients with conventional treatment. The wide variation in sST2 levels (almost all above the cutpoint of 35 ng/ml) in end‐stage heart failure patients delineates the many pathways that feed into the shedding of the decoy receptor sST2. Cardiogenic shock and inflammation (INTERMACS I) are associated with higher levels of sST2. Unloading of the left ventricle by an LVAD results in a rapid decrease of sST2 levels, with complete normalization within 3 months. These findings potentially warrant further research on the role of sST2 as a prognostic and therapeutic marker, not only in moderate heart failure, but also in end‐stage heart failure.
DISCLOSURES
Alan Maisel is consultant for Critical Diagnostics.
Tseng CCS, Huibers MMH, Gaykema LH, et al. Soluble ST2 in end‐stage heart failure, before and after support with a left ventricular assist device. Eur J Clin Invest. 2018;48:e12886 https://doi.org/10.1111/eci.12886
REFERENCES
- 1. Bayes‐Genis A, Zhang Y, Ky B. ST2 and patient prognosis in chronic heart failure. Am J Cardiol. 2015;115(7 Suppl):64B‐69B. [DOI] [PubMed] [Google Scholar]
- 2. Dieplinger B, Mueller T. Soluble ST2 in heart failure. Clin Chim Acta. 2015;443:57‐70. [DOI] [PubMed] [Google Scholar]
- 3. Broch K, Ueland T, Nymo SH, et al. Soluble ST2 is associated with adverse outcome in patients with heart failure of ischaemic aetiology. Eur J Heart Fail. 2012;14:268‐277. [DOI] [PubMed] [Google Scholar]
- 4. De Boer RA, Daniels LB, Maisel AS, Januzzi JL Jr. State of the Art : newer biomarkers in heart failure. Eur J Heart Fail. 2015;17:559‐569. [DOI] [PubMed] [Google Scholar]
- 5. Boisot S, Beede J, Isakson S, et al. Serial sampling of ST2 predicts 90‐day mortality following destabilized heart failure. J Card Fail. 2008;14:732‐738. [DOI] [PubMed] [Google Scholar]
- 6. Breidthardt T, Balmelli C, Twerenbold R, et al. Heart failure therapy‐induced early ST2 changes may offer long‐term therapy guidance. J Card Fail. 2013;19:821‐828. [DOI] [PubMed] [Google Scholar]
- 7. Simera I, Moher D, Hoey J, Schulz KF, Altman DG. A catalogue of reporting guidelines for health research. Eur J Clin Invest. 2010;40:35‐53. [DOI] [PubMed] [Google Scholar]
- 8. Chen W‐Y, Hong J, Gannon J, Kakkar R, Lee RT. Myocardial pressure overload induces systemic inflammation through endothelial cell IL‐33. Proc Natl Acad Sci USA. 2015;112:7249‐7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martinez‐Martinez E, Cachofeiro V, Rousseau E, et al. Interleukin‐33/ST2 system attenuates aldosterone‐induced adipogenesis and inflammation. Mol Cell Endocrinol. 2015;411:20‐27. [DOI] [PubMed] [Google Scholar]
- 10. Broch K, Andreassen AK, Ueland T, et al. Soluble ST2 reflects hemodynamic stress in non‐ischemic heart failure. Int J Cardiol. 2015;179:378‐384. [DOI] [PubMed] [Google Scholar]
- 11. Daniels LB, Clopton P, Iqbal N, Tran K, Maisel AS. Association of ST2 levels with cardiac structure and function and mortality in outpatients. Am Heart J. 2010;160:721‐728. [DOI] [PubMed] [Google Scholar]
- 12. Kohli P, Bonaca MP, Kakkar R, et al. Role of ST2 in non–ST‐elevation acute coronary syndrome in the MERLIN‐TIMI 36 trial. Clin Chim Acta. 2012;58:257‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pascual‐Figal DA, Januzzi JL. The biology of ST2: the international ST2 consensus panel. Am J Cardiol. 2015;115:3B‐7B. [DOI] [PubMed] [Google Scholar]
- 14. Wu AHB, Wians F, Jaffe A. Biological variation of galectin‐3 and soluble ST2 for chronic heart failure: implication on interpretation of test results. Am Heart J. 2013;165:995‐999. [DOI] [PubMed] [Google Scholar]