

Abstract
Quantitative pharmacology (QP) applications in translational medicine, drug‐development, and therapeutic use were crowd‐sourced by the ASCPT Impact and Influence initiative. Highlighted QP case studies demonstrated faster access to innovative therapies for patients through 1) rational dose selection for pivotal trials; 2) reduced trial‐burden for vulnerable populations; or 3) simplified posology. Critical success factors were proactive stakeholder engagement, alignment on the value of model‐informed approaches, and utilizing foundational clinical pharmacology understanding of the therapy.
Quantitative pharmacology (QP) discovers and confirms the key drug characteristics to provide clear, reproducible, and predictive evidence for optimizing drug development plans, enabling critical decision‐making, and eventually bringing safe and effective medicines to patients.1 These modeling approaches include empirical, semimechanistic, or quantitative systems pharmacology modeling techniques with the aim to integrate current knowledge regarding the drug, disease, and mechanism, and then to predict (interpolate or extrapolate) outcomes under new conditions such as untested doses and regimens, populations, or diseases.2 As part of the model‐informed drug discovery and development paradigm, QP methods can be employed in all phases of the drug development process from biomarker selection in translational medicine to dose/regimen selection, evidence generation for regulatory approval, and for extrapolation or pharmacoeconomic assessment during therapeutic use.2
Application of QP has been advocated to play an important role in delivering new therapies to patients faster by increasing confidence in decision‐making during drug development and by increasing efficiency through eliminating costs or reducing cycle times. However, most reports to date have summarized the return on investment of using QP approaches primarily in the domain of cost savings and efficiency gains.2, 3, 4 Here, we describe the potential of QP approaches to accelerate patient access to innovative therapies.
Although QP approaches are ardently supported by QP practitioners and their department heads, widespread and consistent appreciation of the value of QP in bringing novel therapies quicker to patients at the right dose is still lacking in the wider community of the pharmaceutical industry and healthcare providers. We think that this gap in appreciation and adoption by the wider community of stakeholders and decision‐makers can be bridged through better communication designed to convey the value of such QP approaches from a patient access and healthcare perspective,2 coupled with wider adoption of good practices among the communities of practitioners.5
IMPACT AND INFLUENCE INITIATIVE
With this goal in mind, the Impact and Influence Initiative of the QP Network of the American Society for Clinical Pharmacology and Therapeutics (ASCPT) crowd‐sourced a compendium of case‐examples demonstrating the innovative applications of QP throughout the drug development process. Thirty‐seven case‐examples were received and grouped into five application areas: Translational Medicine, Drug Development Decision‐making, Regulatory Decision‐making, Therapeutic Use, and Cost‐effectiveness & Differentiation, illustrating the positive impact and tangible value of applied QP in an easily understood and educational format (details are provided in Supplementary Material). From this compendium, eight examples were identified in which QP enabled faster access to innovative medicines for patients through rational dose regimen selection for pivotal registration trials, reducing trial burden for vulnerable populations, or simplification of dosing (posology) for patients and healthcare providers during therapeutic use expansion of the novel therapy (see Table 1 for details on case studies, and Figure 1 as a graphical illustration).
Table 1.
