

Abstract
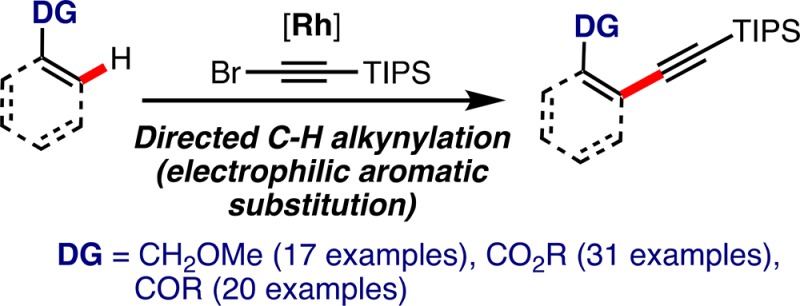
We report the alkynylation of C(sp2)–H bonds with bromoalkynes (inverse-Sonogashira reaction) directed by synthetically useful ester, ketone, and ether groups under rhodium catalysis. Other less common directing groups such as amine, thioether, sulfoxide, sulfone, phenol ester, and carbamate are also suitable directing groups. Mechanistic studies indicate that the reaction proceeds by a turnover-limiting C–H activation step via an electrophilic-type substitution.
Keywords: alkynylation, rhodium catalysis; C−H functionalization; inverse Sonogashira; metallacycle; Hammett correlation; DFT
Introduction
Alkynes are among the most versatile functional groups1 and are widely present in natural products,2 drugs,3 and organic materials.4 The chemistry of alkynes has gained particular momentum in recent years by the discovery of a wide variety of catalytic transformations triggered by gold(I), platinum(II), and other alkynophilic Lewis acids.5 Therefore, the development of methods for the introduction of alkyne groups onto organic molecules is of high importance. To this end, the Sonogashira coupling reaction is the most general method for the formation of C(sp)–C(sp2) bonds from aryl or alkenyl (pseudo)halides and terminal alkynes.6
The main limitation of the Sonogashira coupling reaction resides in the synthetic availability of the required (pseudo)halides. An alternative approach that is better suited for the late-stage functionalization of complex molecules involves the alkynylation of C(sp2)–H bonds with terminal alkynes or activated acetylenes such as ethynylbenziodoxolone (EBX) reagents or haloalkynes using transition-metal catalysts.7 Often named inverse-Sonogashira coupling, this methodology relies on the reactivity of electronically activated (hetero)arenes8 or on a chelating group to assist a C–H activation process.9 The former strategy is restricted to aromatic C(sp2)–H bonds, which need in addition to be acidic or electron-rich enough to undergo deprotonation or a Friedel–Crafts type reaction. The latter has been achieved for both arenes and alkenes9b with a variety of directing groups, typically amides or nitrogen coordinating groups such as heterocycles or imine derivatives (oxime, nitrone, azomethine).9c The applicability of this strategy in multistep synthesis is however limited, as in most cases the directing groups need to be installed and/or removed. Therefore, to render this approach useful, the development of new protocols using instead widely used functional groups serving as synthetic handles is highly desirable.10
Toward this goal, we recently reported a general peri-alkynylation of naphthols using ruthenium catalysis.11 Benzoic acids can also be alkynylated at the ortho position,11,12 although the use of other versatile O functionalities13,14 as directing groups is still limited, mainly due to the challenging formation of a weakly coordinated metallacyclic intermediate.15 In particular, despite intense efforts in the field of catalytic C(sp2)–H functionalization, only two examples of the use of benzyl ether as a directing group have been reported in the context of C–H borylation.16
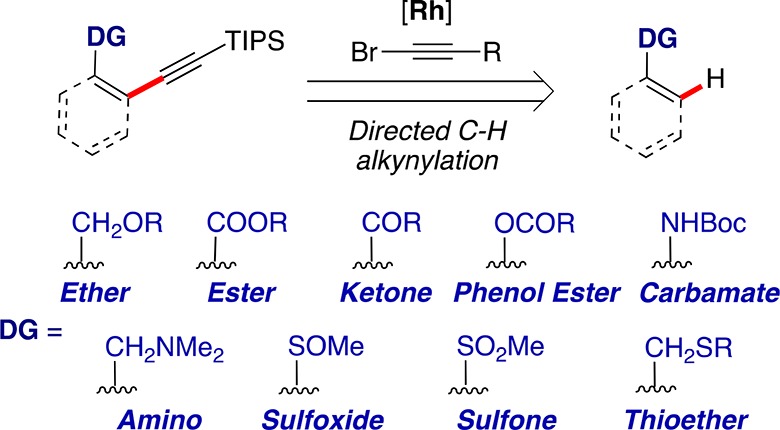
Here, we report the use of synthetically useful ether, ester, and ketone as directing groups for the direct alkynylation of C(sp2)–H bonds with bromoalkynes under rhodium catalysis (Scheme 1).17 We also demonstrate for the first time that amine,18 thioether,19 sulfoxide,20 sulfone,21 carbamate,22 and phenol esters23 are suitable directing groups in this transformation. Furthermore, our experimental and theoretical mechanistic study shows that this Rh-catalyzed alkynylation occurs by a turnover-determining C–H activation in which a five-membered ring metallacycle is formed by an electrophilic aromatic substitution type process.
Scheme 1. C(sp2)–H Alkynylation with Bromoalkynes Directed by a Broad Range of Coordinating Groups under Rhodium Catalysis.
Results and Discussion
Reaction Scope
Our studies began by evaluating the reactions of TIPS-protected bromoacetylene (1) with ethyl benzoate (2a) and benzyl methyl ether (4a). We discovered that a combination of [Cp*RhCl2]2 (2.5 mol %), AgSbF6 (20 mol %), Ag2CO3 (1 equiv), and LiOAc (20 mol %) in 1,2-dichloroethane (DCE) at 45 °C provided 3a in 69% yield (Table 1, entry 1). Control experiments showed the essential role of all reaction components (Table 1, entries 2–11). Thus, lower yields of 3a were obtained at temperatures lower or higher than 45 °C (Table 1, entries 2 and 3). Similar results were obtained by decreasing the amount of Ag2CO3 to 0.5 equiv or replacing this silver salt by K2CO3 (Table 1, entries 4 and 5). Solvents different from DCE led to poor results (Table 1, entries 6–11). The use of other bromoalkynes, such as (bromoethynyl)benzene and 1-bromooctyne, led to no conversion.24
Table 1. Rh-Catalyzed o-C–H Alkynylation of Ethyl Benzoate and Benzyl Methyl Ether: Optimization Conditions24.
