

Abstract
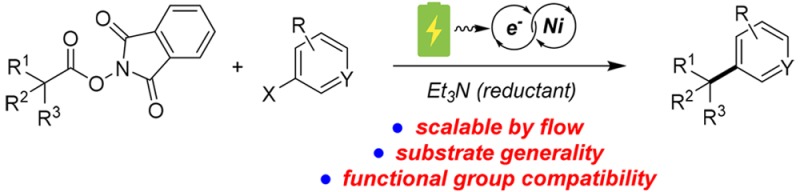
An electrochemically driven, nickel-catalyzed reductive coupling of N-hydroxyphthalimide esters with aryl halides is reported. The reaction proceeds under mild conditions in a divided electrochemical cell and employs a tertiary amine as the reductant. This decarboxylative C(sp3)–C(sp2) bond-forming transformation exhibits excellent substrate generality and functional group compatibility. An operationally simple continuous-flow version of this transformation using a commercial electrochemical flow reactor represents a robust and scalable synthesis of value added coupling process.
The formation of Csp3–Csp2 bonds is a powerful means of synthesizing high-value chemicals.1 Nickel catalysis has offered numerous avenues to Csp3–Csp2 bond assembly via cross coupling of Csp3 electrophiles. Most methods make use of organometallic compounds as transmetalating agents, which are reminiscent of classic Kumada, Negishi, and Suzuki couplings.2 The needs of using these highly sensitive, less functional group compatible organometallic reagents necessitated the development of additional modes of nickel-catalyzed Csp3–Csp2 bond formation using native functional groups as latent nucleophiles. These include cross-coupling methods that combine single-electron-transfer catalytic cycles of nickel with iridium photoredox cocatalysts3 and cross-electrophile reductive couplings relying on an exogenous, stoichiometric metallic powder reductant (e.g., Zn, Mn) to transfer electrons to nickel catalysts.4 Herein, we report a novel and powerful approach for electrochemical generation of highly reactive radicals and their incorporation into a Ni-catalyzed Csp3–Csp2 coupling manifold to construct structurally diverse molecules in both batch and continuous flow.
One seminal example in this field reported by MacMillan and Doyle elegantly utilized an Ir-photoredox/Ni dual catalyst system to achieve decarboxylative Csp3–Csp2 coupling (Figure 1). The requirement of an α-heteroatom in the starting material carboxylic acid limits its broad use of inexpensive and readily available aliphatic carboxylic acids. To utilize more general carboxylic acids, Weix group demonstrated a complementary approach of Ni-catalyzed cross coupling of N-hydroxyphthalimide (NHP) esters in the presence of zinc powder as a reducing agent.5 Nonetheless, reactive metal powders are generally challenging to work with, in particular on large scales due to purity, surface oxidation, and safety issues. Mechanistically, many electron-transfer steps in photoredox-catalytic cycle or chemical reduction parallels electrochemical processes. Instead of intrinsically less efficient electron-photon-electron conversion (e.g., LED light) or stoichiometric amounts of metal reductants, we thought direct reduction of NHP esters using electric current would be a much more energy efficient and safer method to introduce reactive radical species into a catalytic cycle.6
Figure 1.
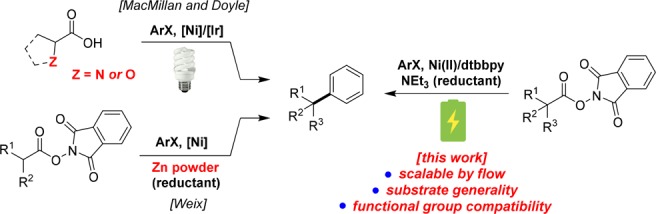
Nickel-catalyzed sp3–sp2 cross-couplings using carboxylic acids/derivatives and aryl halides as coupling partners.
Interest in electrochemical methods has recently been revived as an attractive solution to lingering problems in organic synthesis.7 By tuning the applied potential and current, electrochemistry offers precise, selective formation and regulated generation of reactive species, enabling predictable and controllable chemoselectivity.8 Such advantages render electrochemistry a sustainable, economically practical, and environmentally benign technique for chemical synthesis.
