

Abstract
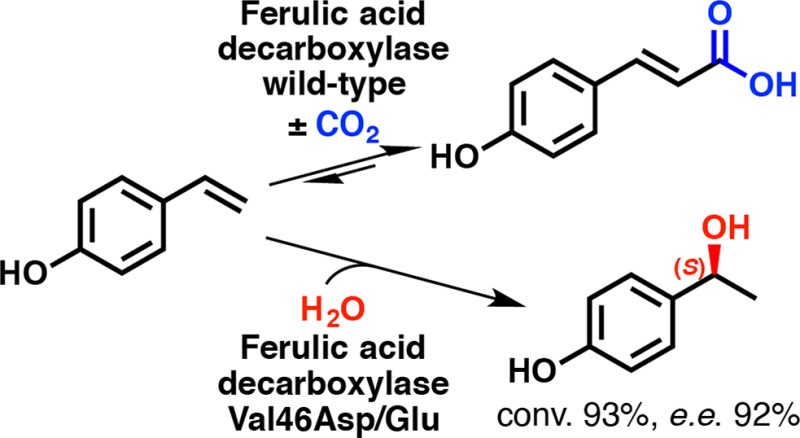
The promiscuous regio- and stereoselective hydration of 4-hydroxystyrenes catalyzed by ferulic acid decarboxylase from Enterobacter sp. (FDC_Es) depends on bicarbonate bound in the active site, which serves as a proton relay activating a water molecule for nucleophilic attack on a quinone methide electrophile. This “cofactor” is crucial for achieving improved conversions and high stereoselectivities for (S)-configured benzylic alcohol products. Similar effects were observed with simple aliphatic carboxylic acids as additives. A rational redesign of the active site by replacing the bicarbonate or acetate “cofactor” with a newly introduced side-chain carboxylate from an adjacent amino acid yielded mutants that efficiently acted as C=C hydratases. A single-point mutation of valine 46 to glutamate or aspartate improved the hydration activity by 40% and boosted the stereoselectivity 39-fold in the absence of bicarbonate or acetate.
Keywords: biocatalysis, hydration, enzyme engineering, decarboxylase, hydratase
The ability of an enzyme to catalyze a reaction other than its annotated “natural” activity is known as catalytic promiscuity1−3 and is the result of evolutionary processes upon the encounter of “non-natural” substrates and the organism’s striving for survival.4−6 This phenomenon is an important criterion when selecting enzymes for the development of biocatalysts for the selective transformation of synthetic compounds, using rationally guided or randomly based directed evolution protocols.7 Coumaric acids and their derivatives constitute monomeric units of lignin in plant cell walls8 and are a major waste product of palm oil manufacturing.9,10 In nature, their degradation is achieved by ferulic and phenolic acid decarboxylases (FDCs and PADs, respectively). These enzymes cleave their substrates into CO2 and their respective 4-hydroxystyrenes [1 (Scheme 1a)], which constitute undesired off-flavor components in beer and wine11 but have also found application as renewable building blocks for polymers with interesting dielectric properties.12−14 By supplying an excess of CO2 in the form of bicarbonate, researchers can perform the process as the reverse β-carboxylation for the production of substituted coumaric acid derivatives (Scheme 1a).15−17 During studies of regioselective carboxylation, the promiscuous hydration of 4-hydroxystyrene derivatives by FDCs and PADs in the presence of bicarbonate was discovered (Scheme 1b), and the best results were obtained with a ferulic acid decarboxylase from Enterobacter sp. (FDC_Es).18 The nucleophile scope apart from water was extended to methoxyamine, cyanide, and propanethiol, which are added via an analogous mechanism to furnish (S)-configured benzylic amines, nitriles, and thioethers, however without the need for bicarbonate (Scheme 1b).19
Scheme 1. (a) Reversible (De)carboxylation of Coumaric Acids and (b) Promiscuous Nucleophile Addition to 4-Hydroxystyrenes Catalyzed by Phenolic and Ferulic Acid Decarboxylases.