| entry | DG | variation from the “standard conditions”a | yield (%)b |
|---|---|---|---|
| 1 | ester | none | 58–69 |
| 2 | ester | at 25 °Cc | 35 |
| 3 | ester | at 65 °Cc | 16 |
| 4 | ester | with Ag2CO3 (0.5 equiv)d | 41 |
| 5 | ester | with K2CO3 (1 equiv)d | 5 |
| 6 | ester | in dichloromethanee | 8–14 |
| 7 | ester | in toluenee | 0 |
| 8 | ester | in t-AmOHe | 0 |
| 9 | ester | in Et2Oe | 4 |
| 10 | ester | in EtOAce | 18 |
| 11 | ester | in MeOHe | 0 |
| 12 | ether | none | 0 |
| 13 | ether | at 100 °Cc | 50–64 |
| 14 | ether | without [Cp*RhCl2]2 | 0 |
| 15 | ether | without Ag2CO3 | 0 |
| 16 | ether | without LiOAc | 0 |
| 17 | ether | without AgSbF6 | 0 |
| 18 | ether | with AgOAc (1.2 equiv)f | <1.5 |
| 19 | ether | AgOAc (1 equiv) + Ag2CO3 (0.2 equiv)g | 12 |
| 20 | ether | in toluenee | 0 |
| 21 | ether | in tert-amOHe | 0 |
| 22 | ether | in 1,4-dioxanee | 0 |
| 23 | ether | with TIPS-acetyleneh | 0 |
| 24 | ether | with [Cp*IrCl2]2i | 0 |
| 25 | ether | with Pd(OAc)2i | 0 |
| 26 | ether | with [RuCl2(p-cymene)]2i | <3 |
Standard reaction conditions: 2a or 4a (0.2 mmol), 1 (2 equiv), [Cp*RhCl2]2 (2.5 mol % for DG = ester, 3 mol % for DG = ether), Ag2CO3 (1 equiv), AgSbF6 (0.2 equiv), LiOAc (0.2 equiv), DCE, 16 h, 45 °C.
Yield of the monoalkynylated product determined by 1H NMR using bromomesitylene as internal standard.
Instead of 45 °C.
Instead of Ag2CO3 (1 equiv).
Instead of DCE.
Instead of Ag2CO3 and LiOAc.
Without LiOAc.
Instead of 1.
Instead of [Cp*RhCl2]2.
Although treatment of benzyl methyl ether (4a) with bromoacetylene 1 under essentially the same conditions did not lead to the product of alkynylation (Table 1, entry 12), simply increasing the temperature to 100 °C led to 5a in 64% yield (Table 1, entry 13). Using ethynyltriisopropylsilane instead of 1 did not afford 5a (Table 1, entry 23). Replacing [Cp*RhCl2]2 with other metal catalysts typically used in C–H functionalization did not lead to alkynylated product (Table 1, entries 24–26). The alternative hydroxy-directed alkynylation of primary, secondary, or tertiary benzyl alcohol led to oxidation, decomposition, or unproductive reaction.
Different alkyl benzoates 2a–d could be ortho-alkynylated, with ethyl benzoate 2a giving the highest yield (Scheme 2). Electron-donating alkyl or methoxy groups and electron-withdrawing substituents such as NO2, CF3, and different halides at the ortho, meta, and para positions were well tolerated, affording alkynylated products 3e–w in 23–90% yield. In the case of meta-substituted substrates 2i,k,m, the alkynylation occurred at the least sterically hindered site. However, fluoro and methoxy derivatives 2j,l favor formation of the 1,2,3-trisubstituted compounds 3j,l, respectively.
Scheme 2. Rh-Catalyzed o-C–H Alkynylation of Alkyl Benzoates.
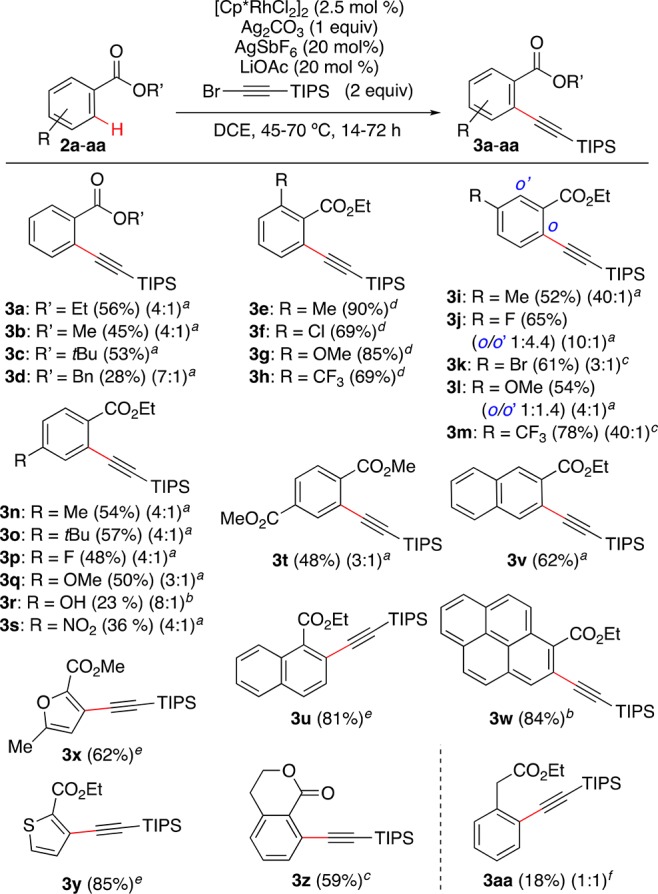
Legend to conditions: (a) 45 °C, 16–24 h; (b) 45 °C, 48 h; (c) 45 °C, 72 h; (d) 60 °C, 48 h; (e) 70 °C, 24–72 h; (f) 90 °C, 72 h (0.2 mmol scale). Yields of isolated monoalkynylated products are shown. In cases in which diakynylated products were also formed, mono- vs dialkynylation selectivity is shown in parentheses.
The alkynylation of ethyl 1-naphthoate (2u) and ethyl pyrene-1-carboxylate (2w) does not take place at the peri position, leading instead to ortho-fuctionalized products 3u,w, respectively. Reaction of ethyl 2-naphthoate (2v) afforded exclusively the product of alkynylation at C-3 (3v). Furan and thiophene esters were also alkynylated to give 3x (62%) and 3y (85%), respectively. The carbonyl group of isochroman-1-one is also an effective directing group, affording 3z in 59% yield. On the other hand, the alkynylation of ethyl phenylacetate required heating at 90 °C and was less efficient, leading to 3aa in 18% yield along with an equivalent amount of the dialkynylated product.
Whereas the alkynylation of 4a leads to 5a in 64% yield, substrates 4b–d with bulkier alkyl or silyl groups failed to give the expected products (Scheme 3). Similarly, MOM-protected benzyl alcohol 4e and esters 4f,g were unreactive substrates. On the other hand, methyl benzyl ethers bearing diverse substituents at the ortho, meta, or para positions such as i-Pr, CF3, fluoro, chloro, bromo, and iodo led to o-alkynylated products 5h–u in 32–71% yields. As observed for the benzoates, the alkynylation of meta-substituted substrates 4n,o occurred at the least sterically hindered site, whereas fluoro derivative 4m led to a mixture of ortho-alkynylated derivatives 5m, favoring the formation of the 1,2,3-trisubstituted product. Again, the alkynylation of naphthyl derivative 4v takes place at C-3 to form 5v in 70% yield. The reaction of thiophene 4w provided 5w, the product of C-2 alkynylation, which was isolated in 31% yield.
Scheme 3. Rh-Catalyzed o-C–H Alkynylation of Benzyl Ethers.