We reasoned that cathodic reduction of the redox active NHP ester 1(9,10) would result in a decarboxylative fragmentation resulting in generation of the Csp3 radical 2 (Figure 2). Observation of an irreversible reduction using cyclovoltametry of model substrate 4 at −1.2 V (vs Ag/AgCl) supported this initial hypothesis. Alkyl radical 2 is then intercepted by a homogeneous nickel catalyst, which could be either Ni(0) or Ni(II) generated from oxidative addition of aryl halides. The challenge in this case is whether or not a highly reactive radical species generated on a solid-state electrode surface would have sufficient lifetime to interact with nickel catalyst before Kolbe dimerization or sequential over reduction into carbanion. Upon generation of Ni(III) species 3, the alkyl and aryl groups are expected to undergo reductive elimination to form the Csp3–Csp2 coupling product. The resulting Ni(I) would be reduced at the cathode to Ni(0), thus closing the catalytic cycle. Notably, we propose to use electron-rich tertiary amines as sacrificial reductants, which would easily undergo anodic oxidation and donate electrons to the electrochemical system.11
Figure 2.
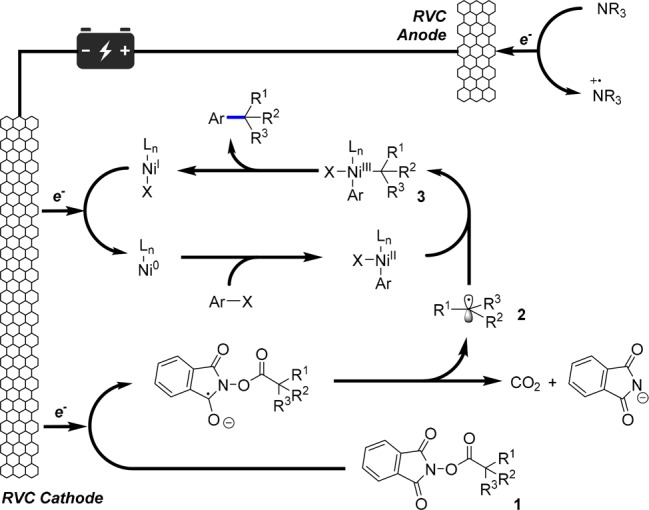
Proposed electrochemical-driven nickel-catalyzed decarboxylative arylation unit steps.
With this mechanistic hypothesis in mind, we set out to explore the feasibility of our proposed decarboxylative arylation reaction (Table 1). Our investigations began with the coupling between NHP ester 4 and iodobenzene using triethylamine as the sacrificial reductant, reticulated vitreous carbon (RVC) as cathode and anode material, and with maximum potential (V) and current (A) output set at 10 V and 20 mA, respectively. Initial experiments (Table 1) indicate that the reaction proceeds with superior chemoselectivity for the desired coupling pathway versus Kolbe-type coupling in a divided H-cell over an undivided cell. The superior performance in a divided cell is likely due to competitive anodic oxidation of low-valent nickel species in an undivided cell (entries 1 and 2). Diminished yield and chemoselectivity (entry 3) occurred when nickel foam was used as the cathode material compared to using RVC. A cursory solvent and ligand screening did not afford improved reaction outcome (entries 4–7) from the original condition. Increasing the catalyst loading of NiII/dtbbpy combination to 30 mol % promoted the yield of 5 (entry 8). Further optimization of the electrolyte counteranion identified PF6– as providing an optimal yield of 5 and high chemoselectivity (74%, > 10:1, entry 10).12 As expected, a control experiment performed in the absence of electron current resulted in no product formation. Performing the reaction without a nickel catalyst present resulted in formation of Kolbe coupling dimer 6 along with a significant amount of hydrocinnamic acid 7 (entries 11 and 12). It is worth noting that formation of 7 was observed under all productive conditions (entries 1–10, 12). Water content of the reaction mixture (∼100 ppm) only accounted for ∼6 mol % of 7. The results suggest a competitive electrochemically mediated pathway is presumably responsible for the formation of 7.
