

Abstract
Blepharocheilodontic syndrome (BCDS) consists of lagophthalmia, ectropion of the lower eyelids, distichiasis, euryblepharon, cleft lip/palate and dental anomalies and has autosomal dominant inheritance with variable expression. We identified heterozygous variants in two genes of the cadherin–catenin complex, CDH1, encoding E-cadherin, and CTNND1, encoding p120 catenin delta1 in 15 of 17 BCDS index patients, as was recently described in a different publication. CDH1 plays an essential role in epithelial cell adherence; CTNND1 binds to CDH1 and controls the stability of the complex. Functional experiments in zebrafish and human cells showed that the CDH1 variants impair the cell adhesion function of the cadherin–catenin complex in a dominant-negative manner. Variants in CDH1 have been linked to familial hereditary diffuse gastric cancer and invasive lobular breast cancer; however, no cases of gastric or breast cancer have been reported in our BCDS cases. Functional experiments reported here indicated the BCDS variants comprise a distinct class of CDH1 variants. Altogether, we identified the genetic cause of BCDS enabling DNA diagnostics and counseling, in addition we describe a novel class of dominant negative CDH1 variants.
Introduction
Blepharocheilodontic (BCD) syndrome (OMIM 119580) is a rare, but possibly underdiagnosed disorder [1]. The main clinical features are bilateral cleft lip/palate, eyelid abnormalities, such as lagophthalmia (incomplete closure of the eyelids), distichiasis (double row of eyelashes) of the upper eyelids, and ectropion of the lower eyelids and ectodermal defects like hair and dental abnormalities as was reported by Allanson and McGillivray in 1985 [2] and was recognized as a distinct craniofacial syndrome by Korula et al. [3]. The term Blepharocheilodontic syndrome (BCDS [MIM 119580]) was proposed by Gorlin et al. in 1996 [4]. In addition to the characteristic dental anomalies, including hypodontia and conical tooth shape, ankyloblepharon, hair abnormalities, dysmorphic facial features, syndactyly, imperforate anus, hypothyroidism, dermoid cysts, and neural tube defects were also reported [4–6]. To date, the condition is reported to occur in sporadic patients but also to segregate in an autosomal dominant manner with variable expression.
Materials and methods
Sequencing and validations
After referral for routine diagnostic exome sequencing, exomes of child and parents (U1a, U2 and U3a) were enriched using the SureSelect XT Human All Exon V5 kit (Agilent) and sequenced in rapid run mode on the HiSeq2500 sequencing system (Illumina) at a mean target depth of 100×. Reads were aligned to hg19 using BWA (BWA-MEM v0.7.5a) and variants were called using the GATK haplotype caller (v2.7-2). For R1 Whole exome sequencing was done with SureSelect version 2 and 4, further as described previously [7]. Detected variants were annotated, filtered and prioritized using the Bench NGS Lab platform (Cartagenia, Leuven, Belgium). Reported variants were validated by Sanger sequencing, primer sequences are available upon request.
Data availability
The genetic and phenotypical data belonging to this study has been submitted to the Leiden Open Variation Database (LOVD; http://www.lovd.nl/3.0/home) as www.LOVD.nl/CDH1 (patient IDs 106657, 110508-110510, 110512-110515, 110523-110528, and 110531-110534) and www.LOVD.nl/CTNND1 (patient IDs 110511 and 110516-110522). Patient IDs 110529 and 110530 carry no variant in either CDH1 or CTNND1.
cDNA analysis of CDH1
RNA was isolated from fibroblasts followed by cDNA analysis as described previously [8]. Primers used for splicing analysis are: CDH1_RNA_ex7F: 5′-CAGGAACACAGGAGTCATC-3′ and CDH1_RNA_ex11R: 5′-CAAAATCCTCCCTGTCC-3′.
cDNA analysis of CTNND1
Fibroblasts (patient and controls) were cultured with Cycloheximide (final conc. 0.25 mg/ml) or 0.25 % DMSO for 4.5 h followed by RNA isolation with RNeasy mini kit (Qiagen) and conversion to cDNA with iScript cDNA Synthesis kit (Bio-Rad). Primers used for Sanger sequencing and qPCR are resp.: CTNND1_HD2_F: 5′-CCAAAAAGGAAGTGCACCTTGG-3′ and CTNND1_HD2_R: 5′-CATGGGATGAAAGATTCCACAGG-3′; CTNND1_HD12 _F: 5′-CATCTGGAGCACTGAGAAACC-3′ and CTNND1_HD12 _R: 5′-CCTCCTGGCAGATTCTTTACC-3′. Relative expression levels were measured on CFX 96 real-time system (Bio-Rad) with qPCR Syberselect mastermix (Applied Biosystems) using reference genes RNF111 and CLK2.
