

Abstract
Background
Esophageal sarcomatoid carcinoma (ESC) is a rare disease with a mixture of both carcinomatous and sarcomatous components in the tumor. Its genetic background and mechanisms of oncogenesis remain largely unknown.
Methods
Here we performed targeted next generation sequencing (NGS) on a pan-cancer gene panel in 15 ESC tumors to explore their genetic alterations, and aimed to identify clinically actionable mutations for future treatment instructions.
Results
TP53 alterations were identified in all patients. Alterations in receptor tyrosine kinases (RTK) were identified in 10 out of 15 patients. Members of downstream RAS and PI3-kinase pathways are also mutated in 10 patients, and PIK3CA is the top mutated gene in these pathways. In addition, we identified mutations on histone modification genes in 5 patients, including histone acetyltransferase gene EP300 and its homologue CREBBP, lysine methyltransferase genes KMT2A and KMT2B, and lysine demethylase gene KDM5A. Finally, mismatch repair (MMR) genes and proofreading gene POLE all together were mutated in one third of the ESC patients.
Conclusions
This is the first study to unravel the mutational profile of ESC tumors. Our findings could match 9 patients to the targeted therapies currently available in clinical practice or in active clinical trials, suggesting the potential utility of targeted therapies for this rare disease in the future.
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4159-2) contains supplementary material, which is available to authorized users.
Keywords: Esophageal sarcomatoid carcinoma, Next generation sequencing, Mutation profiling, Targeted therapy
Background
Esophageal sarcomatoid carcinoma (ESC) is a rare malignant tumor with a reported incidence of below 2% in esophageal carcinomas [1, 2]. The majority of the patients were male at the age of 60 and the correlation with smoking behavior is not clear. The histological pattern of sarcomatoid carcinoma is characterized by a biphasic appearance of both carcinomatous and sarcomatous components, most frequently a mixture of the malignant spindle cells and squamous cells [3]. Molecular characterization now supports the theory that these two components are originated from the same tissue precursor rather than two independent precursors [4, 5]. For diagnostic pathology, two types of immunohistochemical biomarkers, including epithelial biomarkers (e.g. cytokeratin and epithelial membrane antigen) and mesenchymal biomarkers (e.g. vimentin, smooth muscle actin and S100) are used to identify the carcinomatous and sarcomatous components, respectively [6]. However, the etiology of this rare disease is unclear due to the lack of genetic information.
Previous reports of ESC are often sporadic case studies without in-depth investigation of genetic characteristics. As a result, surgery to dissect tumors and nearby lymph nodes, complemented with adjuvant chemotherapy and/or radiotherapy before or afterwards, is the most adoptive treatment to this disease, and targeted therapies still remain unavailable. The 3-year survival rate of ESC is close to 60%, which is much higher than that of esophageal squamous cell carcinoma (ESCC) [6, 7]. However, its 5-year survival rate is close to that of ESCC, likely due to a lack of effective treatments to recurrent diseases.
To explore the genomic features of ESC and provide evidences for developing targeted therapies, we performed targeted next generation sequencing (NGS) with a customized NGS panel, which covers exons of 416 cancer-relevant genes and introns of 16 fusions genes, to interrogate genomic alterations in 15 dissected primary ESC tumors. All tumor samples were sampled at the time of diagnosis through esophagoscopy, or from surgical dissections of tumor. Due to the lack of normal controls for these tissue samples, public and in-house databases were used to filter out germline mutations and identify somatic mutations. The sequencing results not only depicted multiple molecular pathways that were commonly influenced in this disease, but also revealed the genetic heterogeneity between individuals. Moreover, clinically actionable mutations were identified in the majority of patients, suggesting the potential of involving targeted drugs into future treatment to improve the long-term survival rates of this disease.