Comprehensive summary of case studies that enabled faster access to innovative medicines for patients (references to the case studies are included in the Supplemental Material)
| Quantitative pharmacology (QP) application area | Compound and title of case study | Key question addressed in case study | Conclusion | Impact on decision making | Estimated time savings by case study owner | Enablers of successful QP application |
|---|---|---|---|---|---|---|
| Model‐informed dose selection for pivotal trials by integrated analysis of totality of data. | Secukinumab: Model‐based selection of the secukinumab doses and dosing regimens for psoriasis patients for the pivotal trials. | What is the optimal dosing regimen to be used in phase III? | Model‐based integration of phase I/II data allowed the selection of two dosing regimens for phase III which had not been tested previously. Phase III confirmed the positive benefit‐risk for those doses and dosing regimens, and these were subsequently approved (150 and 300 mg once weekly for 5 injections, followed by a dose every 4 weeks). | Without M&S, a suboptimal regimen would have been selected that would have led to an inferior benefit‐risk profile, or required further iterations of finding optimal dosing regimen. | > 1 y representing one phase III study duration. Note that the phase III program consisted of 6 studies. In the case of use of inferior dosing regimens, the program may not have delivered a successful outcome that could have been fixed with a single additional study. | Long‐term collaboration with team that created trust and understanding. Comprehensive and diverse phase II data combined with iterative modeling and prospectively predicting study readouts, leading to an iterative learning and confirming cycle. |
| Naloxegol: Modeling and simulation to support Naloxegol dose selection for Phase 3 studies. | What is the optimal dose of Naloxegol and best trial design for clinical phase III trial to confirm efficacy and safety? | The exposure–response analysis at phase II demonstrated that 25 mg was an effective dose with updated primary endpoint. Model‐based simulations suggested that doses of 12.5 mg and higher would provide a promising clinical benefit over placebo. This was subsequently confirmed in the phase III studies, followed by approval. | Prediction of the dose with an optimal risk/benefit ratio. | ∼0.5 year through not generating additional Phase 2 data on the 12.5 mg dose. | Early and active involvement was a key enabler. Continuous engagement with study clinician and statisticians was important to win their trust. | |
| Pembrolizumab: Modeling and simulation drove the selection of pembrolizumab efficacious dose of 2 mg/kg. | What is the efficacious dose and dosing regimen of pembrolizumab? | Translational, clinical pharmacokinetics and exposure–response analyses demonstrated that 2 mg/kg Q3W achieved a maximal response and that 10 mg/kg Q3W dose and regimen did not produce further improvement. The 2 mg/kg Q3W was subsequently approved for patients with advanced metastatic melanoma. This dose, however, was not studied in the pivotal trial (KEYNOTE‐006) but justified through integrated analysis of the totality of the data of other trials (KEYNOTE‐001,002 and 006). | M&S enabled approval at a lower dose (2 mg/kg Q3W) than studied in the pivotal trial (10 mg/kg Q3W) for ipilimumab naïve metastatic melanoma patients through integrated analysis of clinical data. | ∼ 6 months to 1 year by avoiding addition of a dose arm of 2 mg/kg Q3W to the pivotal trial. | Studying a wide dose range in initial trials allowed bridging through understanding of the exposure response relationship. | |
| Reducing trial burden for vulnerable populations by model‐informed trial design, dose selection and replacement. | Raltegravir: Adaptive trial design to define raltegravir dosing regimen to treat neonates from birth up to 6 weeks of age. | How should the dramatic increase of clearance due to UGT‐1A1 maturation be addressed in in a 6‐week dosing regimen for neonates? | The dramatic increase in raltegravir clearance as the result of UGT‐1A1 enzyme activity in neonates requires 2 dose changes over the first 6 weeks of life to meet efficacy and safety PK criteria. | Traditional paradigm of dose ranging followed by confirmatory study was changed into a single study in which M&S was used to integrate data to adapt dosing recommendations and update predictions. | ∼3–4 y by removing 1 study in a vulnerable population that would be very slowly in recruitment. | Both company and regulator supported the use of modeling and simulation approaches, which resulted in shortened development time. |