Zebrafish husbandry
Tübingen Longfin (TL) wild-type fish used in this study were kept under standard laboratory conditions.
mRNA functional assay in Zebrafish
Human wild-type and patient CDH1 variants’ cDNA were cloned into pCS2GW by Gateway cloning (Life Technologies BV). The resulting pCS2-based constructs were linearized by NotI-HF (NEB) restriction and used as template for in vitro synthesis of capped mRNA with mMESSAGE mMACHINE SP6 Ultra Kit (Life Technologies BV). 1-Cell stage TL embryos were microinjected with ~50 pg mRNA and kept at 28.5 °C in E3 medium. Phenotypical analysis was subsequently carried out within 28 h post fertilization.
Cartilage staining (Alcian blue)
5 d.p.f. (days post fertilization) zebrafish larvae were incubated overnight at 4 °C in fixative solution (76% ethanol; 20% acetic acid; 4% formaledhyde supplemented with 15 mg Alcian Blue). Larvae were subsequently washed in 70%-100 Ethanol, briefly transferred to 100% Methanol and finally imaged in Murray’s (v/v: 2:1 Benzylbenzoate: Benzylalcohol).
Plasmids
Human CDH1-GFP was obtained from Addgene (plasmid 28009). SNAP-tagged CDH1 was generated by PCR-subcloning SNAP from nSNAP (New England Biolabs) between the mouse H2-Kb signal sequence and CDH1. The indicated variations were generated via site-directed mutagenesis using a QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA).
Cell culture, transfection, and immunofluorescence
MCF7 CDH1 KO cells [9] were cultured in DMEM-F12 medium (Life Technologies) supplemented with 12% fetal calf serum (GE health care), 100 units/ml penicillin and 100 μg/ml streptomycin (Life Technologies). For transfection, cells were grown on glass coverslips and transfected with the indicated constructs using Polyethylenimine (PEI) according to the manufacture’s protocol. 24 h after transfection, cells were labeled with 1 μM SNAP surface 549 (New England Biolabs) for 15 min at RT, washed with phosphate buffered saline (pH 7.4) containing 1 mM of CaCl2 and MgCl2 and fixed in ice-cold methanol. Coverslips were mounted in Prolong Gold (Life Technologies) and analyzed using a Zeiss LSM510 confocal microscope.
Results and discussion
To identify the genetic cause of BCDS, we performed exome sequencing and validations in a cohort of 28 individuals (mean age 18 years) from 17 different families diagnosed with BCDS, of which 3 patients had been reported previously [4–6] (Fig. 1 (pictures), Table 1 (symptoms), Supplementary Table S1 (extensive symptoms), Supplementary Fig. S1 (pedigrees)). We noticed the features of the eye phenotype, like distichiasis, ectropion and euryblepharon were diminishing when individuals became older (Supplementary Fig. S1).
Fig. 1.

Facial features of BCDS subjects. Pictures of 24 BCD patients, showing the variability of the phenotype at different ages. The patient identification below the picture corresponds with the patient identification in the tables and pedigrees
Table 1.
Pivotal clinical features of BCDS patients of our study and the study of Ghoumid et al. [1] and Nishi et al. [22], with and without mutation
| Clinical features | CDH1 (n = 26) | CTNND1 (n = 10) | No (n = 2) |
|---|---|---|---|
| Male gender | 6/26 | 5/10 | 1/2 |
| Hypertelorism | 13/18 | 6/7 | 2/2 |
| Euryblepharon | 20/22 | 9/10 | 2/2 |
| Lagophthalmos | 20/22 | 8/10 | 2/2 |
| Ectropion | 186/22 | 8/10 | 0/2 |
| Lacrimal duct abnormalities | 3/14 | 0/7 | 0/2 |
| Distichiasis | 17/22 | 8/10 | 0/2 |
| Ankyloblepharon | 3/14 | 4/7 | 0/2 |
| CL/P | 22/24 | 6/10 | 2/2 |
| Hypodontia | 20/22 | 9/10 | 2/2 |
| Delayed dentition | 8/10 | 4/5 | 2/2 |
| Abnormal crown form | 19/22 | 8/10 | 1/2 |
| Sparse hair | 15/22 | 6/10 | 1/2 |
| High frontal hairline | 13/16 | 2/7 | 2/2 |
| Broad forehead | 10/18 | 1/7 | 2/2 |
| Malformed ears | 7/15 | 1/7 | 0/2 |
| Everted lower lip | 11/16 | 0/7 | 0/2 |
| Hypothyroidism | 6/23 | 1/10 | 0/2 |
| Imperforate anus | 4/26 | 0/10 | 0/2 |
| Dermoid cysts | 2/14 | 0/7 | 0/2 |
| Neural tube defect | 6/25 | 0/10 | 0/2 |
The CTNND1 mosaic patient (U3b) is not included this comparison.
