

Abstract
Molecular characterization of balanced chromosomal abnormalities constitutes a powerful tool in understanding the pathogenic mechanisms of complex genetic disorders. Here we report a male with severe global developmental delay in the presence of a complex karyotype and normal microarray and exome studies. The subject, referred to as DGAP294, has two de novo apparently balanced translocations involving chromosomes 1 and 14, and chromosomes 4 and 10, disrupting several different transcripts of adhesion G protein-coupled receptor L2 (ADGRL2) and protocadherin 15 (PCDH15). In addition, a maternally inherited inversion disrupts peptidyl arginine deiminase types 3 and 4 (PADI3 and PADI4) on chromosome 1. None of these gene disruptions explain the patient’s phenotype. Using genome regulatory annotations and chromosome conformation data, we predict a position effect ~370 kb upstream of a translocation breakpoint located at 14q12. The position effect involves forkhead box G1 (FOXG1), mutations in which are associated with the congenital form of Rett syndrome and FOXG1 syndrome. We believe the FOXG1 position effect largely accounts for the clinical phenotype in DGAP294, which can be classified as FOXG1 syndrome like. Our findings emphasize the significance of not only analyzing disrupted genes by chromosomal rearrangements, but also evaluating potential long-range position effects in clinical diagnoses.
Introduction
Balanced chromosomal abnormalities (BCAs) in humans constitute an unparalleled opportunity to improve our understanding of genes involved in genetically complex disorders. The precise disruption of a chromosome region can lead to discovery of haploinsufficient genes and regulatory elements that can confer disease risks in humans. The Developmental Genome Anatomy Project (DGAP, dgap.harvard.edu) is an established research endeavor in the genomic and functional characterization of individuals with congenital anomalies harboring cytogenetically visible BCAs [1–6].
With the development of the chromosome conformation capture technique [7] and its multiple adaptations to interrogate whole genome chromatin organization [8, 9], comprehensive maps are available now of regulatory chromatin interactions and modular partitioning of chromosomes by topologically associating domains (TADs) in several human cell lines and tissues [10–15]. With these maps, BCA pathogenic outcomes can be analyzed at the level of chromosome structure. Recent studies have experimentally examined the disruption of regulatory chromatin contacts and TADs by pathogenic structural variants causative of limb malformations [16], cancer [17, 18], and autosomal dominant adult-onset demyelinating leukodystrophy [19]. Such observations emphasize the importance of not only studying gene sequences disrupted by structural variants, but also analyzing their impact on local chromatin structure, which may result in long-range position effects that can affect neighboring gene expression and cause disease. Such position effects can oftentimes serve as important predictors for pathogenicity and better inform clinical diagnosis [4, 20].
The present work is a study of DGAP294, a karyotypically complex DGAP subject [4] exhibiting severe global developmental delay with a normal clinical microarray and no detected pathogenic exome variants. His two de novo apparently balanced translocations, ((t(1;14)(p21.2;q11.2)dn and t(4;10)(p13;q11.2)dn), disrupt several transcript isoforms of adhesion G protein-coupled receptor L2 (ADGRL2) and protocadherin 15 (PCDH15). A maternally inherited inversion on chromosome 1, inv(1)(p36.13p36.3)mat, disrupts PADI3 and PADI4 encoding peptidyl arginine deiminase types 3 and 4, respectively. None of these four disrupted genes are known to be associated with an abnormal phenotype that explains the clinical findings of DGAP294. Using regulatory annotations and chromatin conformation data, we predict a long-range position effect from one of the rearrangement breakpoints detected on chromosome 14 involving forkhead box G1 (FOXG1). Several FOXG1 variants are associated with the congenital form of Rett syndrome (RTT) [21] and FOXG1 syndrome [22–27]. DGAP294 presents with a similar clinical phenotype to both RTT and FOXG1 syndromes, and we therefore predict that dysregulation of FOXG1 due to a position effect may be causative for DGAP294′s clinical phenotype.
Patient and methods
DGAP294 was recruited into DGAP after identification of two independent BCAs. Informed consent, medical records, and blood samples were obtained through the DGAP protocol approved by the Partners HealthCare System Institutional Review Board.