| Ivabradine: Modeling and simulation for clinical studies in pediatric populations. | How to determine the starting dose in children suffering from chronic heart failure? | This work emphasizes the importance of modeling and simulation for internal decision‐making such as the design of clinical studies in pediatric populations. With this work, it was possible to determine the starting dose in children and to define a lower weight‐normalized dose in younger children since they presented a higher exposure compared to adults and thus avoiding the risk of excessive bradycardia. | M&S supported to pick a higher weight‐normalized dose in children older than 1 year and thus quicker titration avoiding the risk of insufficient efficacy in children. Inclusion of dry‐blood spot sampling reduced additional trial burden. | ∼ 1 y through reduced need for more extensive dose ranging. | Early interaction with the pediatric committee at the European Medical Agency. In addition, good examples needed to be shared to convince stakeholders. | |
| Eslicarbazepine acetate (ESL): Extrapolation strategy for ESL dosing in pediatric patients with partial‐onset seizures (POS). | Which doses of ESL provide exposures in 4‐17 y patients with POS that are similar to those determined to be safe and effective in adult patients for ESL adjunct therapy or monotherapy? | Extrapolation obviated the need to conduct a US‐based clinical trial in pediatric patients aged ≥4 y. Benefits of this strategy were to reduce the number of pediatric patients exposed to clinical trials and to allow for earlier availability of ESL for clinical use in pediatric patients. | Reduced number of patients exposed in clinical trial, earlier availability for children. | ∼2–3 years by obviating a confirmatory efficacy trial in pediatric patients. | Early clear and transparent interaction with stakeholders. Inclusion of experts to discussions in project team. The collection of sufficient pharmacokinetic data in children in age range of interest. | |
| Simplified dosing for patients and physicians by model‐informed bridging of weight‐based and flat dosing regimens. | Nivolumab: Quantitative clinical pharmacology analyses conducted in support of 240 mg nivolumab flat dose approval. | Can quantitative clinical pharmacology approaches be used to switch body weight based dosing (3 mg/kg Q2W) to simplified, flat dose (240 mg Q2W) of nivolumab? | The quantitative clinical pharmacology approach provided evidence for regulatory decision‐making on dose modification, obviating the need for an independent clinical study. | Demonstration of efficacy and safety was similar for flat dose as mg/kg dose for wide range of body weight. | >2 y through avoiding a flat dose trial and simplifying study design for expanding therapeutic use to other indications. | Thorough understanding of pharmacokinetics, pharmacodynamics, efficacy and covariates across multiple tumor types and multiple endpoints. Being proactive, demonstrating potential value through exploratory analysis in time. |
| Ixazomib: Switch from body‐surface area (BSA)‐based to fixed dosing led to simplified dosing guidance and clinical development. | Can modeling guide switching from BSA‐based to fixed dosing without conducting a standalone study to compare fixed dosing vs BSA‐based dosing? | Clinical development switched posology from BSA‐based to fixed dosing, simplifying capsule strength manufacture and dosing in global clinical trials. Fixed dose of 4 mg was subsequently used in the phase III (TOURMALINE MM1) study that formed basis for approval of ixazomib by FDA and EMA. | Modeling helped challenge the established practice of weight based dosing in oncology. Simplified dosing for patients and physicians reducing pill burden and avoiding dosing errors. | ∼1–2 years through avoiding a trial for fixed dose. | Early engagement with team, good understanding of intrinsic/extrinsic factors and consistent education of all stakeholders (including oncology key opinion leaders) was critical for success. |
Figure 1.
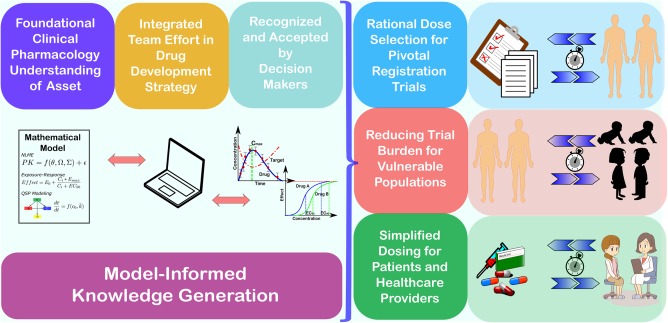
Quantitative pharmacology (QP) is an enabler for faster access to innovative medicines as described through case studies. For the first registration of a novel therapy, QP can accelerate patients' access through rational dose regimen selection for the pivotal registration trials by a self‐estimated ∼0.5–1 year. Moreover, QP can facilitate reducing trial burden for vulnerable populations through effective translation of efficacious concentrations between adult and children and bringing the treatment options ∼1–4 years earlier to children. Finally, QP can enable simplification of dosing (posology) during therapeutic use expansion of a novel therapy. Solid understanding of the clinical pharmacology of the novel therapy, integrated into model‐informed knowledge, effective communication of QP learnings and results to project teams and stakeholders, and alignment on the value of model‐informed learnings by decision makers are all critical factors for QP to impact and influence the drug development process and ultimately bringing novel therapies faster to patients at the right dose.
RATIONAL DOSE SELECTION FOR PIVOTAL REGISTRATION TRIALS
QP played a critical role in accelerating access to novel medicines by incorporating information from all available trials, allowing to select optimal doses and dosing regimens for pivotal confirmatory trials based on exposure‐related benefit/risk analysis. In addition, QP approaches were utilized to seek regulatory approval for a dose that had not been tested in a pivotal trial.