In 15 of the 17 index cases (88%), we identified a mutation in either CDH1 (n = 12) or CTNND1 (n = 3; Table 2), as was recently described [1]. The mutation occurred de novo in 9 patients and low level mosaicism was detected in one parent. The familial mutation could be detected in 11 additional affected relatives (Table 2). Detected variants were not present in population frequency reference data sets (NCBI dbSNP Build 137 for Human, EVS, 1000Genomes [10], GoNL [11], or ExAc [12]).
Table 2.
BCDS-associated variants in CDH1 and CTNND1
| Index | Gene | Mutation | Protein | Inheritance | Carriers (N) |
|---|---|---|---|---|---|
| U1a | CDH1 | c.760 G > A | p.(Asp254Asn) | Inherited | 2 |
| R5a | CDH1 | c.760 G > A | p.(Asp254Asn) | Inherited | 3 |
| USA1a | CDH1 | c.760 G > A | p.(Asp254Asn) | Inherited | 2 |
| R20 | CDH1 | c.768 T > A | p.(Asn256Lys) | De novo | 1 |
| R19 | CDH1 | c.768 T > G | p.(Asn256Lys) | De novo | 1 |
| S4 | CDH1 | c.862 G > C | p.(Asp288His) | De novo | 1 |
| USA2a | CDH1 | c.1118 C > G | p.(Pro373Arg) | Inherited | 3 |
| R11 | CDH1 | c.1320 + 1 G > A | p.(Tyr380_Lys440del) | De novo | 1 |
| S1/R6 | CDH1 | c.1320 + 1 G > A | p.(Tyr380_Lys440del) | De novo | 1 |
| R2 | CDH1 | c.1320 + 1 G > A | p.(Tyr380_Lys440del) | De novo | 1 |
| U2 | CDH1 | c.1320 + 1 G > T | p.Tyr380_Lys440del | De novo | 1 |
| S2 | CDH1 | c.1320 + 5 G > A | p.(Tyr380_Lys440del) | De novo | 1 |
| R1a | CTNND1 | c.1372 C > T | p.Arg458* | Inherited | 5 |
| U3a | CTNND1 | c.1595 G > A | p.(Gly532Asp) | Inherited | 2* |
| S3 | CTNND1 | c.2489 G > A | p.(Trp830*) | De novo | 1 |
See Supplementary Fig. 1 for pedigrees of the families with inherited BCDS. The mosaic parent (U3b) is included as a carrier in this table, noted with an asterisk (*). See Supplementary Fig. 1 for pedigrees. Used reference sequences are NM_004360.3, NG_008021.1 (CDH1) and NM_001085458.1, NG_029078.1 (CTNND1).
CDH1 and CTNND1 form the epithelial junction complex. CDH1 or E-cadherin (epithelial cadherin) is a single-pass transmembrane protein, expressed primarily in epithelial cells where it forms the core of the adherens junction (AJ). E-cadherin and the AJ are essential for a cell to form a tight and polarized cell layer that can perform barrier and transport functions. CTNND1 (p120/catenin) is an armadillo-repeat-containing protein that interacts with the juxtamembrane cytoplasmic tail of CDH1 and has a regulatory function in the stability and turnover of the epithelial junction complex [13]. Loss of the CDH1/CTNND1 complex is a driving event in tumor development and progression [14].
We identified 7 different variants in CDH1 (Fig. 2a, Table 2), including missense variants at residues 254, 256, 288, and 373. The c.760 G > A; p.(Asp254Asn) variant was observed in three families originating from different geographic regions, namely Brazil, Morocco and the Netherlands. Combined with the absence of this allele in the > 60,000 exomes of ExAc12, common ancestry is unlikely. The c.768 T > G and c.768 T > A; p.(Asn256Lys) variant was observed in two persons, however caused by different codon changes. The extracellular region of CDH1 consists of five cadherin-type repeats that are bound together by Ca2+ ions to form stiff, rod-like proteins. All missense variants occur at highly conserved amino acid residues in the extracellular cadherin-type repeats in CDH1 that directly interact with a Ca2+ ion [15]. Cell adhesion mediated by CDH1 is dependent on Ca2+ [13], all BCDS-associated missense variants are predicted to destabilize or disturb the interaction with the interacting Ca2+ ion [15] (Fig. 2b).