The biocatalytic hydration of 4-hydroxystyrenes allows easy access to the (S)-1-(4-hydroxyphenyl)ethanol structural motif (2) from renewable resources in a stereoselective fashion without the need for protecting groups. The product hydrate is a substructure of bioactive molecules exerting herbicidal,20 insecticidal,21,22 antiproliferative,23 and (hepatitis C) protease inhibition activities.24,25 Though little is known about the active enantiomeric forms of these congeners, an improvement in biocatalytic styrene hydration would be of practical interest to facilitate the exploitation of the benzylic alcohols as chiral building blocks.
Experimental data18 and quantum mechanical calculations26 suggest that the promiscuous hydration of 4-hydroxystyrene requires a bicarbonate anion as a “cofactor”, which is bound to Arg49 in the active site of the decarboxylase to achieve good conversion and a high enantiomeric excess (ee) of the benzylic alcohol product, (S)-2 (Scheme 2). Potassium bicarbonate (0.5 M, pH 8.5) promotes the hydration of 4-vinylphenol (1) with ∼65% conversion and ∼80% ee using FDC_Es as a biocatalyst.18 In the absence of bicarbonate, however, a water molecule occupies its position, resulting in (almost complete) loss of stereoselectivity.26 Consequently, supplementation of bicarbonate (>0.5 M) is required to maintain stereoselectivity, which renders this process economically and operationally less attractive.
Scheme 2. Mechanism of the FDC_Es-Catalyzed Hydration of 4-Hydroxystyrene with Bicarbonate Acting as a “Cofactor” for Proton Transfer.

In initial studies, the compensability of bicarbonate as a “cofactor” was investigated by replacing it with different (anionic) additives (Figure 1). Potassium phosphate buffer (50 mM) was used as a reaction buffer, and the pH was adjusted to 8.0 after dissolving the additive salts to avoid bicarbonate decomposition (pKaH = 7.7) and to allow for direct comparison of additive effects. Control reactions in neat phosphate buffer gave moderate conversion (∼60%) but completely lacked stereoselectivity. Addition of Cl–, HPO42–, or SO42– improved the conversion slighty, while imidazole had adverse effects, again without stereoinduction. Borate gave a low ee (15%), while the conversion and ee were significantly improved by bicarbonate. Remarkably, the best values for conversion and ee were obtained in the presence of acetate. Next, the concentration of bicarbonate and acetate was varied within a range of 10–500 mM (Figure S3). While the conversion profiles for both ions showed a similar behavior at increasing ion concentrations (leveling off at ∼90–95% conversion and 250 and 300 mM), the product ee profile with bicarbonate differs from that with acetate in a shallow maximum of 33% ee (at ∼100 mM), while the ee rises in a linear fashion with an increased acetate concentration, reaching an ee of 45% at 500 mM. Because the carboxylate moiety appears to be an ideal promoter for stereoselective hydration, a set of carboxylic acid sodium salts with varying chain lengths, substituents, and branching patterns was compared at neutral pH (7.0) (Figure 1b). Significant positive effects on the stereoselectivity were detected with all aliphatic carboxylates, depending on the side-chain size (Et > iPr > tBu ≈ Me ≈ H) with a maximum for propanoate (74% ee). Trifluoroacetate enhanced neither conversion nor the ee, because of its low pKa, which disables it to act as a catalytic acid in proton transfer (cf. Scheme 2). Because of the formation of an unidentified side product, only 60% of the starting material was recovered in the presence of formate.
Figure 1.