Clinical description
DGAP294 is the second child of healthy unrelated parents. His delivery was induced at 38 weeks due to polyhydramnios and a potential fetal seizure with a birth weight of 2.920 kg (10%ile) and a head circumference of 35 cm (50%ile). After birth, DGAP294 presented with feeding difficulty and low body temperature, and was diagnosed with pneumonia. The feeding difficulty continued for the first month of life, along with increased fatigue, prolonged periods of sleep, daytime colic, hypotonia, and a marked disinterest in the world around him with failure to engage in eye contact. From the sixth week through his third month, DGAP294 had frequent episodes of reflux, vomiting, choking, and gagging, as well as eczema, which were attributed to milk allergy. At 6 months, acquired microcephaly was confirmed (below 0.4%ile), with magnetic resonance imaging showing an underdeveloped brain. He began to roll over at 7 months, and smile and control head movements at 14 months, indicative of developmental delay. After 11 months of age there was no developmental progression and a diagnosis of severe cortical visual impairment was rendered. Despite a normal electroencephalogram at 8 months, starting at 9 months DGAP294 suffered frequent apneic and tonic seizures up to 30 times monthly. Following a ketogenic diet, the frequency of seizures was relatively controlled at 2 years of age. Currently, at 5 years of age, DGAP294 has a severe impairment of expressive and receptive language, spinal deformity, psychomotor retardation, ataxia, and displays stereotypic hand movements. Chromosome analysis by G-banded karyotyping showed a complex and apparently balanced male karyotype. One chromosome 1 homolog has a small paracentric inversion within the short arm, whereas the other chromosome 1 is involved in an apparently balanced translocation between its short arm and the proximal long arm of one copy of chromosome 14. A second apparently balanced translocation was detected between the short arm of chromosome 4 and the long arm of chromosome 10. Parental karyotyping revealed the paracentric inv(1) to be maternally inherited and therefore unlikely to be contributing to the clinical findings observed in DGAP294. Microarray analysis was performed using a Bluegnome 8 × 60 K International Standard Cytogenomic Array. No evidence of genomic imbalances at any of the breakpoints identified by the G-banded chromosome analysis was observed. His exome was sequenced as part of the Deciphering Developmental Disorders (DDD, www.ddduk.org) project at the Wellcome Trust Sanger Institute with an Agilent SureSelect Exome Plus and HiSeq sequencing; [28, 29] no pathogenic variants were indicated in the DDD exome case report. DGAP294′s DECIPHER accession number is 349672.
Lymphoblastoid cell line generation
Epstein–Barr virus transformation of DGAP294’s peripheral blood was performed at the Genomics and Technology Core in the Center for Human Genetic Research at Massachusetts General Hospital (Boston, MA).
Base pair sequence definition of BCA breakpoints
Whole-genome sequencing was performed using a custom large-insert jumping library protocol with a targeted insert size of 3,000 bp [30, 31]. The library was sequenced on an Illumina HiSeq 2500 rapid run mode using a paired-end 25-cycle protocol, to assure minimum insert coverage of 40×. Sequencing achieved a raw yield of 85,007,504 read pairs. After postprocessing, 94.3% of the read pairs were aligned to the human genome reference GRCh37 v71 sourced from Ensembl (http://useast.ensembl.org/Homo_sapiens/Info/Index), with an insert size median of 3,212 bp (±470 bp median absolute deviation) yielding a resulting haploid insert coverage of 67.1× (Supplementary Table 1).
All computational analyses have been described previously [6, 32, 33]. Mapping against the human genome reference version hg19 revealed eight groups of chimeric read pairs underlying eight BCA breakpoints that were distributed between a complex de novo translocation involving chromosomes 1 and 14, which included a 127 bp de novo insertion, a de novo reciprocal translocation between chromosomes 4 and 10, and a maternally inherited inversion in the non-translocated chromosome 1 (Fig. 1) [4]. All identified BCA breakpoints were validated through PCR amplification and Sanger sequencing, except the maternally inherited inversion (Supplementary Table 2) [4].
Fig. 1.