Methods
Sample collection and sequencing library preparation
Formalin-fixed paraffin-embedded (FFPE) blocks from 15 ESC patients between 2009 and 2016 were retrospectively collected from Zhejiang Cancer Hospital in China. Tumor tissues were taken either at the time of diagnosis through esophagoscopy, or from surgically removed tumors. Only patient #3 received neoadjuvant therapy (2 cycles of docetaxel anhydrous and cisplatin-based therapy) prior sample acquisition. FFPE sections containing both carcinoma and sarcoma components of the tumor were collected for genomic DNA extraction using QIAamp FFPE DNA kit (Qiagen, Hilden, Germany) according to manufacturer’s protocol. We were unable to collect paired normal DNA controls for each patient due to the death of the patients or their release from the hospital. All DNA was qualified using Nanodrop 2000 and quantified using Qubit 2.0 fluorometer with Qubit dsDNA HS Assay Kit (Thermo Fisher, Waltham, MA).
500 ng~ 1 μg of extracted DNA for each sample was sheared into 350 bp fragments using Covaris M220 instrument (Covaris, Woburn, MA), followed by library preparation using KAPA Hyper DNA Library Prep Kit (KAPA Biosystems, Wilmington, MA). In brief, fragmented DNA underwent sequential steps of end-repairing, A-tailing and ligation with indexed adapters, followed by size selection with Agencourt AMPure XP beads (Beckman Coulter, Mississauga, Canada) and PCR amplification.
A customized NGS panel targeting exons of 416 cancer-relevant genes and introns of 16 fusions genes was used for hybridization enrichment (Additional file 1: Table S1). In brief, indexed DNA libraries were pooled together to a total amount of 2 μg and subjected to probe-based hybridization using IDT xGen Lockdown reagents (IDT, Coralville, IA) and Dynabeads M-270 (Thermo Fisher). Captured libraries were on-beads amplified with Illumina p5 and p7 primers in KAPA HiFi HotStart ReadyMix (KAPA Biosystems). The final library was quantified by KAPA Library Quantification kit (KAPA Biosystems) per manufacturer’s instructions. Bioanalyzer 2100 (Agilent, Stanta Clara, CA) was used to determine the fragment size distribution of the final library.
Sequencing and data processing
FFPE samples of dissected primary tumors were sequenced to an average sequencing coverage of at least 300× on Illumina Hiseq 4000 platform (Illumina, San Diego, CA). The sequencing data were first demultiplexed by bck2fastq and then subjected to Trimmomatic [8] for FASTQ file quality control (QC). Leading/trailing low quality (base phred score below 15) or N bases were removed. Qualified reads were mapped to the reference human genome hg19 using Burrows-Wheller Aligner (BWA-mem, v0.7.12) [9]. Genome Analysis Toolkit (GATK 3.4.0) was employed to apply the local realignment around indels and base quality score recalibration. PCR duplicates were removed by Picard (available at: https://broadinstitute.github.io/picard/). VarScan2 was employed for the detection of single-nucleotide variations (SNVs) and insertion/deletion mutations with the following parameters: minimum read depth = 20, minimum base quality = 15, minimum variant allele frequency (VAF) = 0.01, minimum variant supporting reads = 5, variant supporting reads mapped to both strands, and strand bias no greater than 10% [10]. ADTEx (http://adtex.sourceforge.net) was used to identify copy number variations (CNVs) using a normal human HapMap DNA sample NA18535, and the cutoff of log2 ratio was set at ±0.6 for copy number changes (corresponding to 1.5-fold copy number gain and 0.65-fold copy number loss).
Variant filtering and annotation
The vcf files contain both single-nucleotide polymorphism (SNPs) and small insertions/deletions (indels) were annotated by ANNOVAR against the following databases: dbSNP (v138), 1000Genome, ExAC, COSMIC (v70), ClinVAR, and SIFT. Only missense, stopgain, frameshift and non-frameshift indel mutations were kept. Mutations were removed if they were present in > 1% population frequency in the 1000 Genomes Project or 65,000 exomes project (ExAC). The resulted mutation lists were filtered through an internally collected list of recurrent sequencing errors on the same sequencing platform, which is summarized from the sequencing results of 53 normal samples with a minimum average sequencing depth of 700×. Specifically, if a variant was detected (i.e. ≥3 mutant reads and > 1% VAF) in > 20% of the normal samples, it was considered a likely artifact and was removed. Mutations occurred within the repeat masked regions were also removed. In a further filtering step, the mutation was only called out when the VAF is above 1% with a minimum of 5 mutant reads for COSMIC mutations, or above 2% with a minimum of 8 mutant reads for non-COSMIC mutations.