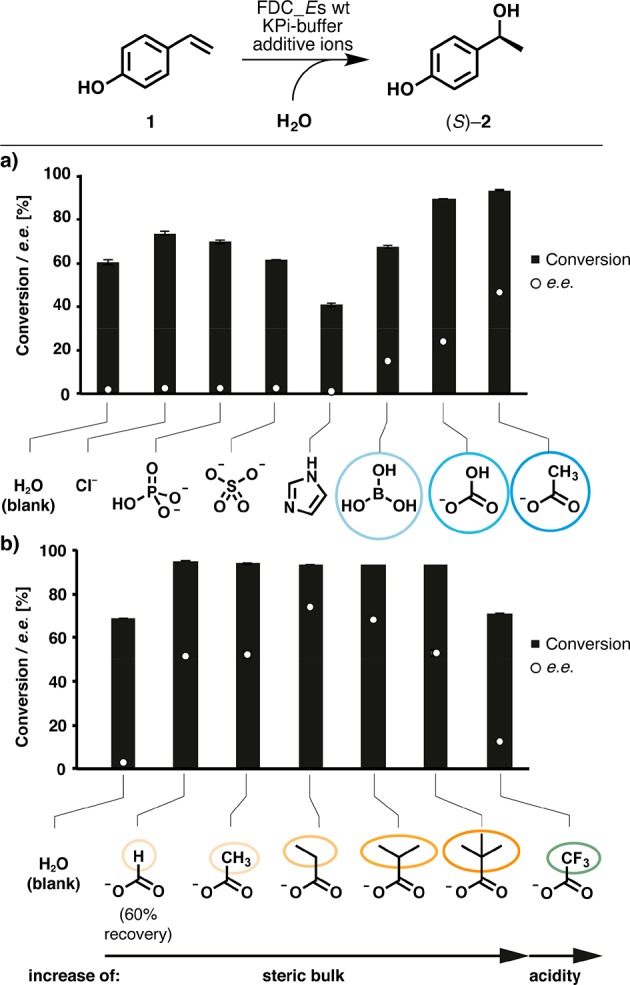
Asymmetric hydration in the presence of (anionic) additives. Conditions: substrate 1 [10 mM, from a 10% (w/w) propylene glycol solution] in potassium phosphate buffer (50 mM), and lyophilized Escherichia coli cells containing wild-type FDC_Es (20 mg/mL, 32 units). Effect of (a) various anions (0.5 M, pH 8.0) and (b) different carboxylates (0.5 M, pH 7.0) on enzymatic hydration (additives are shown in the respective protonation state at the corresponding pH).
On the basis of these promising results, we envisioned the design of mutants bearing a “tethered carboxylate” acting as a catalytic residue in the active site to mediate the proton transfer,27 thereby waiving the need for an external bicarbonate/carboxylate “cofactor”. The amino acid residues introduced should meet two crucial requirements. (i) The spatial arrangement in the active site needs to mimic bound bicarbonate, suggesting residues near the bicarbonate binding site as targets for mutagenesis, and (ii) residues must be chemically competent surrogates of bicarbonate/carboxylate to shuttle protons between the substrate Cβ atom and Glu72.
After detailed inspection of active-site residues of FDC_Es, Val46 located on a mobile loop in the vicinity to the bicarbonate binding site was selected for mutations. On the basis of their ability to act as a three-atom proton shuttle, Val46 was exchanged with Glu, Gln, Asp, His, and Arg (Figure 2). The validity of this strategy was proven by the fact that the introduction of a carboxylate group at position 46 with aspartate (Val46Asp) or glutamate (Val46Glu) enhanced the stereoselectivity 39-fold and boosted the conversion by ∼40%. The slightly higher stereoselectivity (∼6%) for the Asp versus the Glu mutant may be explained by the tether that is one CH2 unit shorter and pulls the water nucleophile farther out of the plane of symmetry toward the si face of the substrate (Figure 3). Apparently, only a carboxylate moiety is an efficient bicarbonate/acetate mimic, because mutations to histidine, glutamine, and arginine were not beneficial (maximum 57% conversion for Val46Arg). However, despite its lower activity, the Val46His mutant showed slight stereoselectivity (up to 17% ee).
Figure 2.

FDC_Es variants with tethered bicarbonate/acetate surrogates in positions Val46 and Arg49 (indicated by dashed circles). The quinone methide form of substrate 4-vinylphenol (green) was docked into the active site of FDC_Es (Protein Data Bank entry 4UU3) using the UCSF Chimera AutoDock Vina plug-in. Catalytic key residues are colored blue; Val46 is colored red, and others are colored gray. Dotted red spheres indicate water molecules in the superimposed apo structure. Surrogate residues at positions 46 and 49 are shown in their protonation state at pH 7.0. Conditions: substrate 1 (10 mM) in potassium phosphate buffer (50 mM, pH 7.0), and lyophilized E. coli whole cells containing FDC_Es variants (20 mg/mL).
Figure 3.