Fig. 2.

BCDS variants in CDH1 and CTNND1. a Schematic overview of protein alterations in CDH1 identified in BCDS subjects, combined the number of observations in independent index cases. b Protein structures of CDH1 highlighting the fact that all missense variants alter amino acid that directly interact with a Ca2+ ion. The wild-type residues are depicted in green, the mutated form in red. c Splicing analysis on by PCR analysis of cDNA from a subject with c.1320 + 1 G > T; p.Tyr380_Lys440del, an exon 9 splice donor mutation using primers on exons 7 and 11. The BCDS subject (U2) shows a lower band on agarose gel (indicated by the arrow) compared to the control cDNA (C). Sanger sequencing of this band showed absence of the entire exon 9, predicted to result in an in-frame deletion of 61 amino acids (p.Tyr380_Lys440del). d Schematic overview of protein alterations in CTNND1 identified in BCDS subjects. e Relative expression levels of CTNND1 cDNA of the patient and three controls and f Sanger sequencing data of CTNND1 patient cDNA showing nonsense mediated decay of the mutated transcript. Variants identified in this study are reported as circles, variants identified by Ghoumid et al. [1] as triangles and variants identified by Nishi et al. [22] as squares
Besides missense variants at Ca2+ interacting residues, we identified five de novo variants at the splice donor site of exon 9 in individuals with BCDS. Splicing analysis on RNA isolated from fibroblasts from patient U2 (mutation c.1320 + 1 G > T), identified an aberrant splicing product (Fig. 2c). Sequencing of this aberrant product revealed complete absence of exon 9, predicted to cause an in-frame deletion of 183 bp and partial deletion of the 3rd cadherin domain of the CDH1 protein. The splice donor variants of exon 9 most likely result in a shortened form of the protein with lower protein stability and decreased cell adhesion.
We identified three different variants in CTNND1 (Fig. 2d, Table 2), one segregating with BCDS in a three generation family, one de novo and one inherited from a mosaic parent. Quantitative cDNA analysis of CTNND1 in fibroblasts from subjects with the c.1372 C > T; p.Arg458* allele revealed an expression level of about 60% of the controls’ value. This reduction was reversed by cyclohexamide treatment indicating that the allele is subjected to nonsense mediated decay (Fig. 2e and f). The c.1595 G > A; p.(Gly532Arg) missense mutation affects the highly conserved H3 helix in the 5th armadillo repeat of CTNND1, changing glycine 532 for an arginine. The armadillo repeats in CTNND1 and this H3 helix specifically are essential for interaction with E-cadherin/CDH1 cytoplasmic tail [16].
The genetic data firmly link mutations in CDH1 and CTNND1 to BCDS. Germline variants in CDH1 have previously been linked to hereditary diffuse gastric cancer (HDGC) [17], with or without cleft lip and/or palate (OMIM 137215) and invasive lobular breast cancer (ILBC) [18], however there is no overlap between BCDS and the cancer-related CDH1 variants. Although gastric or breast cancer is not mentioned in the BCDS families included in this study (n = 28) and those reported before (n = 81), cancer risk among carriers of these variants needs to be defined. Germline variants in CDH1 have been observed in subjects with non-syndromic orofacial clefting [19–21], in total five germline CDH1 missense variants have been reported [19, 20]. One BCDS variant, c.760 G > A; p.(Asp254Asn) has also been described in two families with non-syndromic clefting. Another mutation, c.1108G- > T; p.(Asp370Tyr), is located near the BCDS mutation c.C1118G; p.(Pro373Arg), but this mutation does not interact directly with the calcium ion as seen for all BCDS missense variants thus far. Three variants (c.88 C > A; p.(Pro30Thr), c.2351 G > A p.(Arg784His), c.2413G- > A p.(Asp805Asn)) were located in different domains than the BCDS variants. The absence of pivotal BCDS features in the reported non-syndromic cleft patients might be explained by the variability in expression, vanishing eye phenotype, non-recognition of the syndrome or non- or reduced penetrance. Some of the relatives of the BCDS probands (n = 3), identified in this study as carriers of a CDH1 or CTNND1 variants, showed very mild features (patient U1b, U3b, and R1e).