Table 1. Initial Studies toward Nickel-Catalyzed Electrochemical Decarboxylative Arylation Reactions.
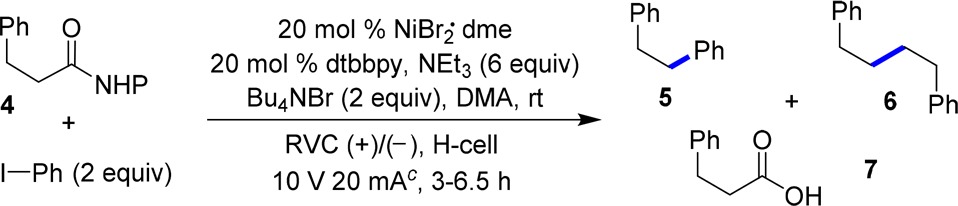
| yieldb (%) |
||||
|---|---|---|---|---|
| entrya | deviation from above | 5 | 6 | 7 |
| 1 | none | 54 | 11 | 22 |
| 2 | undivided cell | 6 | 39 | 18 |
| 3 | nickel foam cathode | 41 | 23 | 21 |
| 4 | DMF instead of DMA | 49 | 20 | 21 |
| 5 | CH3CN instead of DMA | 48 | 26 | 19 |
| 6 | bpy instead of dtbbpy | 47 | 23 | 24 |
| 7 | dmebpy instead of dtbbpy | 49 | 14 | 25 |
| 8 | 30 mol % NiII/dtbbpy | 67 | 9 | 18 |
| 9 | 30 mol % NiII/dtbbpy, Bu4NClO4 | 65 | 10 | 15 |
| 10 | 30 mol % NiII/dtbbpy, Bu4NPF6 | 74 | 6 | 14 |
| 11 | no electricity | 0 | 0 | 0 |
| 12 | no NiBr2·glyme | 0 | 44 | 38 |
2 mmol scale.
Yield determined by calibrated HPLC assay.
Maximum potential and current output set at 10 V and 20 mA.
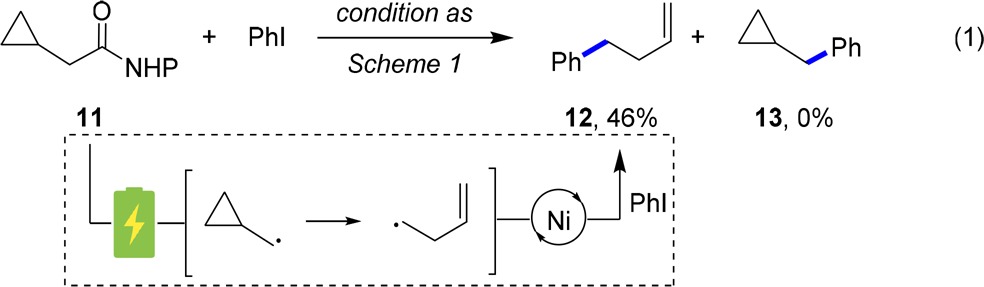
With optimized conditions identified, we then explored the reaction scope (Scheme 1). The reaction tolerated a range of primary and secondary NHP esters, including esters derived from natural products (e.g., 10q, 10r). NHP esters with constrained α-carboxyl quaternary sp3 centers can be incorporated with good efficiency (10f), but nonconstrained quaternary centers are beyond the range of the present system (10g). Various electron-neutral and electron-deficient (hetero)aryl halides are suitable partners and coupled efficiently (e.g., 10h–l). Electron-rich 4-iodoanisole was also amenable to the current electrochemical condition (10m), albeit in modest yield. The mild nature of the current protocol facilitates broad functional group compatibility, tolerating ketone (10p), esters (10o), alkene (10q), protected nitrogen (10b, 10c, 10r), and boronic ester (10r) moieties. Benzylic alcohol (10n) and terminal alkyne (10s) containing substrates, however, are not compatible with the current reaction conditions.
 |
1 |
Scheme 1. Reaction Scope of Nickel-Catalyzed Electrochemical Decarboxylative Arylations.