Results
Patient information
Of the 15 ESC patients enrolled, 12 were males and 9 of them were heavy smokers (≥ 20 packs/year). All 3 female patients have no smoking history. The median age of all patients is 65, ranging from 46 to 70. Eleven patients were diagnosed with stage I-II diseases, and 4 patients had stage III diseases (Table 1). Primary tumors occurred mostly in the bottom two thirds of esophagus (14 patients in total) and 10 patients were classified as the polypoid type when inspected by esophagoscopy.
Table 1.
Clinical characteristics of patients
| Age at diagnosis (n = 15) | Years |
| Median | 65 |
| Range | 46 - 70 |
| Gender | No. of patients |
| Female | 3 |
| Male | 12 |
| Clinical stage | No. of patients |
| I | 4 |
| II | 7 |
| III | 4 |
| Primary tumor position in esophagus | No. of patients |
| Upper thoracic | 1 |
| Mid-thoracic | 7 |
| Lower thoracic | 7 |
| Macroscopic type | No. of patients |
| Polypoid type | 10 |
| Ulcerating type | 1 |
| Infiltrative type | 4 |
Molecular pathways that were influenced by identified mutations
Of these 15 patients, a total of 161 genetic alterations were identified in 94 genes (Additional file 2: Table S2). Six cases have mutations in histone modification genes. In particular, EP300 was mutated in 3 cases and its homolog CREBBP in 1 case (Fig. 1a). Both proteins encoded by EP300 and CREBBP are suggested as tumor suppressors and indicative of poor prognosis in ESCC [11]. All EP300 mutations are in its histone acetyltransferase (HAT) domain and the mutation Y1414C which has already been reported as an inactivating mutation occurred in two patients [11]. In addition, 4 cases have mutations in KMT2B, KDM5A or KMT2A, all of which are components of histone methyltransferase complex (HMT). These results suggest that epigenetic regulation may play a critical role in ESC oncogenesis.
Fig. 1.
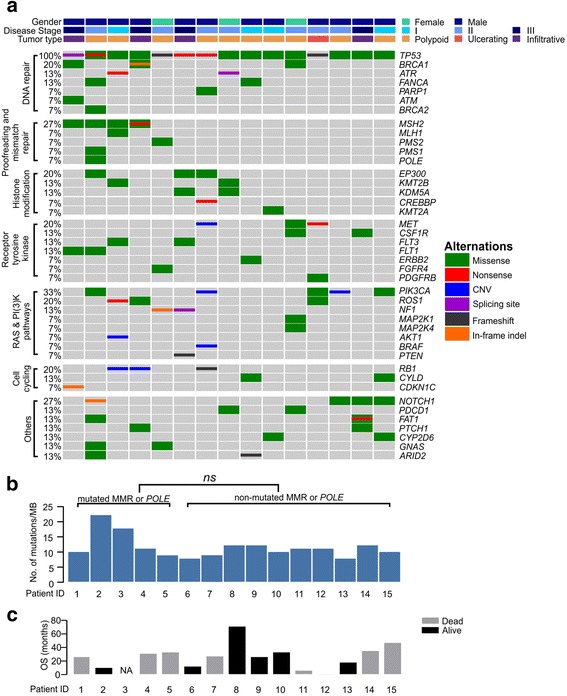
Mutation analysis of ESC patients. a A co-mutation plot of various types of mutations in all patients. Genes were grouped according to their functions. Each column represents one patient. The mutation rates of each gene were marked on the left in percentage and grouped according to their protein functions. Patient characteristics such as gender, disease stage and tumor type were shown at the top with different colors. b TMB in each patient. “ns”, not significant (Student’s t test, p < 0.05). c For overall survival (OS) time, black bars indicate that the patient was still alive at the time of last visit and grey bars indicate patients that were passed away. One patient was lost to follow-up and his OS time was marked as “NA”. All patients were placed in the same order in the 3 panels
We also identified that receptor tyrosine kinases (RTK) and their downstream pathways were frequently mutated in these patients (Fig. 1a). Eleven out of 15 patients have at least one nonsynonymous mutation or structural variation in RTKs, including MET, CSF1R, FLT3, FLT1, ETBB2, FGFR4 and PDGFRB, and their occurrences were mutually exclusive from each other. As to the downstream elements of RTKs, PIK3CA is altered in 5 cases and 2 of them are copy number gain. The other 3 variants are E545K, M1043 V and H1047L, all of which have been reported to increase the catalytic activity of the p110α subunit of PIK3CA [12, 13].