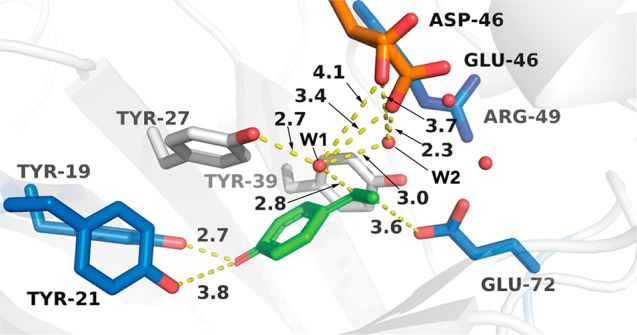
Overlay of the active-site structures of the FDC_Es Val46Glu and Val46Asp variants (Swissmodel Web server). Mutated residues Asp/Glu46 are colored orange, and the docked quinone methide form of the substrate 4-vinylphenol is colored green (Autodock Vina plug-in, UCSF Chimera). Water molecules shown as red spheres are derived from the superimposed wild-type FDC_Es crystal structure (Protein Data Bank entry 4UU3). Distances are given in angstroms.
A homology model of the Val46Asp/Glu mutants reveals that the additional carboxylate groups are well positioned in the proximity of a water molecule (W1) to activate it as nucleophile within the hydrogen bond network of Tyr27, Glu72, and Arg49 (Figure 3). Changing the latter to a glutamate residue (thereby inverting the positive charge at this position into a negative one) could yield some additional hydration activity (86% conversion) but yielded a racemic product. Tyr39 was suggested to dictate the (S)-stereoselectivity through steric repulsion between its aromatic ring and the Cβ atom of the quinone methide intermediate, which consequently presents its si face to the water nucleophile located above the plane (Scheme 2).26 A double variant having both the beneficial Val46Glu mutation and a neutral Arg49Met mutation did not affect the conversion but showed a decrease in stereoselectivity compared to that of the single mutant. Removal of the hydrogen bond donor Arg49 weakens the bonding network and renders Tyr39 less rigid, which is responsible for the lower ee of the product. The Val46Glu mutant was also tested with other nucleophiles (methoxyamine and cyanide),19 which gave improved stereoselectivities compared to that of the wild-type enzyme (Table S3).
The reaction conditions for the rationally designed “hydratases” were optimized in terms of pH, and effects of various organic co-solvents were examined (Figure S5). Furthermore, the stereoselectivity of the process was found to increase with a decrease in temperature (Figure S4), indicating a major contribution of the enthalpy difference (ΔΔH⧧) to the free energy difference between the enantiomeric transition states (ΔΔG⧧).28,29
Finally, hydration of 1 was achieved with the Val46Glu and Val46Asp variants with a maximum turnover number of ∼220 and a product ee of ≤91% with 50 mM substrate loading at pH 6.0 and 25 °C (Tables S1 and S2). The process was subsequently performed on a 100 mg scale with 0.8 mol % of FDC_Es V46E to afford 68 mg of (S)-hydrate product (60% yield) with a high enantiomer purity (96% ee after recrystallization) underpinning the usability of this reaction on a preparative scale.
In conclusion, prompted by the observation of bicarbonate- and acetate-assisted asymmetric hydration of hydroxystyrenes catalyzed by ferulic acid decarboxylase, we rationally designed a hydratase from this decarboxylase through mutation of an Asp- or Glu-carboxylate moiety into the active site, which efficiently functions as a proton shuttle. The mutants showed 40% higher activity and 39-fold improved stereoselectivity, which allowed preparative-scale transformations with turnover numbers of ≤220.
Acknowledgments
The authors thank Georg Steinkellner for his advice with respect to structural biology and Fahmi Himo for fruitful discussions about the reaction mechanism and thermodynamics. Funding by the Austrian Science Fund (FWF Project P26863) and the Austrian BMWFW, BMVIT, SFG, Standortagentur Tirol, Government of Lower Austria, and ZIT through the Austrian FFG-COMET-Funding Program is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b04293.