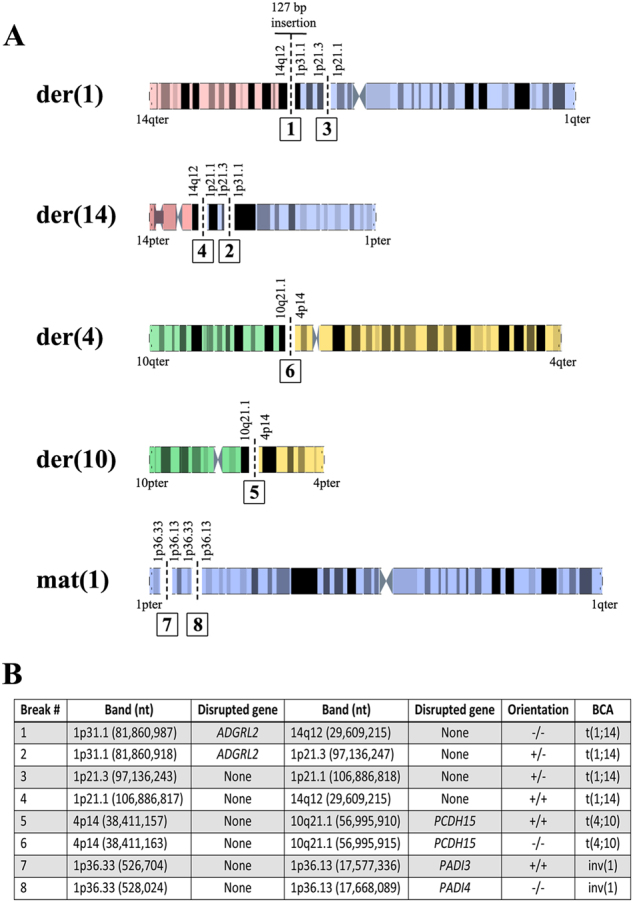
a Karyogram of DGAP294’s derivative chromosomes. Chromosomes 1, 4, 10, and 14 are depicted in blue, yellow, green, and red, respectively, with their corresponding G-bands (obtained from http://grch37.ensembl.org/Homo_sapiens/Location/Genome). BCA breakpoints are indicated with dashed black lines and bold numbers within solid squares (breakpoint numbers correspond to those shown in b). b DGAP294 BCA breakpoints' genomic coordinates and disrupted genes locations are reported in human genome version GRCh37/hg19
BCAs breakpoint genomic analysis
Overlap analyses between regulatory or structural genomic elements and the BCA breakpoints were performed using custom Perl scripts and their significance calculated with 1,000 simulations performed by the Genome Association Tester (GAT) program [34]. Genomic features from the hg19 human genome build were downloaded from the UCSC genome browser [35], enhancer positions were obtained from Andersson et al. [36] and the VISTA Enhancer browser [37], enhancer and DNaseI hypersensitive sites (DHSs) were obtained from the ENCODE project [38], and clinical variants from additional patients were obtained from ClinVar [39, 40] and the DECIPHER database [41].
Quantitative real-time PCR
RNA from three independent lymphoblastoid cell lines (LCLs) was isolated. Cell lines DGAP244–02m (MIN#31173) and DGAP245–02m (MIN#31356) were used as karyotypically normal male controls. DGAP294’s LCL (MIN#35293) was used to test differential gene expression for the genes ADGRL2, NTNG1, and FOXG1, using quantitative PCR (qPCR). GAPDH, TBP, and GUSB were used as housekeeping gene controls. qPCR experiments were performed in the Harvard Biopolymers Facility (https://genome.med.harvard.edu/) using TaqMan probes Hs00202347_m1 (ADGRL2), Hs00263709_m1 (PCDH15), Hs01850784_s1 (FOXG1), Hs01552822_m1 (NTNG1), Hs04420697_g1 (GAPDH), Hs00427620_m1 (TBP), and Hs00939627_m1 (GUSB). Data were analyzed using the ΔCT method for three sample replicates per LCL and four technical replicates per sample, and fold changes were calculated accordingly.
Results
The initial clinical karyotype for DGAP294 reported a de novo apparently balanced translocation between chromosomes 1 and 14, a de novo apparently balanced translocation between chromosomes 4 and 10, and a maternally inherited apparently balanced inversion in the non-translocated chromosome 1, and was designated 46,XY,inv(1)(p36.13p36.3)mat,t(1;14)(p21.2;q11.2)dn,t(4;10)(p13;q11.2)dn. Using large-insert whole genome sequencing [4, 31], eight pairs of breakpoints were mapped for the two de novo translocations (t(1;14) and t(4;10)) and the maternally inherited inv(1); the t(1;14) translocation was determined to be a more complex rearrangement containing four breaks and a 187 bp de novo insertion (Fig. 1). All breakpoints were validated with additional Sanger sequencing with the exception of the inv(1)mat. Following sequence analysis, the karyotype was reinterpreted and is described using next-gen cytogenetic nomenclature [42] as 46,XY,inv(1)(p36.13p36.3)mat,t(1;14)(p21.2;q11.2)dn,t(4;10)(p13;q11.2)dn.seq[GRCh37/hg19](1,4,10,14)cx,inv(1)(pter- > p36.33(526,704~)::p36.13p36.33(17,577,336~−528,024~)::p36.13(17,668,089~)− > qter)mat,der(1)(14qter- > 14q12(29,609,215)::TGTATGAGATATCACA::1p31.1(81,860,923–81,860,982)::TTTTCGTATACTTCTTGGCCACTTTCATATACTTTCATATACTTTCGTATAC::1p31.1p21.3(81,860,983–97,136,24{2–3})::1p21.1(+)(106,886,81{8–9})- > 1qter),der(4)(10qter- > 10q21.1(56,995,91{5})::4p14(38,411,16{3})- > 4qter),der(10)(10pter- > 10q21.1(56,995,9{09–10})::4p14(38,411,15{7–6})- > 4pter),der(14)(14pter- > ?::?14q12(-)(29,609,21{5})::1p21.1p21.3(106,886,81{7}−97,136,328)::ACGNNNANAGACAGNTNCCACTCAAGTTATGTGGACATAAACA::1p21.3(97,136,285–97,136, 247)::1p31.1(81,860,918)- > 1pter)dn. The Human Genome Variation Society nomenclature [43] for this complex rearrangement is inv(1):g.