Although the total number of patients, described here and in previous studies [1], [22] is limited, we could not clinically distinguish between index cases (Fig. 1, Table 1 and Supplementary Table S1) with a CDH1 (U1a, R5a, USA1a, R19, R20, S4, USA2a, U2, R2, R11, and S1/R6,S2), CTNND1 (R1a, U3a, and S3) variant or without a variant (R3 and R4), nor could we relate phenotypic differences to the type of mutation or distinguish index patients with a de novo variant and a familial-inherited maternal or paternal variant. However, as a group, the features seem less striking in the patients with a variant in CTNND1, compared to the patients with a CDH1 variant, in which clefting was less frequent. Some dysmorphic features, lacrimal duct abnormalities and associated features like hypothyroidism, neural tube defects, and imperforate anus were absent or less frequent in this group of patients. However, ankyloblepharon was more frequently found in patients with a CTNND1 mutation. The features in the two patients not carrying a variant in either CDH1 or CTNND1, seem even milder compared to CDH1 or CTNND1 patients: absence of ectropion, distichiasis, ankyloblepharon, lacrimal duct abnormalities, and some dysmorphic features additionally to the absence of associated features. There was no clinical difference between male and female patients, although there is an unexplained female preponderance (67%).
To assess the effect of the BCDS variants on CDH1 function, we performed in vivo assays in zebrafish embryos. Microinjection of mRNA of the CDH1 variants (Fig. 3a) included in this study resulted in the phenotypes presented in Fig. 3. BCDS-related CDH1 variants induced profound developmental defects in zebrafish embryos including different degrees of head hypoplasia, which ranged from incomplete development to total absence of head structures (Fig. 3b and c compare class II, III and V), severely dysmorphic trunk and failure for the tailfin to develop normally. Furthermore, in embryos expressing BCDS-related variants we observed severely delayed and defective early embryonic development (Fig. 3d). During somitogenesis embryos additionally displayed detaching cells and in some cases a dramatically defective dorsal midline phenotype (Fig. 3e, f). At 1 d.p.f., numerous round, detaching cells at the surface of the neural tube and embryonic midline (Fig. 3d) could be observed. These phenotypes overlap remarkably accurately with those of known zebrafish CDH1 mutants carrying missense variants in one of the extracellular (EC) domains of CDH1 (half baked; hab [23, 24]), illustrating the importance of the EC domains for proper CDH1 function. In larvae surviving to 5 d.p.f., we could observe abnormal craniofacial development and defects in palate formation (Fig. 3g). Hence, these phenotypes carry a sensible level of overlap with the features displayed by BCDS patients, such as neural tube defects or orofacial clefting. Neural tube defects have been observed in BCDS previously [25] and in our cohort, namely anencephaly (family R5) and spinal lipomas (family USA2). The expression of a CDH1 mutations associated with hereditary diffuse gastric cancer (HDGC; p.Trp638*, [26] or p.(Pro373Leu) [27]) had comparatively little or reduced effect on embryo development, suggesting distinct mechanisms for BCDS- and gastric cancer-associated variants in CDH1.
Fig. 3.