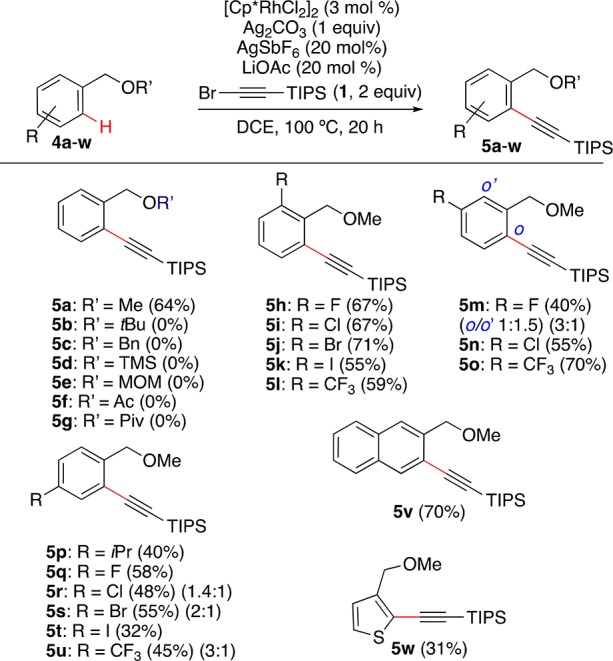
Yields of isolated monoalkynylated products are shown. In cases in which diakynylated products were also formed, mono- vs dialkynylation selectivity is shown in parentheses.
Under conditions similar to those used for the reaction of the ester derivatives, a wide variety of aryl ketones 6a–p could be alkynylated in a general manner to give 7a–p in good to excellent yield (Scheme 4). Bis(alkynylated)acetophenone 7k was obtained in quantitative yield from acetophenone at room temperature, while bulkier alkyl substituents allowed a monoselective alkynylation, affording products 7a–c in 50–95% yield. Diverse substituents at the ortho position of acetophenone were well tolerated to give products 7d–i in 81–95% yield. 2-Acetyl derivatives N-methylpyrrole (6n), furan (6o), and thiophene (6p) were alkynylated at C-3 in 75–95% yield. The double alkynylation of 1,5-dichloroanthraquinone (6q) proceeded at 100 °C to give dialkynylated product 7q in 82% yield.
Scheme 4. Rh-Catalyzed o-C–H Alkynylation of Aryl Ketones.
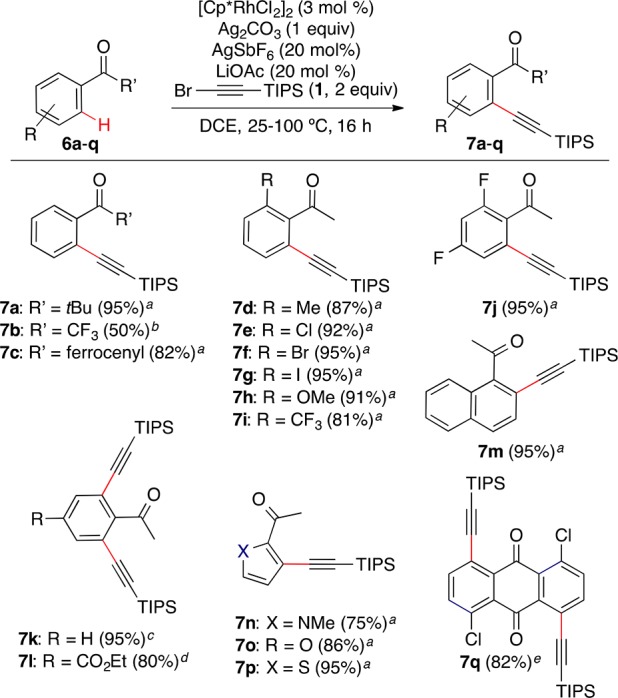
Legend to conditions: (a) 45 °C, (1 equiv 1); (b) 90 °C, (1 equiv 1); (c) 25 °C, (2 equiv 1); (d) 45 °C, (2 equiv 1); (e) 100 °C, (2 equiv 1).
As an example of late-stage functionalization of a pharmaceutical compound, fenofibrate (6r) was alkynylated in 35% yield for the major product (Scheme 5).
Scheme 5. Late-Stage Alkynylation of Fenofibrate.
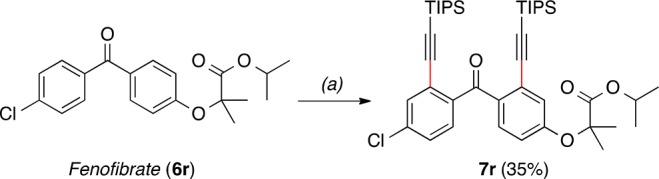
Standard conditions for the Rh-catalyzed reaction using 2 equiv of bromoalkyne, at 50 °C, 14 h.
Stereocontrolled synthesis of conjugated enynes or acyclic tri- and tetrasubstituted alkenes is a longstanding challenge in organic chemistry.25 We were pleased to find that the alkynylation of vinyl C–H bonds of α,β-unsaturated esters 8a–e and ketones 8f,g proceeded under the standard conditions at 45–85 °C to afford a series of Z-configured 1,3-enynes 9a–g in 44–84% yield, with total control of the stereoselectivity (Scheme 6).
Scheme 6. Alkynylation of Vinyl C–H Bonds.
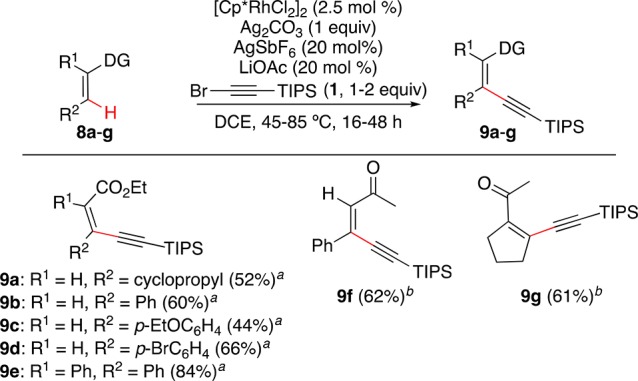
Legend to conditions: (a) 85 °C 48 h, (2 equiv 1); (b) 45 °C, 16 h (1 equiv 1).
Other Directing Groups
With slight modification of the reaction conditions, we discovered that other functional groups are viable chelating groups (Scheme 7). As rare examples of the use of a simple phenol ester as a directing group,23 the ortho alkynylation of phenol pivalate (10a) and 1-naphthol acetate (10b) led to 11a,b in moderate yields. Although they are considered to bind too tightly to metals to be involved in catalytic processes, strongly coordinating groups could also be used under similar conditions. Thus, the reaction proceeds on substrates bearing sulfoxide, thioether, thioacetal, sulfone, and tertiary amine functional groups, giving products 11c–h in 53–75% yield. Boc-protected pyrrole 10i could also be dialkynylated to give product 11i in 66% yield.
Scheme 7. Rhodium-Catalyzed C(sp2)–H Alkynylation with Other Directing Groups.