(526705_528023)del(528024_17577336)inv(17577337_17668088)del,der(1):g.[chr14:29609215::TGTATGAGATATCACA::chr1:81860923_81860982::TTTTCGTAT ACTTCTTGGCCACTTTCATATACTTTCATATACTTTCGTATAC::81860983_97136243::106886819_cen_qter]dn,der(14):g.[chr14:pter_cen_?::(29609215_?)inv::chr1:97136328_ 106886817inv::ACGNNNANAGACAGNTNCCACTCAAG TTATGTGGACATAAACA::97136247_97136285inv::pter_ 81860918inv]dn,der(4):g.[chr10:56995916_qterinv::chr4: 38411163_cen_qter]dn, der(10):g.[chr10:pter_cen_56995908::chr4:pter_38411157inv]dn. For both nomenclature systems, the question mark (?) indicates a missing genomic segment not found by the sequencing analysis; for the next-gen cytogenetic nomenclature according to Ordulu et al. [42], the ~ symbol represents approximate coordinates. The underlined chromosome 1 indicates the homologue that does not contain the maternal inversion. Part of the insertion sequence at 1p31.1 belongs to a LINE element with locations in chromosomes 3 and 5, and is probably derived from a replication-based mechanism that generated the rearrangement; [44] the second insertion sequence did not have any match with the rest of the genome.
Of the Sanger-validated breakpoints, none overlapped important genomic structural elements that could distally affect gene function, such as binding sites for the CCCTC-binding factor (CTCF), known to be a boundary element of TADs [15, 45], or regulatory functional elements such as enhancers (Supplementary Tables 3, 4); however, this lack of functional overlap was not statistically significant (GAT p > 0.05). The 4p14 breakpoint overlapped a single DHS in H1-ESC (Supplementary Table 5) with a reported GAT simulation p = 0.03 (Supplementary Table 6). Interestingly, we observed that five of the validated breakpoint positions overlapped repeated genomic elements in chromosomes 1, 10, and 14 at a significant level (GAT simulation p = 0.0001) (Supplementary Tables 6 and 7), suggesting a non-allelic homologous recombination process in their generation, whereas the translocation in chromosome 4 could represent a non-homologous end-joining event. Although not further validated with Sanger sequencing, one of the maternally inherited inv(1) breakpoints overlapped a segmental duplication with associated partners scattered in diverse chromosomes (Supplementary Table 8).
The Sanger-validated breakpoints disrupted several protein coding transcripts from ADGRL2, PCDH15, PADI3, and PADI4 (Fig. 1). The breakpoint in 1p31.1 disrupted adhesion G protein-coupled receptor L2 (ADGRL2), also known as LPHN2 (OMIM#607018), and truncated several isoforms of ADGRL2 by separating the first exon from the rest of the gene (Supplementary Figure 1). The breakpoint at 10q21.1 disrupted two protein coding transcripts and the 5′-untranslated region of protocadherin 15 (PCDH15, OMIM#605514) (Supplementary Figure 2). PADI3 (OMIM#606755) and PADI4 (OMIM#605347) were separated from one and two of their protein coding transcripts, respectively, by the maternally inherited breakpoints in chromosome 1 (Supplementary Figure 3A, B).