Effect of human CDH1 variants on zebrafish early development. a Schematical representation of the experimental setup. Fertilized 1-cell zebrafish embryos were injected with mRNA corresponding to the human CDH1 and phenotyped at 1 day post fertilization (d.p.f.) b Phenotypical classification of 1 d.p.f. zebrafish embryos microinjected with hCDH1 variants. Arrowheads point at head hypoplasia in Class III and severe tail defect in Class IV, respectively. c Quantification of the phenotypical classes described in b for different hCDH1 variants. The p.(Pro373Leu) and p.Trp638* variants are found in hereditary diffuse gastric cancer patients. For all categories, number of embryos analyzed n: 112 < n < 276 d At 10 hpf (bud stage; wild type, upper panel), embryos expressing BCDS-related CDH1 variants often display severe developmental delay, denoted by absence of the tail bud (lower panel; arrowhead) e During somitogenesis (upper left panel; 13 somites) embryos expressing BCDS-related CDH1 variants may display severely delayed development with rough surface and detaching cells at the posterior end of the midline (right panels; lower panel is magnification of upper panel). Major midline defects (lower left panel) are also observed. f Numerous detaching cells (arrowheads) can be observed at the midline of affected embryos at 1 d.p.f. (Class II-V; embryo and magnifications shown are representative of the observed phenotype). Note abnormal development of the neural tube. g 5 d.p.f. zebrafish larvae microinjected with mRNA from BCDS-related CDH1 variants (p.(Asp254Asn) in the representrative picture) display severe jaw and craniofacial defects (arrowhead). Cartilage staining reveals major defective patterning and interrupted cartilage formation at the base of the palate (arrow)
Our results are consistent with a dominant negative effect of the BCDS-related alleles. Once localized to the membrane, cadherins carry out their cellular adhesion function by forming dimers through interaction of their EC domains [28, 29]. Missense alleles associated with BCDS would dimerize with wild-type (WT) proteins at the cellular membrane and interfere with normal cadherin function, thus exert a dominant negative effect. To get more insight into the consequences of the BCDS-inducing missense variants at the cellular level and test this hypothesis, we used a breast cancer MCF7 cell line in which CDH1 was previously knocked out by CRISPR/Cas9. We co-expressed wild-type GFP-labeled CDH1 with different SNAP-tagged forms of CDH1 in which BCDS-or HDGC-specific variants had been introduced (Fig. 4). Upon co-expression with wild-type CDH1 we observed enriched localization to the adherens junctions (Fig. 4a). In contrast, co-expression of SNAP-Ecad c.760 G > A; p.(Asp254Asn) prevents GFP-Ecad WT to form adherens junctions between transfected cells, resulting in a decreased adherence of cells (Fig. 4b), indicating a dominant negative effect. Co-expression of SNAP-Ecad c.1913G > A p.Trp638* (HDGC variant) does not display a significant effect on GFP-Ecad WT localization. Instead, SNAP-Ecad p.Trp638* is mislocalized via rapid internalization and accumulation in peripheral endocytic structures, suggesting an alternative mode of action (Fig. 4c). The outcome of these in vitro experiments confirms our hypothesis that BCDS variants affect CDH1 in a dominant negative manner and therefore fall in a different functional class than HDGC and ILBC variants. In conclusion, we show that BCDS is caused by variants in CDH1 and CTNND1. Our findings provide important insights in the role of the cadherin–catenin complex in human development and enables diagnostics for BCDS subjects.
Fig. 4.
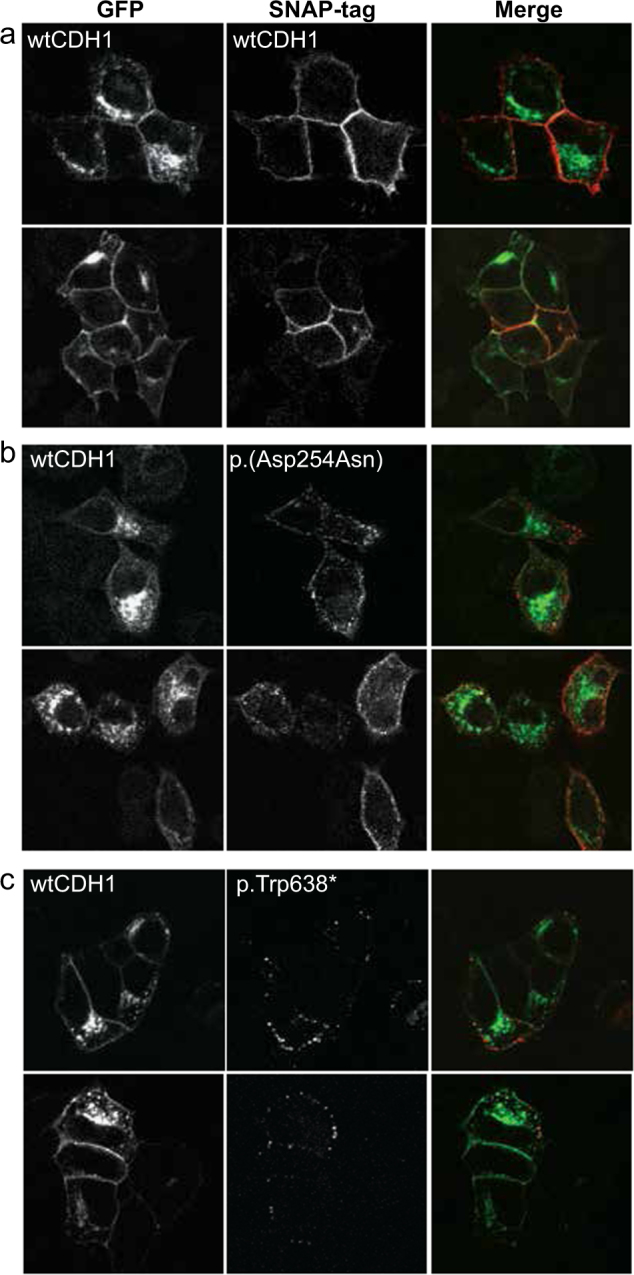
Dominant negative effects of the c.760 G > A; p.(Asp254Asn) BCDS-associated variant. Immunofluorescence images of MCF7 CDH1 KO cells co-expressing a GFP-CDH1 wild type (WT) and SNAP-CDH1 WT, b c.760 G > A; p.(Asp254Asn) or c p.Trp638*. Cells were labeled with SNAP surface 549 before fixation. In contrast to SNAP-CDH1 WT, which highly localizes with GFP-CDH1 at the plasma membrane, SNAP-CDH1 c.760 G > A; p.(Asp254Asn) inhibits the stable localization of GFP-CDH1 to the plasma membrane. SNAP-CDH1 p.Trp638* has no effect on GFP-CDH1 localization