Consider the case of the interleukin‐17A antagonist secukinumab for the treatment of psoriasis, where mathematical modeling of integrated data from all phase I and II studies gave the team the confidence to select dosing regimens for the phase III trials that had not been tested previously. Phase III confirmed the positive benefit–risk for the QP‐informed dosing regimens, which were subsequently approved. A key learning from this successful application of QP was that both long‐term planning, as well as involvement and commitment from colleagues with a range of skill‐sets (clinicians, statisticians, biologists, modelers, etc.) was required for building and applying the model, in combination with building trust with the stakeholders and decision makers about the predictive capabilities of the model.
Similarly, for naloxegol, a selective opioid receptor antagonist for treatment of opioid‐induced constipation, an untested lower dose than what had previously been tested was brought into phase III based on a QP approach that predicted an improved benefit–risk for the lower dose. The modeling approach here reduced the number of doses tested and increased the probability of success of the pivotal trial.
In an even more impactful example, pembrolizumab, a potent antibody against PD‐1 receptor, was approved in the early days for ipilimumab‐naïve metastatic melanoma patients at a 2 mg/kg Q3W dose. This dose, however, was not studied in the pivotal trial, but justified through integrated analysis of the totality of the data from available trials at the time. Since then, the label has been updated with respect to a fixed dose posology of 200 mg and additional indications.
In each of these three case examples, application of a model‐informed approach for dose selection and justification enabled patients to have access to the medicine earlier (∼0.5–1 year, estimated by case‐study owners) than using a solely data‐driven approach.
REDUCING TRIAL BURDEN FOR VULNERABLE POPULATIONS
Special populations such as pediatrics have immensely benefited from QP with model‐informed selection of starting doses and trial designs leading to reduced burden on pediatric patients by optimizing the information gained in small sample sizes. These benefits are clearly demonstrable for drugs like raltegravir (for neonates), ivabradine (in pediatrics), and eslicarbazepine acetate (ESL, for children 4–17 years).
Consider the case of raltegravir, an antiretroviral to treat HIV infections, which is in development for the neonatal population. Neonates undergo dramatic changes in metabolic enzyme maturation in their first weeks of life. In an adaptive design, with very limited pharmacokinetic (PK) data in a small number of neonates, the PK profiles of neonates after dosing were modeled to quantify and understand the impact of a dramatic age‐related change in clearance of raltegravir. With this understanding, the team proposed two changes in dose regimen to achieve the desired exposure in newborns. This approach abrogated the need for sequential dose‐ranging and confirmatory studies which typically take 3–4 years to complete. Thus, an integrative model‐informed approach accelerated the availability of treatment for a vulnerable population that is extremely difficult to study using traditional clinical trial designs.
A similar example with ivabradine shows that modeling defined a lower weight‐normalized starting dose in younger children suffering from chronic heart failure based on drug exposure, avoiding the risk of bradycardia related to supratherapeutic exposure. In older children, a higher weight‐normalized dose with quicker titration was selected that avoided a lengthier study with unnecessary periods of ineffective subtherapeutic exposure.
For eslicarbazepine acetate, a modeling and simulation approach was made to gain approval for use in the pediatric population ranging from 4–17 years through extrapolation of drug exposures. After determining the pharmacokinetics in children, the team predicted the dosing regimen that provides similar drug exposure in pediatric patients 4 years of age and older and in adult patients with Partial Onset Seizures (POS). In such a way, this modeling replaced an independent pediatrics efficacy study, and thereby allowed earlier access for pediatric patients with POS.
In an era where serving the pediatric population is of utmost importance, these examples demonstrate how quantitative pharmacology can help us fulfill that goal more rapidly, while reducing the risk of either over‐ or underdosing these vulnerable patients in the context of the study.
SIMPLIFIED DOSING FOR PATIENTS AND HEALTHCARE PROVIDERS
QP has been instrumental in simplifying dosing for both patients and healthcare providers. For example, the flat dosing regimen for nivolumab, a fully‐human anti‐PD1 monoclonal antibody for the treatment of a variety of cancers, shortens drug preparation time and improves ease of administration. Integrated modeling and simulation of phase I–III data played a critical role in demonstrating that the antitumor activity plateaued at 3 mg/kg, and that the initially approved 3 mg/kg Q2W body weight‐based dosing could be safely replaced with a 240 mg nivolumab flat dose without generating new clinical data. The approval of the flat dose resulted in self‐estimated savings of ∼2 years in bringing simplified prescribing, shortened manufacturing time, and, most important, ease of administration to patients and healthcare providers for several cancer indications. As with other examples, this work required proactive planning and a clear understanding of the PK properties of the drug, together with the relationship of exposure to both efficacy and tolerability.