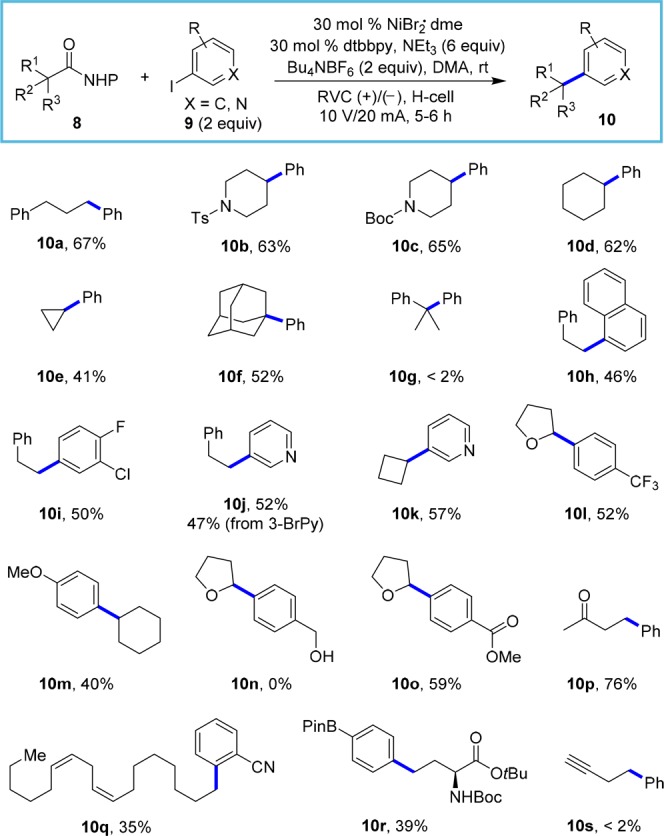
The intermediacy of radical species 2 was supported by a radical-clock experiment (eq 1). Coupling of NHP ester 11 with iodobenzene provided 12 exclusively in 46% yield, which is consistent with rapid ring opening of the intermediate cyclopropylmethyl radical resulting from decarboxylative fragmentation of the carboxyl radical.
The kinetic profile of the reaction of 4 with iodobenzene was examined to gain insight into the decarboxylative coupling reaction (entry 1, Table 1). As shown in Figure 3, very little coupling product 5 was observed during the initial hour of electrolysis. The major cathodic event is likely the reduction of NiII to NiI/Ni0 species. This is supported by the observation of a reduction event at −0.8 V in a cyclic voltammetry experiment (vs Ag/AgCl). Such a voltage is below the reductive potential of both NHP ester 4 at −1.2 V and iodobenzene at −2.3 V (see the Supporting Information). The onset of product 5 formation appears around 70 min, at which time 0.79 mmol of electrons have been passed through the system. This value is very close to the theoretical amount of electrons (i.e., 0.8 mmol) required to completely reduce NiII to Ni0. Interestingly, neither the dimer 6 nor hydrocinnamic acid 7 is observed until about 3 h of electrolysis. This observation suggests that deactivation of the nickel catalyst occurs after ∼3 h of electrolysis. As indicated in Figure 3, the production rate of 6 and 7 increases while the rate of the desired pathway decreases. This regime is reminiscent of the reaction pathway devoid of nickel (entry 12, Table 1).
Figure 3.