Experimental details, preparation of variants, docking and details of structural biology, and supporting screening results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bornscheuer U. T.; Kazlauskas R. J. Catalytic Promiscuity in Biocatalysis: Using Old Enzymes to Form New Bonds and Follow New Pathways. Angew. Chem., Int. Ed. 2004, 43, 6032–6040. 10.1002/anie.200460416. [DOI] [PubMed] [Google Scholar]
- Hult K.; Berglund P. Enzyme Promiscuity: Mechanism and Applications. Trends Biotechnol. 2007, 25, 231–238. 10.1016/j.tibtech.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Humble M. S.; Berglund P. Biocatalytic Promiscuity. Eur. J. Org. Chem. 2011, 2011, 3391–3401. 10.1002/ejoc.201001664. [DOI] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Enzyme Promiscuity: a Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Jensen R. A. Enzyme Recruitment in Evolution of New Function. Annu. Rev. Microbiol. 1976, 30, 409–425. 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Tawfik D. S. Messy Biology and the Origins of Evolutionary Innovations. Nat. Chem. Biol. 2010, 6, 692–696. 10.1038/nchembio.441. [DOI] [PubMed] [Google Scholar]
- Renata H.; Wang Z. J.; Arnold F. H. Expanding the Enzyme Universe: Accessing Non-natural Reactions by Mechanism-guided Directed Evolution. Angew. Chem., Int. Ed. 2015, 54, 3351–3367. 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan N.; Rodríguez-Duran L. V.; Saucedo-Castaneda G.; Madhavan Nampoorthiri K. Review on Technological and Scientific Aspects of Feruloyl Esterases: A Versatile Enzyme for Biorefining of Biomass. Bioresour. Technol. 2015, 193, 534–544. 10.1016/j.biortech.2015.06.117. [DOI] [PubMed] [Google Scholar]
- Zulkarnain A.; Bahrin E. K.; Ramli N.; Phang L. Y.; Abd-Aziz S. Alkaline Hydrolysate of Oil Palm Empty Fruit Bunch as Potential Substrate for Biovanillin Production via Two-Step Bioconversion. Waste Biomass Valorization 2018, 9, 13–23. 10.1007/s12649-016-9745-4. [DOI] [Google Scholar]
- Tang P.-L.; Hassan O.; Maskat M. Y.; Badri K. Production of Monomeric Aromatic Compounds from Oil Palm Empty Fruit Bunch Fiber Lignin by Chemical and Enzymatic Methods. BioMed Res. Int. 2015, 2015, 891539. 10.1155/2015/891539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurrough I.; Madigan D.; Donnelly D.; Hurley J.; Doyle A.-M.; Hennigan G.; McNulty N.; Smyth M. R. Control of Ferulic Acid and 4-Vinyl Guaiacol in Brewing. J. Inst. Brew. 1996, 102, 327–332. 10.1002/j.2050-0416.1996.tb00918.x. [DOI] [Google Scholar]
- Uchiyama M.; Satoh K.; Kamigaito M. Cationic RAFT Polymerization Using ppm Concentrations of Organic Acid. Angew. Chem., Int. Ed. 2015, 54, 1924–1928. 10.1002/anie.201410858. [DOI] [PubMed] [Google Scholar]
- van Nunen J. L. M.; Folmer B. F. B.; Nolte R. J. M. Induction of Liquid Crystallinity by Host–Guest Interactions. J. Am. Chem. Soc. 1997, 119, 283–291. 10.1021/ja9603113. [DOI] [Google Scholar]
- Jin X.; Zhang S.; Runt J. Dielectric Studies of Blends of Poly(ethylene oxide) and Poly(styrene-co-p-hydroxystyrene). Semicrystalline Blends. Macromolecules 2004, 37, 4808–4814. 10.1021/ma049743l. [DOI] [Google Scholar]
- Wuensch C.; Pavkov-Keller T.; Steinkellner G.; Gross J.; Fuchs M.; Hromic A.; Lyskowski A.; Fauland K.; Gruber K.; Glueck S. M.; Faber K. Regioselective Enzymatic beta-Carboxylation of para-Hydroxystyrene Derivatives Catalyzed by Phenolic Acid Decarboxylases. Adv. Synth. Catal. 2015, 357, 1909–1918. 10.1002/adsc.201401028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuensch C.; Glueck S. M.; Gross J.; Koszelewski D.; Schober M.; Faber K. Regioselective Enzymatic Carboxylation of Phenols and Hydroxystyrene Derivatives. Org. Lett. 2012, 14, 1974–1977. 10.1021/ol300385k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuensch C.; Schmidt N.; Gross J.; Grischek B.; Glueck S. M.; Faber K. Pushing the Equilibrium of Regio-complementary Carboxylation of Phenols and Hydroxystyrene Derivatives. J. Biotechnol. 2013, 168, 264–270. 10.1016/j.jbiotec.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Wuensch C.; Gross J.; Steinkellner G.; Gruber K.; Glueck S. M.; Faber K. Asymmetric Enzymatic Hydration of Hydroxystyrene Derivatives. Angew. Chem., Int. Ed. 2013, 52, 2293–2297. 10.1002/anie.201207916. [DOI] [PubMed] [Google Scholar]
- Payer S. E.; Sheng X.; Pollak H.; Wuensch C.; Steinkellner G.; Himo F.; Glueck S. M.; Faber K. Exploring the Catalytic Promiscuity of Phenolic Acid Decarboxylases: Asymmetric, 1,6-Conjugate Addition of Nucleophiles Across 4-Hydroxystyrene. Adv. Synth. Catal. 2017, 359, 2066–2075. 10.1002/adsc.201700247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto A.Uracil Compounds and Use Thereof. U.S. Patent 6,664,214 B1, 2003; CAN134:100884.