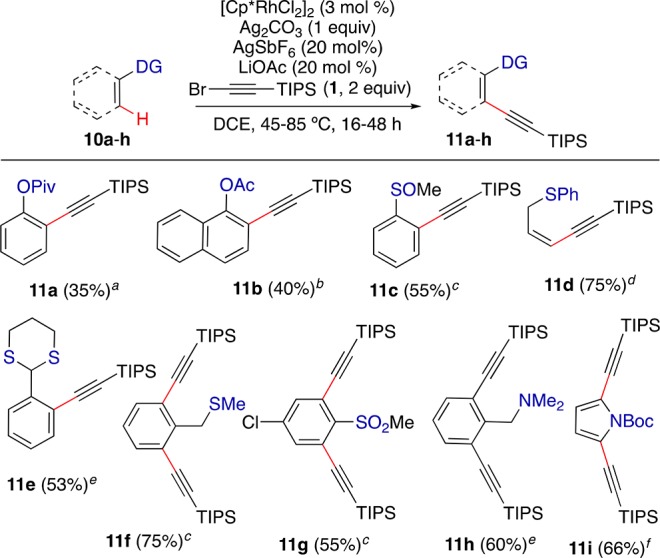
Legend to conditions: (a) 90 °C, 72 h; (b) 70 °C, 24 h; (c) 100 °C, 16 h; (d) 50 °C, (1 equiv 1), 16 h; (e) 90 °C, 16 h; (f) 45 °C, 16 h.
Mechanistic Studies
Several experiments were carried out in order to shed light on the reaction mechanism. First, the C–H functionalization step was found to be irreversible according to the reaction of 2a-d5 in the presence of water and in the absence of bromoalkyne 1 (Scheme 8i). The intermolecular and parallel competition experiments between deuterated and hydrogenated labeled substrates (Scheme 8ii) showed the same kinetic isotope effect (KIE = 3.1) in both cases, indicating that the C–H bond cleavage probably occurs in the rate-determining step of the catalytic cycle,26 which is consistent with related rhodium-catalyzed C–H functionalizations.27 Finally, the intermolecular competition between electron-rich and electron-poor substrates (Scheme 8iii) suggests that substrates bearing electron-donating groups (Me or MeO) at the meta position of the C–H functionalization site are more reactive. This result indicates that the C–H functionalization step might occur through an electrophilic aromatic substitution type mechanism.12b,28
Scheme 8. D/H Exchange, Kinetic, and Competition Experiments,24.

Yield of the monoalkynylated product determined by 1H NMR using bromomesitylene as internal standard.
A Hammett correlation was found (R2 = 0.99 using σp+) for meta-substituted substrates (Figure 1).29 A negative ρ value also suggests that electron density decreases at the aryl ring in the product-determining step, which is in accordance with a C–H functionalization step occurring through an electrophilic aromatic substitution type mechanism.
Figure 1.
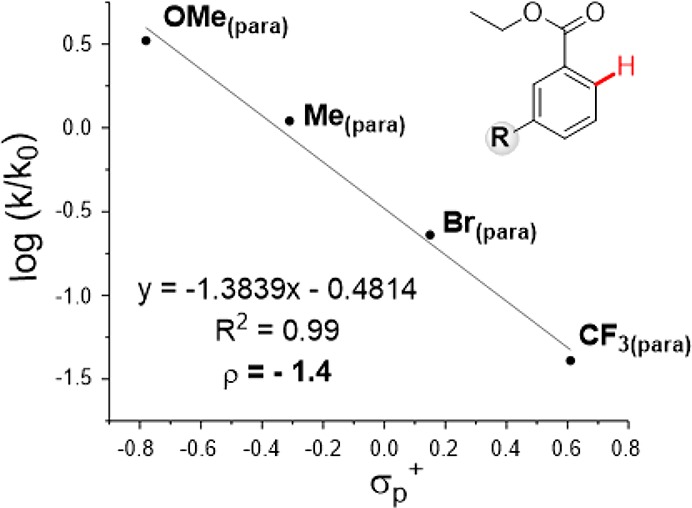
Hammett plot for the reaction of meta-substituted benzoates.24
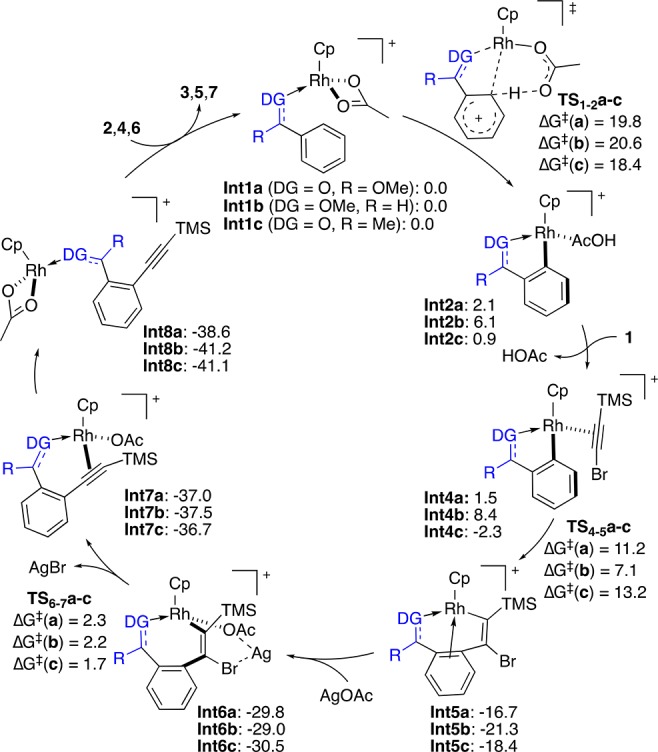
To get a deeper insight into the reaction mechanism, we performed DFT calculations (Scheme 9).30,31 According to our studies, the C–H functionalization of methyl benzoate (2b) proceeds from Int1a by the intramolecular assistance of the acetate ligand through the six-membered cyclic transition state TS1–2a (ΔG⧧ = 19.8 kcal/mol). The alternative four-membered cyclic transition state (ΔG⧧ = 34.6 kcal/mol) or the intermolecular acetate-assisted C–H activation (ΔG⧧ = 51.2 kcal/mol) would require much higher energy barriers.24,32 The resulting Int2a undergoes dissociative ligand exchange with bromoacetylene 1b through Int3a (not shown)24 to form the (η2-alkyne)rhodium complex Int4a. Subsequent alkyne insertion (ΔG⧧ = 11.2 kcal/mol) to give Int5a, followed by AgOAc-assisted bromide elimination (ΔG⧧ = 2.3 kcal/mol) leads to Int7a and then Int8a. The catalytic cycle restarts upon ligand exchange, delivering the final alkynylated product 3ab and regenerating Int1a.
Scheme 9. Proposed Mechanism of the Rh-Catalyzed C(sp2)–H Alkynylation on the Basis of DFT Calculations.
Free energies in kcal/mol.