During the submission and review of this paper a manuscript linking variants in CDH1 and CTNND1 to BCDS was published1. Where none of the variants in the 15 families we describe are completely identical to the variants in 8 families described by Ghoumid et al. [1], there is a clear overlap in mutated domains and predicted effects at the protein level. We both found missense variants in CDH1 at calcium binding sites and splice variants resulting in deletion of exon 9. In addition, Nishi et al. [22] described a patient with a CDH1 variant (c.2028 A > C; p.Asp676Glu) in which some features of BCD syndrome could be identified, such as neural tube defect, hypertelorism, malformed ears and cleft lip/palate. Other features like facial asymmetry, congenital heart defect and corpus callosum agenesis have not been described before in BCD syndrome [1, 22]. Except for the c.1595 G > A; p.(Gly532Asp) variant, both our study and the one published by Ghoumid et al. [1] detected nonsense variants in CTNND1. The functional modeling in zebrafish and cell lines presented here provides the intriguing evidence of dominant negative function of the CDH1 variants in BCDS.
Electronic supplementary material
Acknowledgements
We thank Tom de Vries Lentsch for preparing the pictures and pedigrees of the patients, Milena Simioni for technical assistance at the Department of Medical Genetics, UNICAMP and Sanne Savelberg for assistance for data submission to LOVD. We acknowledge the support from the Netherlands Cardiovascular Research Initiative and the Dutch Heart Foundation grant CVON2014-18 CONCOR-GENES.
Compliance with ethical standards
Conflict of interest
The authors declare that they no competing interest.
Footnotes
Anneke Kievit, Federico Tessadori, Koen L.I. van Gassen, Annelies de Klein, Marie-José van den Boogaard and Gijs van Haaften contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1038/s41431-017-0010-5) contains supplementary material, which is available to authorized users.
Contributor Information
Anneke Kievit, Phone: +31 (0) 107036915, Email: j.a.kievit@erasmusmc.nl.
Gijs van Haaften, Phone: +31 (0) 88 7567747, Email: G.vanhaaften@umcutrecht.nl.
References
- 1.Ghoumid J, Stichelbout M, Jourdain AS, et al. Blepharocheilodontic syndrome is a CDH1 pathway-related disorder due to mutations in CDH1 and CTNND1. Genet Med. 2017;19:1013–21. doi: 10.1038/gim.2017.11. [DOI] [PubMed] [Google Scholar]
- 2.Allanson JE, McGillivray BC. Familial clefting syndrome with ectropion and dental anomaly--without limb anomalies. Clin Genet. 1985;27:426–9. doi: 10.1111/j.1399-0004.1985.tb02288.x. [DOI] [PubMed] [Google Scholar]
- 3.Korula S, Wilson L, Salomonson J. Distinct craniofacial syndrome of lagophthalmia and bilateral cleft lip and palate. Am J Med Genet. 1995;59:229–33. doi: 10.1002/ajmg.1320590221. [DOI] [PubMed] [Google Scholar]
- 4.Gorlin RJ, Zellweger H, Curtis MW, et al. Blepharo-cheilo-dontic (BCD) syndrome. Am J Med Genet. 1996;65:109–12. doi: 10.1002/(SICI)1096-8628(19961016)65:2<109::AID-AJMG5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 5.Gil da Silva Lopes VL, Guion-Almeida ML, de Oliveira Rodini ES. Blepharocheilodontic (BCD) syndrome: expanding the phenotype? Am J Med Genet A. 2003;121A:266–70. doi: 10.1002/ajmg.a.20223. [DOI] [PubMed] [Google Scholar]
- 6.Freitas EL, Martinhago CD, Ramos ES, Murray JC, Gil-da-Silva-Lopes VL. Preliminary molecular studies on blepharocheilodontic syndrome. Am J Med Genet A. 2007;143A:2757–9. doi: 10.1002/ajmg.a.32010. [DOI] [PubMed] [Google Scholar]
- 7.Yavuzyigitoglu S, Koopmans AE, Verdijk RM, et al. Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology. 2016;123:1118–28. doi: 10.1016/j.ophtha.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 8.van Haelst MM, Monroe GR, Duran K, et al. Further confirmation of the MED13L haploinsufficiency syndrome. Eur J Hum Genet. 2015;23:135–8. doi: 10.1038/ejhg.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hornsveld M, Tenhagen M, van de Ven RA, et al. Restraining FOXO3-dependent transcriptional BMF activation underpins tumour growth and metastasis of E-cadherin-negative breast cancer. Cell Death Differ. 2016;23:1483–92. doi: 10.1038/cdd.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genome of the Netherlands C. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818–25. doi: 10.1038/ng.3021. [DOI] [PubMed] [Google Scholar]