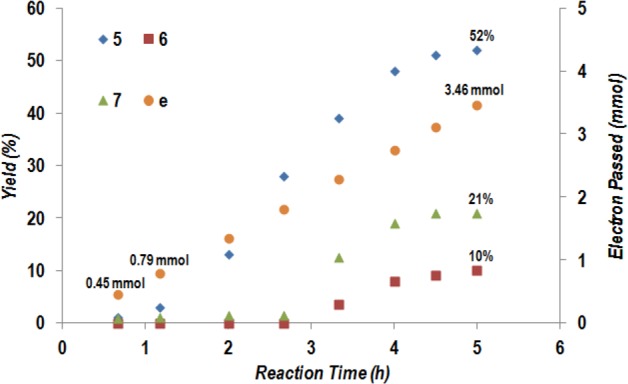
Kinetic profile of decarboxylative coupling between NHP ester and iodobenzene (“e” denotes electron).
Organic electro-synthesis carried out in laboratory batch cells (e.g., H-cells) represents a useful approach for small scale (<5 mmol) electrochemical reactions but presents limitations for larger scale production. One potential approach to address practically and economically the scalability issue is operation under continuous flow conditions.13 Owing to the decreased interelectrode distance and increased interfacial ratio of electrode versus reaction stream within an electrochemical flow cell, the efficiency of mass, energy, and electron transfer may be enhanced significantly.
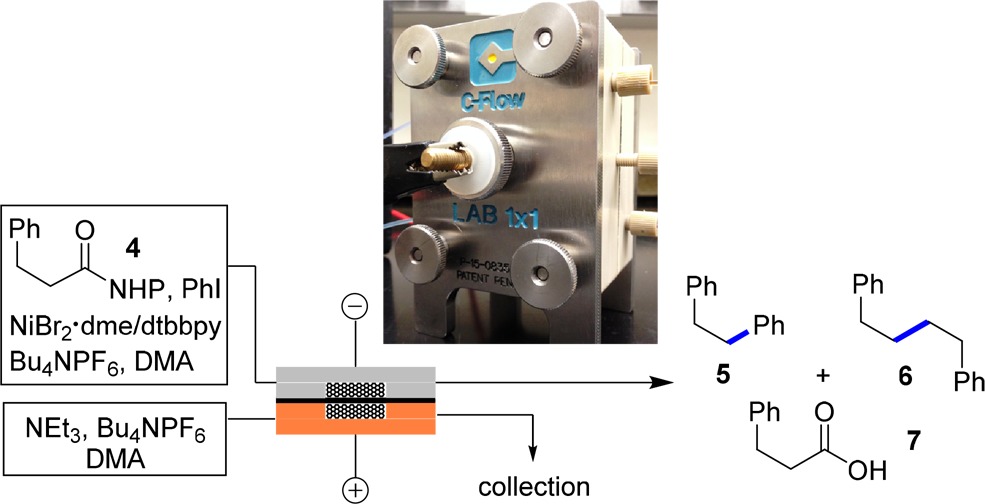
To examine these potential advantages, the electrochemical nickel-catalyzed decarboxylative reactions were evaluated under flow conditions (Table 2). RVC foam pieces were attached to the anode and cathode graphite plates of a C-flow electrochemical flow cell, to provide increased surface area of both electrodes (see SI). A cathodic stream (containing NHP ester 4, iodobenzene, NiII/dtbbpy, and Bu4NPF6) and an anodic stream (containing triethylamine and Bu4NPF6) were flowed into the electrochemical cell. A Nafion membrane was utilized to separate the two streams, allowing cross-membrane flow of ions but not neutral molecules.14 Adjustment of flow rates at a given current density allowed full conversion of NHP ester 4 after a single pass through the cell. Increased flow rates showed a positive effect on the chemoselectivity to favor decarboxylative arylation over Kolbe-type dimerization. This is likely a result of improved mixing at increased flow rates (see the SI). With a current density of 38 mA per cubic centimeter of RVC carbon foam (100 ppi), 55% of decarboxylative arylation product 5 was produced along with significant amount of dimer 6 (33%, entry 1). This indicates that the rate of homobenzyl radical generation is faster than the nickel-based catalytic cycle turnover frequency. We then fine-tuned the current density to match more closely the kinetics of these two chemical events. Indeed, improved chemoselectivity occurred at lower current density (entries 2 and 3). Under the optimized condition, 81% of 5 was furnished at a current density of 14 mA/cm3 and 8.3 min residence time, as compared to 74% yield obtained in batch (H-cells) with 3–4.5 mA/cm3 current density and 410 min residence time (entry 3 vs 4). Interestingly, in continuous flow, the formation of byproduct 7 was suppressed (entries 1–3 vs 4).