Analysis of the Mulliken atomic charges in Int1a, TS1-2a, and Int2a(24) shows that the process involves an ambiphilic metal ligand activation.32e Both an electrophilic metal center and an intramolecular basic ligand are key for the heterolytic scission of the C–H bond and formation of the C–Rh bond (Figure 2). In TS1–2a, the carbon involved in the C–H activation shows a certain sp3 character (the Rh–C–H angle is 73.8°).24 The C–Rh distance (2.23 Å) in TS1-2a is slightly longer than that of the metallacycle Int2a (2.02 Å), whereas the C–H distance is lengthened from 1.09 Å in Int1a to 1.30 Å in TS12a, which suggests that the formation of the Rh–C bond preceeds the cleavage of the C–H bond in a concerted, but asynchronous, process.
Figure 2.
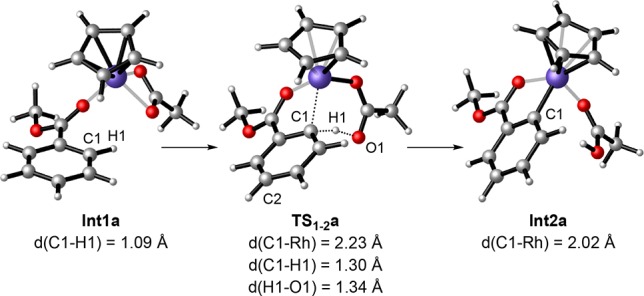
Calculated structures for the C–H activation via TS1-2a.24
Alternative alkynylation pathways were also considered, although they proved to be less favored.24 For instance, the oxidative addition of the C(sp)–Br bond to the metal center in Int4a to form a Rh(V) intermediate33 demands a highly unlikely activation energy of 41.6 kcal/mol. On the basis of the computed energies, the C–H metalation is the rate-determining step, which is in agreement with the experimental results. Similar energy profiles were found in the case of methyl benzyl ether 4a (Scheme 9, pathway b) and acetophenone 6k (Scheme 9, pathway c), which means that the same reaction mechanism presumably operates for them.24 Consistent with the experimental results, among the different substrates, the C–H functionalization of the ketones is the most energetically favored (ΔG⧧ = 18.4 kcal/mol), whereas the corresponding to the benzyl ethers is the most energetically costly (ΔG⧧ = 20.6 kcal/mol).
In addition, the C–H activation step was computed for differently meta-substituted methyl benzoates to study the influence of the electronic effects on the energy barrier. Calculations showed that the more electron-rich the substituent, the lower the activation energy results (Table 2, entries 1–4). This is in total agreement with the experimental results observed for meta-substituted ethyl benzoates (Figure 1) and supports an electrophilic substitution type mechanism for the formation of the five-membered-ring rhodacycle.
Table 2. Substituent Effect in the Activation Energy of the C–H Activation of Benzoatesa.
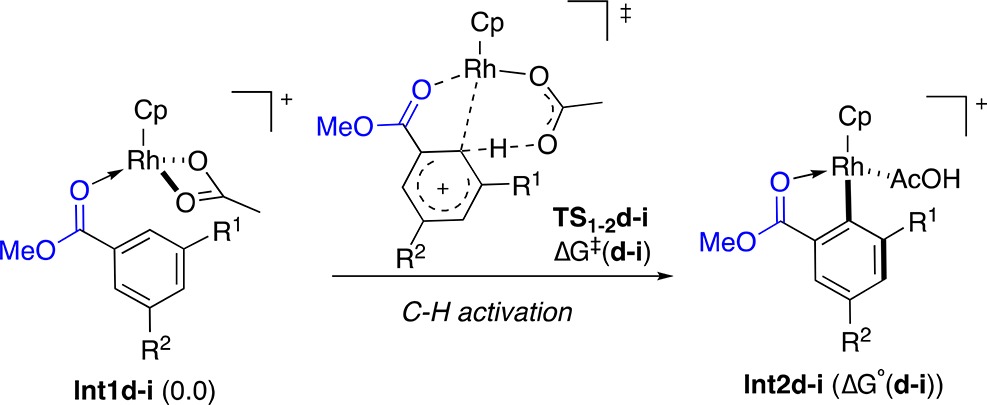
| entry | R1 | R2 | TS1-2d–i | ΔG⧧(d–i) | Int2d–i | ΔG°(d–i) |
|---|---|---|---|---|---|---|
| 1 | H | OMe | TS1-2d | 17.2 | Int2d | 2.9 |
| 2 | H | Me | TS1-2e | 18.9 | Int2e | 3.3 |
| 3 | H | Br | TS1-2f | 20.8 | Int2f | 3.1 |
| 4 | H | CF3 | TS1-2g | 21.5 | Int2g | 3.4 |
| 5 | H | F | TS1-2h | 19.5 | Int2h | 2.5 |
| 6 | F | H | TS1-2i | 17.8 | Int2i | 2.7 |
Free energies in kcal/mol.
In the case of m-fluorobenzoate, the C–H activation preferentially occurs at the ortho (ΔG⧧ = 17.8 kcal/mol, Table 2, entry 6) rather than the para position (ΔG⧧ = 19.5 kcal/mol, Table 2, entry 5) respect to the fluoro substituent. This o-fluorine effect has been experimentally observed with m-fluoro-substituted benzoate 3j (Scheme 2) or benzyl ether compound 5m (Scheme 3), as the metal–carbon bond strength would be increased at this position.34
Conclusions
In summary, we have found that the alkynylation of benzyl methyl ethers, aryl esters, and aryl ketones can be carried out using rhodium catalysis in a general manner. This is the first report of a broad-range o-C–H functionalization of weakly coordinating benzyl ethers. The Rh-catalyzed alkynylation of aryl esters and aryl ketones takes place under milder conditions (45–70 °C for esters and 25–90 °C for ketones) in comparison to those recently reported using Ir catalysis (120 °C). The alkynylation of vinyl C–H bonds of α,β-unsaturated esters and ketones is also possible using rhodium catalysis. Furthermore, other uncommon functional groups such as amine, thioether, thioacetal, sulfoxide, sulfone, phenol ester, and carbamate can also be used as directing groups for the alkynylation. Our mechanistic study shows that the alkynylation reaction proceeds by a turnover-limiting C–H activation step via an electrophilic-type substitution, followed by insertion of the bromoalkyne and bromide elimination.
Acknowledgments
We thank the Agencia Estatal de Investigación (CTQ2016-75960-P MINECO/AEI/FEDER, UE), Severo Ochoa Excellence Acreditation 2014-2018 (SEV-2013-0319 and Severo Ochoa predoctoral fellowship to M.E.d.O.), Juan de la Cierva postdoctoral fellowship (O.Q.), the European Research Council (Advanced Grant No. 321066), the AGAUR (2014 SGR 818), and the CERCA Program/Generalitat de Catalunya for financial support. We also thank CELLEX-ICIQ HTE laboratory.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b04395.