PADI3 has not yet been associated with disease, whereas certain PADI4 variants have been correlated with susceptibility to rheumatoid arthritis, a clinical feature not observed in DGAP294 [46, 47]. PCDH15 has been observed in an autosomal recessive form of Usher syndrome [48], which may be involved in DGAP294’s cortical visual impairment; however, no pathogenic exome variants were detected for PCDH15. On the other hand, ADGRL2 is a highly constrained gene (pLI = 1.00) with a reported haploinsufficiency score of 0.57% [49], predicting a high sensitivity to loss of function (LoF) variants and dosage alterations. ADGRL2 has been classified as a calcium-independent receptor of low affinity for α-latrotoxin and thus proposed to regulate exocytosis. Homozygous ADGRL2-null mice die prenatally at fetal stages, whereas heterozygous mice are hypotonic (MGI:2139714), a shared clinical trait with DGAP294. Interestingly, ADGRL2 has not yet been clearly associated with a disease phenotype. There are seven ClinVar entries involving duplication or deletion of ADGRL2 (Supplementary Table 10). Of these entries, a 4× duplication (four copies) of ADGRL2 was classified as benign (variation ID 151317), despite the observed developmental delay and/or other significant developmental or morphological phenotypes; two duplications of ADGRL2 plus one or two neighboring genes (variation IDs 144500 and 57756) were classified as variants of uncertain significance, respectively, although failure to thrive, developmental delay, and other significant developmental or morphological phenotypes were also observed; finally, a deletion of part of the gene (variant ID 147870) was classified as benign with a phenotype of intellectual disability. It is important to note that for the duplication cases, the duplication region does not encompass the full length of ADGRL2, potentially causing functional loss of the gene similar to that observed in DGAP294’s translocation if the extra copy is present in tandem formation within the coding region. The remaining ClinVar entries were large pathogenic copy number variants involving dozens of genes. Six large deletion/loss cases and two large duplication cases encompassing ADGRL2 and adjacent genes (Supplementary Table 11) are reported in DECIPHER. According to the Genotype-Tissue Expression (GTEx) project [50], the expression of both PCDH15 and ADGRL2 in LCLs is minimal (Supplementary Figures 4 and 5). Significant changes in expression of ADGRL2 were not detected with quantitative real-time PCR experiments in the DGAP294 LCL (Fig. 2) (Mann–Whitney U-test, p = 0.26); however, a 15% reduction in PCDH15 transcript was observed (Fig. 2) (Mann–Whitney U-test, p = 0.00466).
Fig. 2.

Assessment of gene expression changes for DGAP294-derived LCLs. The control genes GAPDH, GUSB, and TBP are shown in blue and genes evaluated are indicated in different colors (legends to the right of each histogram). Each bar represents the ΔCT results of three culture replicates with three technical replicates each, compared with two sex-matched control LCLs
To extend the search of additional genes contributing to DGAP294’s phenotype, we evaluated potential position effects within the TADs in neighboring regions of the DGAP294 breakpoints. DGAP294’s clinical features closely resemble RTT (Phenomizer diagnosis p = 0.0003) [51]. We identified FOXG1, located ~370 kb upstream of the 14q12 breakpoint, and netrin G1 (NTNG1, OMIM#608818), located ~795 kb downstream of the 1p13.3 breakpoint, as potential candidates given their previously reported associations with RTT [52, 53] and the FOXG1 syndrome [23]. Of particular interest was the breakpoint located in 14q12, which fell amidst other rearrangement positions in association with RTT-like phenotypes and reportedly affecting FOXG1 function (Fig. 3) [4, 22–24, 54]. Exome analysis by the DDD project ruled out the contribution of variants in known RTT genes (including methyl-CpG binding protein 2 (MECP2)) leading to the hypothesis that DGAP294’s clinical findings could be attributed to a position effect on FOXG1.
Fig. 3.
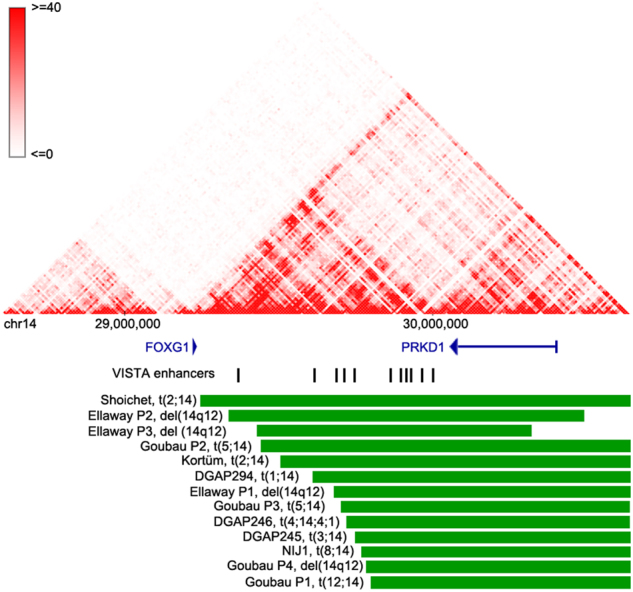
DGAP294’s 14q12 breakpoint and its corresponding TAD structures in fetal brain tissue. TAD structures are derived from the http://promoter.bx.psu.edu/ Hi-C data browser (fetal brain data at 10 kb resolution). Gene positions are obtained from the UCSC Genome Browser and are graphed with blue lines; arrows indicate transcriptional orientation. Enhancer positions are derived from UCSC and are indicated with vertical black lines. Rearrangement positions are shown with green horizontal lines and the type of rearrangement and publication are specified in black text to the left of each rearrangement
As demonstrated in other studies [4, 16–20, 55], TAD disruption may affect expression of genes located within the domain by disrupting long-range promoter/enhancer interactions. FOXG1 is most abundantly expressed in diverse brain regions [50]. Interestingly, the 14q12 BCA breakpoint likely disrupts TAD organization in H1-ESC and fetal brain tissue (Fig. 3 and Supplementary Table 9). Several enhancers within the 14q12 region have been proposed to regulate FOXG1 transcription, as observed by the expression effects of distal rearrangements [24–26]. Similar to these studies, the DGAP294 14q12 breakpoint positions the region’s enhancers onto another chromosome, potentially impacting FOXG1 transcription. Reported GTEx expression for FOXG1 and NTNG1 is very low in LCLs, the only available DGAP294 cell line (Supplementary Figures 6 and 7), and we were not able to detect significant changes in the expression of these genes with qPCR in the DGAP294 LCL (Fig. 2) (Mann–Whitney U-test, p = 0.09 for NTNG1 and p = 0.58 for FOXG1).