- 12.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–34. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 14.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–32. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 15.Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishiyama N, Lee SH, Liu S, et al. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–28. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Hansford S, Kaurah P, Li-Chang H, et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015;1:23–32. doi: 10.1001/jamaoncol.2014.168. [DOI] [PubMed] [Google Scholar]
- 18.Petridis C, Shinomiya I, Kohut K, et al. Germline CDH1 mutations in bilateral lobular carcinoma in situ. Br J Cancer. 2014;110:1053–7. doi: 10.1038/bjc.2013.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogelaar IP, Figueiredo J, van Rooij IA, et al. Identification of germline mutations in the cancer predisposing gene CDH1 in patients with orofacial clefts. Hum Mol Genet. 2013;22:919–26. doi: 10.1093/hmg/dds497. [DOI] [PubMed] [Google Scholar]
- 20.Brito LA, Yamamoto GL, Melo S, et al. Rare Variants in the Epithelial Cadherin Gene Underlying the Genetic Etiology of Nonsyndromic Cleft Lip with or without Cleft Palate. Hum Mutat. 2015;36:1029–33. doi: 10.1002/humu.22827. [DOI] [PubMed] [Google Scholar]
- 21.Bureau A, Parker MM, Ruczinski I, et al. Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics. 2014;197:1039–44. doi: 10.1534/genetics.114.165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishi E, Masuda K, Arakawa M, et al. Exome sequencing-based identification of mutations in non-syndromic genes among individuals with apparently syndromic features. Am J Med Genet A. 2016;170:2889–94. doi: 10.1002/ajmg.a.37826. [DOI] [PubMed] [Google Scholar]
- 23.Kane DA, Hammerschmidt M, Mullins MC, et al. The zebrafish epiboly mutants. Development. 1996;123:47–55. doi: 10.1242/dev.123.1.47. [DOI] [PubMed] [Google Scholar]
- 24.Kane DA, McFarland KN, Warga RM. Mutations in half baked/E-cadherin block cell behaviors that are necessary for teleost epiboly. Development. 2005;132:1105–16. doi: 10.1242/dev.01668. [DOI] [PubMed] [Google Scholar]
- 25.Ababneh FK, Al-Swaid A, Elhag A, Youssef T, Alsaif S. Blepharo-cheilo-dontic (BCD) syndrome: expanding the phenotype, case report and review of literature. Am J Med Genet A. 2014;164A:1525–9. doi: 10.1002/ajmg.a.36465. [DOI] [PubMed] [Google Scholar]
- 26.Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297:2360–72. doi: 10.1001/jama.297.21.2360. [DOI] [PubMed] [Google Scholar]
- 27.Roviello F, Corso G, Pedrazzani C, et al. Hereditary diffuse gastric cancer and E-cadherin: description of the first germline mutation in an Italian family. Eur J Surg Oncol. 2007;33:448–51. doi: 10.1016/j.ejso.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 28.Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–4. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- 29.Takeda H, Shimoyama Y, Nagafuchi A, Hirohashi S. E-cadherin functions as a cis-dimer at the cell-cell adhesive interface in vivo. Nat Struct Biol. 1999;6:310–2. doi: 10.1038/7542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genetic and phenotypical data belonging to this study has been submitted to the Leiden Open Variation Database (LOVD; http://www.lovd.nl/3.0/home) as www.LOVD.nl/CDH1 (patient IDs 106657, 110508-110510, 110512-110515, 110523-110528, and 110531-110534) and www.LOVD.nl/CTNND1 (patient IDs 110511 and 110516-110522). Patient IDs 110529 and 110530 carry no variant in either CDH1 or CTNND1.