Table 2. Continuous-Flow Ni-Catalyzed Electrochemical Decarboxylative Arylations.
| yieldd (%) |
||||||
|---|---|---|---|---|---|---|
| entrya | reactor | j (mA/cm3)b | tRc (min) | 5 | 6 | 7 |
| 1 | flow cell | 38 | 3.8 | 54 | 33 | 8 |
| 2 | flow cell | 29 | 4.2 | 62 | 17 | 10 |
| 3 | flow cell | 14 | 8.3 | 81 | 6 | 8 |
| 4 | H-celld | 3–4.5 | 410 | 74 | 6 | 14 |
Cathode stream: 4 (0.1 M), PhI (2 equiv) NiBr2 dme/dtbbpy (30 mol %), Bu4NPF6 (0.2 M); anode stream: NEt3 (6 equiv), Bu4NPF6 (0.2 M).
j = current density, mA per cubic centimeter of RVC carbon foam (100 ppi).
Internal wet volume of anode or cathode flow cell is 0.5 mL.
Yield determined by calibrated HPLC assay.
In summary, we have demonstrated that electrochemically generated, highly reactive, and short-lived radical species on a solid-state electrode surface can be selectively incorporated into a transition-metal catalysis manifold to form high-complexity molecules productively. These electrochemically driven, Ni-catalyzed decarboxylative Csp3–Csp2 cross-reductive couplings of NHP ester with aryl halides exhibit broad substrate compatibility, tolerance of a range of sterically and electronically diverse coupling partners. Preliminary studies demonstrated improved reaction performance and efficiency when carried out under continuous flow conditions and delivered a method that is amenable to rapid scale-up. The results reported herein provide a new and practical method for the use of carboxylic acids as precursors to C–C bond-forming reactions and allude to a broad range of potential similar reaction manifolds that are currently being pursued in our laboratories.
Acknowledgments
Financial support of this work was partially provided by an SBIR grant from NSF (164576). C.P.B, H.S., and T.F.J. also thank the Bill and Melinda Gates Foundation (“Medicine for All” Initiative) for financial support. H.S. thanks Amgen Graduate Fellowship in Synthetic Chemistry.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.8b00070.
Experimental procedure and analytical data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Metal-catalyzed Cross-coupling Reactions; Diederich F., Stang P. J., Eds.; Wiley-VCH: New York, 1998. [Google Scholar]; b Jana R.; Pathak T. P.; Sigman M. S. Chem. Rev. 2011, 111, 1417. 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Manolikakes G. 3.08 Coupling Reactions Between sp3 and sp2 Carbon Centers A2. In Comprehensive Organic Synthesis II, 2nd ed.; Knochel P., Molander G. A., Eds.; Elsevier: Amsterdam, 2014; pp 392–464. [Google Scholar]
- For representative reviews, see:; a Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tellis J. C.; Kelly C. B.; Primer D. N.; Jouffroy M.; Patel N. R.; Molander G. A. Acc. Chem. Res. 2016, 49, 1429. 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fu G. C. ACS Cent. Sci. 2017, 3, 692. 10.1021/acscentsci.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lucas E. L.; Jarvo E. R. Nature Rev. Chem. 2017, 1, 0065. 10.1038/s41570-017-0065. [DOI] [Google Scholar]