Similarly, modeling of early clinical data for ixazomib, an oral agent for the treatment of multiple myeloma, showed that there was no relationship between clearance and body surface area (BSA), suggesting that a fixed dose could be more appropriate. Based on these findings, phase III studies were conducted with a fixed dose, ultimately reflected in global labeling.
Thus, QP has played a key role in simplified dosing for multiple drugs across a variety of indications, leading to reduced pill burden for patients (for oral therapies) and potentially lower dosing errors through simplification of infusion preparation for healthcare providers. Moreover, both examples also helped challenge the established belief of BSA or a body weight‐based dose being the norm in oncology—a paradigm shift that may further accelerate simplified treatment options for cancer patients.
COMMON ENABLERS OF SUCCESS
As with most success stories in drug development, these examples all took substantial time, expertise, planning, and collaboration. Several common characteristics could be identified that helped the modeling teams to impact decisions by the broader project team, as illustrated in Figure 1. For all case studies, it was crucial for modeling teams to build confidence in the techniques internally, through proactive discussions during strategic planning, clear communication of the value proposition, alignment on the approach, and exploratory analysis to demonstrate potential benefits. Also, early advisory interactions with regulators were found to be critical to achieve alignment on the approach. Furthermore, robust characterization of intrinsic and extrinsic factors impacting relationships between drug levels and efficacy/safety were crucial for developing quantitative frameworks to maximize the value of information and enable extrapolation of benefit/risk across settings of clinical use. Finally, regular, open, and honest communications with team members regarding the strengths and weaknesses of a model‐informed approach were found to be a critical ingredient to the successful outcome demonstrated in these examples. Here it is equally important for modelers to be able to explain their work and its impact in clear, concise language, as it is for decision makers to request project teams to bring forward these approaches to ensure the most cost‐effective and ethical way to bring novel therapies to patients.
The value proposition for QP as a core enabler for patient‐centric therapeutic development is clearer than ever in the context of value‐based healthcare. An organizational culture of commitment to a QP paradigm as an integral component of asset strategy and decision‐making is foundational to accelerating the availability of innovative therapeutics with rational and simplified dosing and optimized benefit/risk across patient subsets and clinical contexts of use
ACKNOWLEDGMENTS
The authors thank each of the many contributors to the individual case studies mentioned in this article. Also, the ASCPT QP network leads, Anne C. Heatherington, Karthik Venkatakrishnan, and Sriram Krishnaswami are gratefully acknowledged for sponsoring the initiative and for their inspiring discussions during this work.
CONFLICT OF INTEREST
All authors are employees of the stated companies, and have nothing to disclose beyond their affiliation. Details of the work described in case studies are referenced in the Supplemental Material.
AUTHOR CONTRIBUTIONS
S.N. and S.V. wrote the article. O.S., N.A.‐H., D.A., A.C., M.C., S.S., S.A., and N.G. were contributors to case study details. All authors carefully reviewed and approved the final article.
Supporting information
Additional supporting information can be found in the online version of this article.
Supplementary Material
References
- 1. Zhang, L. et al Model‐based drug development: the road to quantitative pharmacology. J. Pharmacokinet. Pharmacodyn. 33, 369–393 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Marshall, S. et al Good practices in model‐informed drug discovery and development (MID3): practice, application and documentation. EFPIA MID3 workgroup. CPT Pharmacodynamics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Milligan, P.A. , Brown, M.J. , Marchant, B. , Martin, S.W. , van der Graaf, P.H. & Benson, N. Model‐based drug development: a rational approach to efficiently accelerate drug development. Clin. Pharmacol. Ther. 93, 502–514 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Allerheiligen, S.R. Impact of modeling and simulation: myth or fact? Clin. Pharmacol. Ther. 96, 413–415 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Visser, S.A. , Norton, J. , Marshall, S. & O'Kelly, M. Common best practice in modeling & simulation across quantitative disciplines: a comparison of independently emerging proposals. Stat. Biopharm. Res. doi: 10.1080/19466315.2017.1385520 (2017) [Epub ahead of print]. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.
Supplementary Material