Discussion
The importance of mapping BCA breakpoints at sequence-level resolution has been highlighted in studies of prenatal and postnatal clinical cases [4, 20]. Precise BCA breakpoint mapping and analysis are even more relevant in cases where subjects with congenital diseases have reportedly normal chromosomal microarrays and exomes. DGAP294 is one example of such cases, as his complex combination of clinical features could not be explained by his normal exome and normal chromosomal microarray results.
The mapping and analysis of DGAP294’s chromosomal breakpoints provided new insight into the pathogenic mechanisms, which may be at play. In total, eight pairs of breakpoints were mapped near nucleotide resolution. The BCAs were found to disrupt several protein coding transcripts from ADGRL2, PCDH15, PADI3, and PADI4; clinical features associated with PADI3 and PADI4 are discordant with DGAP294’s phenotype, and although PCDH15 may contribute to DGAP294’s cortical visual impairment, the involvement of PCDH15 in DGAP294′s visual loss would require further evaluation, especially because only an ~15% reduction in PCDH15 expression was detected in the DGAP294 LCL.
ADGRL2 (also known as LPHN2, OMIM#607018), is a promising candidate for explaining DGAP294’s clinical presentation due to its predicted high sensitivity to LoF variants and dosage alterations. Three duplication cases and one deletion case that involve ADGRL2 (plus one of two adjacent genes) in ClinVar were described in subjects with failure to thrive, intellectual disability, developmental delay, and/or other significant developmental or morphological phenotypes similar to the clinical features observed in DGAP294. Importantly, these duplications do not encompass the full sequence of ADGRL2, potentially representing a tandem duplication that mimics the gene disruption caused by one of DGAP294’s translocations within 1p31.1. This raises the possibility that ADGRL2 can contribute to the phenotype observed in DGAP294, although further studies are needed to assess its pathogenicity and predicted neuronal roles in deletion and duplication cases.
In addition to the potential effects of ADGRL2 disruption in generating DGAP294’s phenotype, we analyzed the long-range position effects of DGAP294’s BCAs, which can oftentimes be important pathogenicity contributors [4, 20]. Neighboring genes within ±3 Mb windows surrounding each of DGAP294’s BCA breakpoints were assessed for their potential contribution to DGAP294’s clinical features. Of these, FOXG1 was the most interesting candidate, as it has been associated with RTT [21] and FOXG1 syndrome [22–27], a recognizable phenotype distinct from classical and congenital RTT but sharing many RTT clinical traits [23]. Phenomizer [51] diagnosed DGAP294’s phenotype as RTT (p = 0.0003), but DGAP294 cannot be classified formally as classical RTT because of a lack of evidence of regression, a necessary feature for RTT diagnosis [52]. However, DGAP294’s phenotype can be designated as FOXG1 syndrome-like because of the presence of additional clinical features such as seizures, gastroesophageal reflux, absence of speech, microcephaly, and cortical visual impairment present in FOXG1-syndrome patients in addition to shared classic and atypical RTT clinical features including gait abnormalities, stereotypic hand movements, abnormal muscle tone, scoliosis, growth retardation, and small cold hands and feet. We believe that a FOXG1 position effect greatly contributes to DGAP294’s clinical phenotype. Such an hypothesis is supported by the observation that one of DGAP294’s translocation breakpoints falls within a region in 14q12 for which long-range position effects caused by translocations and submicroscopic 14q12 deletions have been reported in FOXG1 syndrome patients [4, 23–25, 27]. Although the breakpoints described in these studies did not directly disrupt any known important regulatory elements, the translocations putatively removed enhancers from their regulatory neighborhood, thus effectively disrupting FOXG1 cis regulatory control. Quantitative real-time PCR experiments did not reveal FOXG1 downregulation in DGAP294 LCLs; this is in agreement with another study in which a FOXG1 expression change was not detected in LCLs derived from a single patient with a microdeletion near FOXG1 [24]. Although such observations may be due to predicted minimal to null expression of FOXG1 in LCLs (as detected by GTEx), future expression experiments should be focused to assess FOXG1’s transcript levels in the brain, where it is widely expressed and exerts its functional roles, either through a pluripotent stem cell line, neuronal cell line, or mouse model engineered to harbor the described DGAP294’s translocation.