- For representative reviews, see:; a Twilton J.; Le C. C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. Nature Rev. Chem. 2017, 1, 0052. 10.1038/s41570-017-0052. [DOI] [Google Scholar]; For representative examples, see:; b Zuo Z.; Ahneman T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; For representative examples of using organoborons as nucleophiles, see:; c Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For representative reviews, see:; a Weix D. J. Acc. Chem. Res. 2015, 48, 1767. 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selected examples, see:; b Everson D. A.; Shrestha R.; Weix D. J. J. Am. Chem. Soc. 2010, 132, 920. 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; c Wang X.; Wang S.; Xue W.; Gong H. J. Am. Chem. Soc. 2015, 137, 11562. 10.1021/jacs.5b06255. [DOI] [PubMed] [Google Scholar]; For representative example of nonmetallic reductant, see:; d Suzuki N.; Hofstra J. L.; Poremba K. E.; Reisman S. Org. Lett. 2017, 19, 2150. 10.1021/acs.orglett.7b00793. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Anka-Lufford L. L.; Huihui K. M. M.; Gower N. J.; Ackerman L. K. G.; Weix D. J. Chem. - Eur. J. 2016, 22, 11564. 10.1002/chem.201602668. [DOI] [PubMed] [Google Scholar]; f Kuroboshi M.; Waki Y.; Tanaka H. Synlett 2002, 2002, 637. 10.1055/s-2002-22719. [DOI] [Google Scholar]
- Huihui K. M. M.; Caputo J. A.; Melchor Z.; Olivares A. M.; Spiewak A. M.; Johnson K. A.; DiBenedetto T. A.; Kim S.; Ackerman L. K. G.; Weix D. J. J. Am. Chem. Soc. 2016, 138, 5016. 10.1021/jacs.6b01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambiagio C.; Sterckx H.; Maes B. U. W. ACS Cent. Sci. 2017, 3, 686. 10.1021/acscentsci.7b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For pioneering studies, see:; a Durandetti M.; Nedelec J.; Perichon J. J. Org. Chem. 1996, 61, 1748. 10.1021/jo9518314. [DOI] [PubMed] [Google Scholar]; For representative reviews, see:; b Chaussard J.; Folest J.; Nedelec J.; Perichon J.; Sibille S.; Troupel M. Synthesis 1990, 1990, 369. 10.1055/s-1990-26880. [DOI] [Google Scholar]; c Horn E. J.; Rosen B. R.; Baran P. S. ACS Cent. Sci. 2016, 2, 302. 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]; For representative examples, see:; d Perkins R. J.; Pedro D. J.; Hansen E. C. Org. Lett. 2017, 19, 3755. 10.1021/acs.orglett.7b01598. [DOI] [PubMed] [Google Scholar]; e Frankowski K. J.; Liu R.; Milligan G. L.; Moeller K. D.; Aube J. Angew. Chem., Int. Ed. 2015, 54, 10555. 10.1002/anie.201504775. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Horn E. J.; Rosen B. R.; Chen Y.; Tang J.; Chen K.; Eastgate M. D.; Baran P. S. Nature 2016, 533, 77. 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kawamata Y.; Yan M.; Liu Z.; Bao D. – H.; Chen J.; Starr J.; Baran P. S. J. Am. Chem. Soc. 2017, 139, 7448. 10.1021/jacs.7b03539. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Fu N.; Sauer G. S.; Saha A.; Loo A.; Lin S. Science 2017, 357, 575. 10.1126/science.aan6206. [DOI] [PubMed] [Google Scholar]; i Fu N.; Sauer G. S.; Lin S. J. Am. Chem. Soc. 2017, 139, 15548. 10.1021/jacs.7b09388. [DOI] [PubMed] [Google Scholar]; j Yang Q.; Li Y.; Ma C.; Fang P.; Zhang X.; Mei T. J. Am. Chem. Soc. 2017, 139, 3293. 10.1021/jacs.7b01232. [DOI] [PubMed] [Google Scholar]; k Xiong P.; Xu H.; Xu H. J. Am. Chem. Soc. 2017, 139, 2956. 10.1021/jacs.7b01016. [DOI] [PubMed] [Google Scholar]; l Li C.; Kawamata Y.; Nakamura H.; Vantourout J. C.; Liu Z.; Hou Q.; Bao D.; Starr J. T.; Chen J.; Yan M.; Baran P. S. Angew. Chem., Int. Ed. 2017, 56, 13088. 10.1002/anie.201707906. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Yan M.; Kawamata Y.; Baran P. S. Chem. Rev. 2017, 117, 13230. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida J.; Kataoka K.; Horcajada R.; Nagaki A. Chem. Rev. 2008, 108, 2265. 10.1021/cr0680843. [DOI] [PubMed] [Google Scholar]