Additional details, experimental procedures, characterization data for compounds, and computational results (PDF)
Author Contributions
§ E.T. and O.Q. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Acetylene Chemistry: Chemistry, Biology and Material Science; Diederich F., Stang P. J., Tykwinski R. R., Eds.; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]; b Boyd G. V.The Chemistry of Triple Bonded Functional Groups; Patai S., Ed.; Wiley: Hoboken, NJ, 1994; Chapter 5. [Google Scholar]
- a Zhou Z.-F.; Menna M.; Cai Y. S.; Guo Y. W. Chem. Rev. 2015, 115, 1543–1596. 10.1021/cr4006507. [DOI] [PubMed] [Google Scholar]; b Chai Q.-Y.; Yang Z.; Lin H. W.; Han B. N. Mar. Drugs 2016, 14, 216. 10.3390/md14110216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberth Bioorg. Med. Chem. 2009, 17, 4047–4063. 10.1016/j.bmc.2008.11.037. [DOI] [PubMed] [Google Scholar]
- Broggi A.; Tomasi I.; Bianchi L.; Marrocchi A.; Vaccaro L. ChemPlusChem 2014, 79, 486–507. 10.1002/cplu.201400001. [DOI] [PubMed] [Google Scholar]
- a Fürstner A. Chem. Soc. Rev. 2009, 38, 3208–3221. 10.1039/b816696j. [DOI] [PubMed] [Google Scholar]; b Shapiro N. D.; Toste F. D. Synlett 2010, 2010, 675–691. 10.1055/s-0029-1219369. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Obradors C.; Echavarren A. M. Acc. Chem. Res. 2014, 47, 902–912. 10.1021/ar400174p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fensterbank L.; Malacria M. Acc. Chem. Res. 2014, 47, 953–965. 10.1021/ar4002334. [DOI] [PubMed] [Google Scholar]; e Dorel R.; Echavarren A. M. Chem. Rev. 2015, 115, 9028–9072. 10.1021/cr500691k. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Pflästerer D.; Hashmi A. S. K. Chem. Soc. Rev. 2016, 45, 1331–1367. 10.1039/C5CS00721F. [DOI] [PubMed] [Google Scholar]; g Echavarren A. M.; Muratore M. N.; López-Carrillo V.; Escribano-Cuesta A.; Huguet A.; Obradors C. Org. React. 2017, 92, 1. 10.1002/0471264180.or092.01. [DOI] [Google Scholar]
- a Negishi E.; Anastasia L. Chem. Rev. 2003, 103, 1979–2018. 10.1021/cr020377i. [DOI] [PubMed] [Google Scholar]; b Doucet H.; Hierso J.-C. Angew. Chem., Int. Ed. 2007, 46, 834–871. 10.1002/anie.200602761. [DOI] [PubMed] [Google Scholar]; c Chinchilla R.; Nájera C. Chem. Soc. Rev. 2011, 40, 5084–5121. 10.1039/c1cs15071e. [DOI] [PubMed] [Google Scholar]; d Chinchilla R.; Nájera C. Chem. Rev. 2014, 114, 1783–1826. 10.1021/cr400133p. [DOI] [PubMed] [Google Scholar]; e Wang D.; Gao S. Org. Chem. Front. 2014, 1, 556–566. 10.1039/C3QO00086A. [DOI] [Google Scholar]
- For reviews on C(sp2)–H alkynylation, see:; a Colby D. A.; Bergman R. G.; Ellman J. A. Chem. Rev. 2010, 110, 624–655. 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dudnik A. S.; Gevorgyan V. Angew. Chem., Int. Ed. 2010, 49, 2096–2098. 10.1002/anie.200906755. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen Z.; Wang B.; Zhang J.; Yu W.; Liu Z.; Zhang Y. Org. Chem. Front. 2015, 2, 1107–1295. 10.1039/C5QO00004A. [DOI] [Google Scholar]; For a review on EBX, see:; d Waser J. Synlett 2016, 27, 2761–2773. 10.1055/s-0036-1589409. [DOI] [Google Scholar]; For more works in the field of C(sp2)–H alkynylation, see:; e Kim S. H.; Yoon J.; Chang S. Org. Lett. 2011, 13, 1474–1477. 10.1021/ol200154s. [DOI] [PubMed] [Google Scholar]; f Feng C.; Feng D.; Luo Y.; Loh T.-P. Org. Lett. 2014, 16, 5956–5959. 10.1021/ol502984g. [DOI] [PubMed] [Google Scholar]; g Feng C.; Loh T.-P. Angew. Chem., Int. Ed. 2014, 53, 2722–2726. 10.1002/anie.201309198. [DOI] [PubMed] [Google Scholar]; h Feng C.; Feng D.; Loh T.-P. Chem. Commun. 2014, 50, 9865–9868. 10.1039/C4CC04072D. [DOI] [PubMed] [Google Scholar]; i Zhang X.; Qi Z.; Gao J.; Li X. Org. Biomol. Chem. 2014, 12, 9329–9332. 10.1039/C4OB01596G. [DOI] [PubMed] [Google Scholar]; j Zhang Z.-Z.; Liu B.; Wang C.-Y.; Shi B.-F. Org. Lett. 2015, 17, 4094–4097. 10.1021/acs.orglett.5b02038. [DOI] [PubMed] [Google Scholar]; k Sauermann N.; González M. J.; Ackermann L. Org. Lett. 2015, 17, 5316–531. 10.1021/acs.orglett.5b02678. [DOI] [PubMed] [Google Scholar]; l Landge V. G.; Midya S. P.; Rana J.; Shinde D. R.; Balaraman E. Org. Lett. 2016, 18, 5252–5255. 10.1021/acs.orglett.6b02549. [DOI] [PubMed] [Google Scholar]; m Ye X.; Xu C.; Wojtas L.; Akhmedov N. G.; Chen H.; Shi X. Org. Lett. 2016, 18, 2970–2973. 10.1021/acs.orglett.6b01319. [DOI] [PubMed] [Google Scholar]; n Boobalan R.; Gandeepan P.; Cheng C.-H. Org. Lett. 2016, 18, 3314–3317. 10.1021/acs.orglett.6b01281. [DOI] [PubMed] [Google Scholar]; o Tang G.-D.; Pan C.-L.; Xie F. Org. Biomol. Chem. 2016, 14, 2898–2904. 10.1039/C6OB00106H. [DOI] [PubMed] [Google Scholar]; p Liu B.; Wang X.; Ge Z.; Li R. Org. Biomol. Chem. 2016, 14, 2944–2949. 10.1039/C6OB00179C. [DOI] [PubMed] [Google Scholar]; q Landge V. G.; Jaiswal G.; Balaraman E. Org. Lett. 2016, 18, 812–815. 10.1021/acs.orglett.6b00095. [DOI] [PubMed] [Google Scholar]; r Wang S.-B.; Gu Q.; You S.-L. J. J. Org. Chem. 2017, 82, 11829–11835. 10.1021/acs.joc.7b00775. [DOI] [PubMed] [Google Scholar]; s Székely A.; Péter Á.; Aradi K.; Tolnai G. L.; Novák Z. Org. Lett. 2017, 19, 954–957. 10.1021/acs.orglett.7b00259. [DOI] [PubMed] [Google Scholar]; t Hu S.; Lu L.; Zhu T.; Wu Q.; Chen Y.; Li J. J.; Zhao J. Org. Biomol. Chem. 2018, 16, 43–47. 10.1039/C7OB02438J. [DOI] [PubMed] [Google Scholar]