Finally, NTNG1, the other position effect candidate gene in DGAP294, was excluded from further analysis as after its initial proposal as a candidate RTT gene from its disruption by a balanced translocation in a female with a RTT phenotype [53], a subsequent study failed to identify exon variants that affect NTNG1 function in RTT patients [56].
Taken together, we hypothesize that FOXG1 likely accounts for DGAP294’s neurological problems, although contribution from ADGRL2 cannot be dismissed. It is possible that ADGRL2 could add to the multiple clinical presentations of FOXG1 syndrome; however, our study cannot currently distinguish the contribution of ADGRL2 disruption in the setting of FOXG1 position effects in DGAP294 given absence of knowledge of a clear association for ADGRL2 with human disease at present. Transcriptional and chromatin conformation experiments in translocation engineered neuronal cell lines and/or mouse models may be able to tease apart the roles of both mechanisms or confirm their functional synergy.
Overall, DGAP294 highlights the importance of performing comprehensive clinical sequencing studies and their analysis when neither exome analysis nor microarrays detect variants and rearrangements that affect function; such information can be of value not only in the discovery of directly affected genes in a particular disease, but also further complement diagnosis and possible future therapies through assessment of potential position effects.
Electronic supplementary material
Acknowledgements
We offer heartfelt gratitude to DGAP294 for allowing us to explore his DNA and inspiring us with his story; and to his family, especially to his mother, for their ongoing support of our study. We want to acknowledge and spread awareness of SWAN-UK (Syndromes Without A Name), who support undiagnosed families. We first learned of DGAP294 through one of their weekly Twitter chats and we encourage other researchers to engage online with the undiagnosed disease community. We thank HGVS for assistance in the translation of DGAP294’s complex karyotype into their chromosome rearrangement nomenclature. This study was funded by the National Institutes of Health (GM061354 to CCM and MET).
Compliance with ethical standards
Conflict of interests
The authors declare no conflicts of interest.
Footnotes
Cinthya J. Zepeda-Mendoza and Alexandra Bardon contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1038/s41431-017-0068-0) contains supplementary material, which is available to authorized users.
References
- 1.Higgins AW, Alkuraya FS, Bosco AF, et al. Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am J Hum Genet. 2008;82:712–722. doi: 10.1016/j.ajhg.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HG, Kishikawa S, Higgins AW, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu W, Quintero-Rivera F, Fan Y, et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007;3:e80. doi: 10.1371/journal.pgen.0030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redin C, Brand H, Collins RL, et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet. 2017;49:36–45. doi: 10.1038/ng.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiang C, Jacobsen JC, Ernst C, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44:390–397. doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talkowski ME, Rosenfeld JA, Blumenthal I, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 8.de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denker A, de Laat W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev. 2016;30:1357–1382. doi: 10.1101/gad.281964.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillips-Cremins JE, Sauria ME, Sanyal A, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lupianez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Hnisz D, Weintraub AS, Day DS, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giorgio E, Robyr D, Spielmann M, et al. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD) Hum Mol Genet. 2015;24:3143–3154. doi: 10.1093/hmg/ddv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ordulu Z, Kammin T, Brand H, et al. Structural chromosomal rearrangements require nucleotide-level resolution: lessons from next-generation sequencing in prenatal diagnosis. Am J Hum Genet. 2016;99:1015–1033. doi: 10.1016/j.ajhg.2016.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ariani F, Hayek G, Rondinella D, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet. 2008;83:89–93. doi: 10.1016/j.ajhg.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shoichet SA, Kunde SA, Viertel P, et al. Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet. 2005;117:536–544. doi: 10.1007/s00439-005-1310-3. [DOI] [PubMed] [Google Scholar]