- Keserű G. M.; Balogh G. T.; Bokotey S.; Árvai G.; Bertók B. Metalloporphyrin Catalysed Biomimetic Oxidation of Aryl Benzyl Ethers. Implications for Lignin Peroxidase Catalysis. Tetrahedron 1999, 55, 4457–4466. 10.1016/S0040-4020(99)00134-9. [DOI] [Google Scholar]
- Pap L.; Árvai G.; Bertók B.; Ribai Z. K.; Bakonyvári I. Comparative Evaluation of New Synergists Containing a Butynyl-type Synergophore Group and Piperonyl Butoxide Derivatives. Pest Manage. Sci. 2001, 57, 186–190. . [DOI] [PubMed] [Google Scholar]
- Yang Z.-S.; Wang J.-X.; Zhou Y.; Zuo J.-P.; Li Y. Synthesis and Immunosuppressive Activity of New Artemisinin Derivatives. Part 2:2-[12(β or α)-Dihydroartemisinoxymethyl(or 1′-ethyl)]phenoxyl Propionic Acids and Esters. Bioorg. Med. Chem. 2006, 14, 8043–8049. 10.1016/j.bmc.2006.07.038. [DOI] [PubMed] [Google Scholar]
- Renaudet O.; Reymond J. L. Synthesis of Ether Oligomers. Org. Lett. 2004, 6, 397–400. 10.1021/ol036300d. [DOI] [PubMed] [Google Scholar]
- Woodhead A. J.; Chessari G.; Besong G. E.; Carr M. G.; Hiscock S. D.; O’Brien M. A.; Rees D. C.; Saalau-Bethell S. M.; Willems H. M. G.; Thompson N. T.. Substituted Benzylamine Compounds, Their Use in Medicine, and in Particular the Treatment of Hepatitis C Virus (HCV) Infection. Int. Patent WO2013/064538 A1, 2013; CAN158:711128.
- Sheng X.; Himo F. Theoretical Study of Enzyme Promiscuity: Mechanisms of Hydration and Carboxylation Activities of Phenolic Acid Decarboxylase. ACS Catal. 2017, 7, 1733–1741. 10.1021/acscatal.6b03249. [DOI] [Google Scholar]
- A related approach based on an artificial Cu(II)-dependent Michael hydratase was reported during the preparation of the manuscript:Drienovská I.; Alonso-Cotchico L.; Vidossich P.; Lledos A.; Maréchal J.-D.; Roelfes G. Design of an Enantioselective Artificial Metallo-hydratase Enzyme Containing an Unnatural Metal-binding Amino Acid. Chem. Sci. 2017, 8, 7228–7235. 10.1039/C7SC03477F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R. S. Temperature Effects on Stereochemistry of Enzymatic Reactions. Enzyme Microb. Technol. 1992, 14, 417–419. 10.1016/0141-0229(92)90013-E. [DOI] [Google Scholar]
- Liskova V.; Stepankova V.; Bednar D.; Brezovsky J.; Prokop Z.; Chaloupkova R.; Damborsky J. Different Structural Origins of the Enantioselectivity of Haloalkane Dehalogenases Toward Linear beta-Haloalkanes: Open-Solvated versus Occluded-Desolvated Active Sites. Angew. Chem., Int. Ed. 2017, 56, 4719–4723. 10.1002/anie.201611193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.