- a Seregin I. V.; Ryabova V.; Gevorgyan V. J. Am. Chem. Soc. 2007, 129, 7742–7743. 10.1021/ja072718l. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Matsuyama N.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2009, 11, 4156–4159. 10.1021/ol901684h. [DOI] [PubMed] [Google Scholar]; c Besselievre F.; Piguel S. Angew. Chem., Int. Ed. 2009, 48, 9553–9556. 10.1002/anie.200904776. [DOI] [PubMed] [Google Scholar]; d Wei Y.; Zhao H.; Kan J.; Su W.; Hong M. J. Am. Chem. Soc. 2010, 132, 2522–2523. 10.1021/ja910461e. [DOI] [PubMed] [Google Scholar]; e Brand J. P.; Charpentier J.; Waser J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. 10.1002/anie.200905419. [DOI] [PubMed] [Google Scholar]
- a Tobisu M.; Ano Y.; Chatani N. Org. Lett. 2009, 11, 3250–3252. 10.1021/ol901049r. [DOI] [PubMed] [Google Scholar]; b Collins K. D.; Lied F.; Glorius F. Chem. Commun. 2014, 50, 4459–4461. 10.1039/c4cc01141d. [DOI] [PubMed] [Google Scholar]; c Xie F.; Qi Z.; Yu S.; Li X. J. Am. Chem. Soc. 2014, 136, 4780–4787. 10.1021/ja501910e. [DOI] [PubMed] [Google Scholar]; d Ruan Z.; Sauermann N.; Manoni E.; Ackermann L. Angew. Chem., Int. Ed. 2017, 56, 3172–3176. 10.1002/anie.201611118. [DOI] [PubMed] [Google Scholar]
- Gensch T.; James M.; Dalton T.; Glorius F. Angew. Chem., Int. Ed. 2017, 10.1002/anie.201710377. [DOI] [PubMed] [Google Scholar]
- Tan E.; Konovalov A. I.; Fernández G. A.; Dorel R.; Echavarren A. M. Org. Lett. 2017, 19, 5561–5564. 10.1021/acs.orglett.7b02655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chen C.; Liu P.; Tang J.; Deng G.; Zeng X. Org. Lett. 2017, 19, 2474–2477. 10.1021/acs.orglett.7b00581. [DOI] [PubMed] [Google Scholar]; b Mei R.; Zhang S.-K.; Ackermann L. Org. Lett. 2017, 19, 3171–3174. 10.1021/acs.orglett.7b01294. [DOI] [PubMed] [Google Scholar]; c Chen C.; Zeng X. Eur. J. Org. Chem. 2017, 2017, 4749–4752. 10.1002/ejoc.201700788. [DOI] [Google Scholar]
- For examples of ester-directed C(sp2)–H functionnalization, see:; a Sonoda M.; Kakiuchi F.; Kamatani A.; Chatani N.; Murai S. Chem. Lett. 1996, 25, 109–110. 10.1246/cl.1996.109. [DOI] [Google Scholar]; b Kawamorita S.; Ohmiya H.; Hara K.; Fukuoka A.; Sawamura M. J. Am. Chem. Soc. 2009, 131, 5058–5059. 10.1021/ja9008419. [DOI] [PubMed] [Google Scholar]; c Kim J.; Chang S. Angew. Chem. 2014, 126, 2235–2239. 10.1002/ange.201310544. [DOI] [Google Scholar]; d Yang Y.; Lin Y.; Rao Y. Org. Lett. 2012, 14, 2874–2877. 10.1021/ol301104n. [DOI] [PubMed] [Google Scholar]; e Yu W.; Zhang W.; Liu Z.; Zhang Y. Chem. Commun. 2016, 52, 6837–6840. 10.1039/C6CC02468H. [DOI] [PubMed] [Google Scholar]; f Coxon T. J.; Fernandez M.; Barwick-Silk J.; McKay A. I.; Britton L. E.; Weller A.; Willis M. C. J. Am. Chem. Soc. 2017, 139, 10142–10149. 10.1021/jacs.7b05713. [DOI] [PubMed] [Google Scholar]
- For examples of ketone-directed C(sp2)–H functionnalization, see:; a Murai S.; Kakiuchi F.; Sekine S.; Tanaka Y.; Kamatani A.; Sonoda M.; Chatani N. Nature 1993, 366, 529–531. 10.1038/366529a0. [DOI] [Google Scholar]; For a review, see:; b Huang Z.; Lim H. N.; Mo F.; Young M. C.; Dong G. Chem. Soc. Rev. 2015, 44, 7764–7786. 10.1039/C5CS00272A. [DOI] [PubMed] [Google Scholar]; c Shang R.; Ilies L.; Nakamura E. J. Am. Chem. Soc. 2016, 138, 10132–10135. 10.1021/jacs.6b06908. [DOI] [PubMed] [Google Scholar]; d Zhao Y.; Li S.; Zheng X.; Tang J.; She Z.; Gao G.; You J. Angew. Chem., Int. Ed. 2017, 56, 4286–4289. 10.1002/anie.201612147. [DOI] [PubMed] [Google Scholar]; e Zhang B.; Wang H.-W.; Kang Y.-S.; Zhang P.; Xu H.-J.; Lu Y.; Sun W.-Y. Org. Lett. 2017, 19, 5940–5943. 10.1021/acs.orglett.7b02931. [DOI] [PubMed] [Google Scholar]
- Engle K. M.; Mei T. S.; Wasa M.; Yu J.-Q. Acc. Chem. Res. 2012, 45, 788–802. 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of ether-directed C–H functionalization, see:; a Kawamorita S.; Ohmiya H.; Hara K.; Fukuoka A.; Sawamura M. J. Am. Chem. Soc. 2009, 131, 5058–5059. 10.1021/ja9008419. [DOI] [PubMed] [Google Scholar]; b Liskey C. W.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 12422–12425. 10.1021/ja305596v. [DOI] [PubMed] [Google Scholar]; c Li G.; Leow D.; Wan L.; Yu J.-Q. Angew. Chem., Int. Ed. 2013, 52, 1245–1247. 10.1002/anie.201207770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During the preparation of this paper, the ortho alkynylation of aryl ketones was reported using [IrCp*Cl2]2 (4 mol %), AgNTf2 (16 mol %), NaOAc (30 mol %), and AgOAc (2 equiv) in DCE at 120 °C:Li X.; Wu G.; Liu X.; Zhu Z.; Huo Y.; Jiang H. J. Org. Chem. 2017, 82, 13003–13011. 10.1021/acs.joc.7b01489. [DOI] [PubMed] [Google Scholar]; The alkynylation of esters (six examples) and one example of alkynylation of an α,β-unsaturated ketone were also reported in this study.