- 23.Kortum F, Das S, Flindt M, et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet. 2011;48:396–406. doi: 10.1136/jmg.2010.087528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellaway CJ, Ho G, Bettella E, et al. 14q12 microdeletions excluding FOXG1 give rise to a congenital variant Rett syndrome-like phenotype. Eur J Hum Genet. 2013;21:522–527. doi: 10.1038/ejhg.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allou L, Lambert L, Amsallem D, et al. 14q12 and severe Rett-like phenotypes: new clinical insights and physical mapping of FOXG1-regulatory elements. Eur J Hum Genet. 2012;20:1216–1223. doi: 10.1038/ejhg.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mencarelli MA, Kleefstra T, Katzaki E, et al. 14q12 Microdeletion syndrome and congenital variant of Rett syndrome. Eur J Med Genet. 2009;52:148–152. doi: 10.1016/j.ejmg.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 27.Takagi M, Sasaki G, Mitsui T, Honda M, Tanaka Y, Hasegawa T. A 2.0 Mb microdeletion in proximal chromosome 14q12, involving regulatory elements of FOXG1, with the coding region of FOXG1 being unaffected, results in severe developmental delay, microcephaly, and hypoplasia of the corpus callosum. Eur J Med Genet. 2013;56:526–528. doi: 10.1016/j.ejmg.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Firth HV, Wright CF, Study DDD. The Deciphering Developmental Disorders (DDD) study. Dev Med Child Neurol. 2011;53:702–703. doi: 10.1111/j.1469-8749.2011.04032.x. [DOI] [PubMed] [Google Scholar]
- 29.Wright CF, Fitzgerald TW, Jones WD, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–1314. doi: 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanscom C, Talkowski M. Design of large-insert jumping libraries for structural variant detection using Illumina sequencing. Curr Protoc Hum Genet. 2014;80:21–29. doi: 10.1002/0471142905.hg0722s80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talkowski ME, Ernst C, Heilbut A, et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet. 2011;88:469–481. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brand H, Pillalamarri V, Collins RL, et al. Cryptic and complex chromosomal aberrations in early-onset neuropsychiatric disorders. Am J Hum Genet. 2014;95:454–461. doi: 10.1016/j.ajhg.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brand H, Collins RL, Hanscom C, et al. Paired-duplication signatures mark cryptic inversions and other complex structural variation. Am J Hum Genet. 2015;97:170–176. doi: 10.1016/j.ajhg.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heger A, Webber C, Goodson M, Ponting CP, Lunter G. GAT: a simulation framework for testing the association of genomic intervals. Bioinformatics. 2013;29:2046–2048. doi: 10.1093/bioinformatics/btt343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andersson R, Gebhard C, Miguel-Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ordulu Z, Wong KE, Currall BB, et al. Describing sequencing results of structural chromosome rearrangements with a suggested next-generation cytogenetic nomenclature. Am J Hum Genet. 2014;94:695–709. doi: 10.1016/j.ajhg.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taschner PE, den Dunnen JT. Describing structural changes by extending HGVS sequence variation nomenclature. Hum Mutat. 2011;32:507–511. doi: 10.1002/humu.21427. [DOI] [PubMed] [Google Scholar]
- 44.Nilsson D, Pettersson M, Gustavsson P, et al. Whole-genome sequencing of cytogenetically balanced chromosome translocations identifies potentially pathological gene disruptions and highlights the importance of microhomology in the mechanism of formation. Hum Mutat. 2017;38:180–192. doi: 10.1002/humu.23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Narendra V, Rocha PP, An D, et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science. 2015;347:1017–1021. doi: 10.1126/science.1262088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iwamoto T, Ikari K, Nakamura T, et al. Association between PADI4 and rheumatoid arthritis: a meta-analysis. Rheumatol (Oxf) 2006;45:804–807. doi: 10.1093/rheumatology/kel023. [DOI] [PubMed] [Google Scholar]
- 47.Lee YH, Bae SC. Association between susceptibility to rheumatoid arthritis and PADI4 polymorphisms: a meta-analysis. Clin Rheumatol. 2016;35:961–971. doi: 10.1007/s10067-015-3098-4. [DOI] [PubMed] [Google Scholar]
- 48.Ahmed ZM, Riazuddin S, Bernstein SL, et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohler S, Schulz MH, Krawitz P, et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85:457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borg I, Freude K, Kubart S, et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with Rett syndrome. Eur J Hum Genet. 2005;13:921–927. doi: 10.1038/sj.ejhg.5201429. [DOI] [PubMed] [Google Scholar]
- 54.Goubau C, Devriendt K, Van der Aa N, et al. Platelet defects in congenital variant of Rett syndrome patients with FOXG1 mutations or reduced expression due to a position effect at 14q12. Eur J Hum Genet. 2013;21:1349–1355. doi: 10.1038/ejhg.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ibn-Salem J, Kohler S, Love MI, et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014;15:423. doi: 10.1186/s13059-014-0423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Archer HL, Evans JC, Millar DS, et al. NTNG1 mutations are a rare cause of Rett syndrome. Am J Med Genet A. 2006;140:691–694. doi: 10.1002/ajmg.a.31133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.