- For pioneering studies, see:; a Okada K.; Okamoto K.; Oda M. J. Am. Chem. Soc. 1988, 110, 8736. 10.1021/ja00234a047. [DOI] [Google Scholar]; b Okada K.; Okamoto K.; Morita N.; Okubo K.; Oda M. J. Am. Chem. Soc. 1991, 113, 9401. 10.1021/ja00024a074. [DOI] [Google Scholar]
- For selected recent examples, see:; a Schnermann M. J.; Overman L. E. Angew. Chem., Int. Ed. 2012, 51, 9576. 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cornella J.; Edwards J. T.; Qin T.; Kawamura S.; Wang J.; Pan C.-M.; Gianatassio R.; Schmidt M. A.; Eastgate M. D.; Baran P. S. J. Am. Chem. Soc. 2016, 138, 2174. 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qin T.; Cornella J.; Li C.; Malins L. R.; Edwards J. T.; Kawamura S.; Maxwell B. D.; Eastgate M. D.; Baran P. S. Science 2016, 352, 801. 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang J.; Qin T.; Chen T.-E.; Wimmer L.; Edwards J. T.; Cornella J.; Vokits B.; Shaw S. A.; Baran P. S. Angew. Chem., Int. Ed. 2016, 55, 9676. 10.1002/anie.201605463. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Toriyama F.; Cornella J.; Wimmer L.; Chen T.-G.; Dixon D. D.; Creech G.; Baran P. S. J. Am. Chem. Soc. 2016, 138, 11132. 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Edwards J. T.; Merchant R. R.; McClymont K. S.; Knouse K. W.; Qin T.; Malins L. R.; Vokits B.; Shaw S. A.; Bao D.-H.; Wei F.-L.; Zhou T.; Eastgate M. D.; Baran P. S. Nature 2017, 545, 213. 10.1038/nature22307. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhao W.; Wurz R. P.; Peters J. C.; Fu G. C. J. Am. Chem. Soc. 2017, 139, 12153. 10.1021/jacs.7b07546. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Candish L.; Teders M.; Glorius F. G. C. J. Am. Chem. Soc. 2017, 139, 7440. 10.1021/jacs.7b03127. [DOI] [PubMed] [Google Scholar]
- For representative examples of photoredox/nickel catalysis with nonmetallic terminal reductants, see:; a Duan Z.; Li W.; Lei A. Org. Lett. 2016, 18, 4012. 10.1021/acs.orglett.6b01837. [DOI] [PubMed] [Google Scholar]; b Paul A.; Smith M. D.; Vannucci A. K. J. Org. Chem. 2017, 82, 1996. 10.1021/acs.joc.6b02830. [DOI] [PubMed] [Google Scholar]; c Zhang P.; Le C.; MacMillan D. W. C. J. Am. Chem. Soc. 2016, 138, 8084. 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Using DIPEA as an anodic sacrificial reductant furnished comparable results.
- a Pletcher D.; Green R. A.; Brown R. C. D. Chem. Rev. 2017, 10.1021/acs.chemrev.7b00360. [DOI] [PubMed] [Google Scholar]; b Plutschack M. B.; Pieber B.; Gilmore K.; Seeberger P. H. Chem. Rev. 2017, 117, 11796. 10.1021/acs.chemrev.7b00183. [DOI] [PubMed] [Google Scholar]
- HPLC analysis has shown that <1% substrate/product is present in the outflow anodic stream.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.