- For N,N-dimethylamino-directed C(sp2)–-H functionalization, see:; a Kakiuchi F.; Igi K.; Matsumoto M.; Hayamizu T.; Chatani N.; Murai S. Chem. Lett. 2002, 31, 396–397. 10.1246/cl.2002.396. [DOI] [Google Scholar]; b Cai G.; Fu Y.; Li Y.; Wan X.; Shi Z. J. Am. Chem. Soc. 2007, 129, 7666–7673. 10.1021/ja070588a. [DOI] [PubMed] [Google Scholar]; c Gao D.-W.; Shi Y.-C.; Gu Q.; Zhao Z.- L.; You S.-L. J. Am. Chem. Soc. 2013, 135, 86–89. 10.1021/ja311082u. [DOI] [PubMed] [Google Scholar]
- For thioether-directed C(sp2)–H functionalization, see:; a Yu M.; Xie Y.; Xie C.; Zhang Y. Org. Lett. 2012, 14, 2164–2167. 10.1021/ol3006997. [DOI] [PubMed] [Google Scholar]; b Yao J.; Yu M.; Zhang Y. Adv. Synth. Catal. 2012, 354, 3205–3210. 10.1002/adsc.201200447. [DOI] [Google Scholar]; c Xu B.; Liu W.; Kuang C. Eur. J. Org. Chem. 2014, 2014, 2576–2583. 10.1002/ejoc.201400096. [DOI] [Google Scholar]; d Villuendas P.; Urriolabeitia E. P. Org. Lett. 2015, 17, 3178–3181. 10.1021/acs.orglett.5b01552. [DOI] [PubMed] [Google Scholar]; e Zhang X.-S.; Zhang Y.-F.; Li Z.-W.; Luo F.-X.; Shi Z.-J. Angew. Chem., Int. Ed. 2015, 54, 1–6. [Google Scholar]
- For sulfoxide-directed C(sp2)–H functionalization, see:; a Samanta R.; Antonchick A. P. Angew. Chem., Int. Ed. 2011, 50, 5217–5220. 10.1002/anie.201100775. [DOI] [PubMed] [Google Scholar]; b Hazra C. K.; Dherbassy Q.; Wencel-Delord J.; Colobert F. Angew. Chem., Int. Ed. 2014, 53, 13871–13875. 10.1002/anie.201407865. [DOI] [PubMed] [Google Scholar]; c Nobushige K.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2014, 16, 46–49. [DOI] [PubMed] [Google Scholar]; d For a review, see:Pulis A. P.; Procter D. J. Angew. Chem., Int. Ed. 2016, 55, 9842. 10.1002/anie.201601540. [DOI] [PubMed] [Google Scholar]
- For an example of sulfone-directed C(sp2)–H functionalization, see:Nobushige K.; Hirano K.; Satoh T.; Miura M. Tetrahedron 2015, 71, 6506–6512. 10.1016/j.tet.2015.03.046. [DOI] [Google Scholar]
- For examples of carbamate-directed C(sp2)–H functionalization, see:; a Hoshino Y.; Shibata Y.; Tanaka K. Adv. Synth. Catal. 2014, 356, 1577–1585. 10.1002/adsc.201300884. [DOI] [Google Scholar]; b Zhang X.; Si W.; Bao M.; Asao N.; Yamamoto Y.; Jin T. Org. Lett. 2014, 16, 4830–4833. 10.1021/ol502317c. [DOI] [PubMed] [Google Scholar]; c Morita T.; Satoh T.; Miura M. Org. Lett. 2017, 19, 1800–1803. 10.1021/acs.orglett.7b00569. [DOI] [PubMed] [Google Scholar]
- For an example of phenol ester directed C(sp2)-H functionalization, see:Xiao B.; Fu Y.; Xu J.; Gong T.-J.; Dai J.-J.; Yi J.; Liu L. J. Am. Chem. Soc. 2010, 132, 468–469. 10.1021/ja909818n. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for additional details.
- Wang J.Stereoselective Alkene Synthesis; Springer-Verlag: Berlin, Heidelberg, 2012. [Google Scholar]
- Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 3066–3072. 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- Stuart D. R.; Alsabeh P.; Kuhn M.; Fagnou K. J. Am. Chem. Soc. 2010, 132, 18326–18339. 10.1021/ja1082624. [DOI] [PubMed] [Google Scholar]
- Ma W.; Mei R.; Tenti G.; Ackermann L. Chem. - Eur. J. 2014, 20, 15248–15251. 10.1002/chem.201404604. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- DFT calculations were performed using the Gaussian 09 suite of programs, using wB97XD. Rh, Ag, and Br atoms were described by ECP with the LANL2DZ basis set. Polarization functions were added for Rh (ζf = 1.35), Ag (ζf = 1.611) and Br (ζd = 0.428). The 6-31G(d) basis set was employed for all remaining atoms. Full geometry optimizations were carried out in 1,2-dichloroethane, through an implicit solvent SMD.24
- Sperger T.; Sanhueza I. A.; Kalvet I.; Schoenebeck F. Chem. Rev. 2015, 115, 9532–9586. 10.1021/acs.chemrev.5b00163. [DOI] [PubMed] [Google Scholar]
- Selected discussions of C–H activation mechanisms:; a Qi X.; Li Y.; Bai R.; Lan Y. Acc. Chem. Res. 2017, 50, 2799–2808. 10.1021/acs.accounts.7b00400. [DOI] [PubMed] [Google Scholar]; b Roudesly F.; Oble J.; Poli G. J. Mol. Catal. A: Chem. 2017, 426, 275–296. 10.1016/j.molcata.2016.06.020. [DOI] [Google Scholar]; c Ackermann L. Chem. Rev. 2011, 111, 1315–1345. 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; d Lapointe D.; Fagnou K. Chem. Lett. 2010, 39, 1118–1126. 10.1246/cl.2010.1118. [DOI] [Google Scholar]; e Boutadla Y.; Davies D. L.; Macgregor S. A.; Poblador-Bahamonde A. I. Dalton Trans. 2009, 5820–5831. 10.1039/b904967c. [DOI] [PubMed] [Google Scholar]; f Gorelsky S. I.; Lapointe D.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 10848–10849. 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]; g Oxgaard J.; Tenn W. J. III; Nielsen R. J.; Periana R. A.; Goddard W. A. III Organometallics 2007, 26, 1565–1567. 10.1021/om061189b. [DOI] [Google Scholar]; h García-Cuadrado D.; de Mendoza P.; Braga A. A. C.; Maseras F.; Echavarren A. M. J. Am. Chem. Soc. 2007, 129, 6880–6886. 10.1021/ja071034a. [DOI] [PubMed] [Google Scholar]; i Li L.; Brennessel W. W.; Jones W. D. Organometallics 2009, 28, 3492–3500. 10.1021/om9000742. [DOI] [Google Scholar]
- Vásquez-Céspedes S.; Wang X.; Glorius F. ACS Catal. 2018, 8, 242–257. 10.1021/acscatal.7b03048. [DOI] [Google Scholar]
- a Clot E.; Mégret C.; Eisenstein O.; Perutz R. N. J. Am. Chem. Soc. 2009, 131, 7817–7827. 10.1021/ja901640m. [DOI] [PubMed] [Google Scholar]; b Evans M. E.; Burke C. L.; Yaibuathes S.; Clot E.; Eisenstein O.; Jones W. D. J. Am. Chem. Soc. 2009, 131, 13464–13473. 10.1021/ja905057w. [DOI] [PubMed] [Google Scholar]; c Clot E.; Besora M.; Maseras F.; Mégret C.; Eisenstein O.; Oelckers B.; Perutz R. B. Chem. Commun. 2003, 490–491. 10.1039/b210036n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.