Genes related to cell cycling and DNA repair is another commonly mutated category in ESC, but the mutation spectrum was not entirely the same as in ESCC. The mutation rate of TP53 was quite similar between ESCC (93%) [11] and ESC (100%) (Fig. 1a), but much higher than the reported 73% in gynaecological carcinosarcoma, which is also a mixture of both carcinomatous and sarcomatous components with gynaecological origin [14]. Meanwhile, the loss of RB1 gene was observed in 3 cases (20%) either by copy number loss or frameshift alteration, which is higher than its prevalence in ESCC (9%) [11]. We also observed that CCND1 and CDKN2A, encoding two key molecules for cell cycling, were rarely altered in ESC (Additional file 2: Table S2), while CCND1 amplification and CDKN2A mutations were found in 30% and 20% of ESCC patients, respectively [15].
The NOTCH pathway is also frequently dysregulated in ESC patients as we identified four NOTCH1 mutations (27%) (Fig. 1a) and one FBXW7 inactive mutation H460R (Additional file 1: Table S1) [16]. NOTCH1 encodes a ligand-activated transcription factor in regulating cell differentiation, proliferation and apoptosis [17]. Mutations of NOTCH1 are commonly identified in ESCC (13%) [11], acute and chronic lymphoblastic leukemia and suggested as oncogenic [18]. On the other hand, FBXW7 is a tumor suppressor and its inactivation could result in the constitutive activation of NOTCH1, cyclin E, c-Myc and other oncogenic factors [19, 20].
The tumor mutation burden (TMB) is defined as the number of non-synonymous and indel mutations per mega base (Mb) and it is ranged from 7.8 to 22.2 with a median of 11.1 in these patients (Fig. 1b). Notably, one third of the patients have either missense mutations or truncations in the mismatch repair (MMR) genes (MSH2, MLH1, PMS2, PMS1) and DNA polymerase POLE (Fig. 1a), which are all related to genomic stability [21, 22]. During variant annotation in ANNOVAR, SIFT tool was used to predict the influence of these mutations to their protein functions and ClinVar was used for the clinical significance annotation. We found that half of these mutations (patient #3, 4, 5) were predicted to be neutral in SIFT, and ClinVar only identified MSH2 R929X (patient #3) as a pathogenic variant (Table 2). Correspondingly, TMB of these cases is not significantly higher than those without mutations in these genes (Fig. 1b). Patient #1 harbored one predicted-deleterious MSH2 mutation, but demonstrated similar TMB as others. Only patient #2, whom was predicted to have functionally deleterious mutations in PMS1, MSH2 and POLE, had a TMB of 22 mutations per MB, the highest among all studied cases.
Table 2.
SNVs in MMR and proofreading genes
| Patient ID | Gene | Mutation | SIFT | CLIN_SIG |
|---|---|---|---|---|
| 1 | MSH2 | p.V78I (c.G232A) | Deleterious | – |
| 2 | POLE | p.E991Q (c.G2971C) | Deleterious | – |
| PMS1 | p.A6V (c.C17T) | Deleterious | – | |
| MSH2 | p.A600T (c.G1798A) | Deleterious | – | |
| 3 | MSH2 | p.Q629R (c.A1886G) | Neutral | Not_provided;benign |
| MLH1 | p.Q701K (c.C2101A) | Neutral | Likely_benign;pathogenic | |
| 4 | MSH2 | p.R929X (c.C2785T) | – | Pathogenic |
| MSH2 | p.T564A (c.A1690G) | Neutral | Benign | |
| 5 | PMS2 | p.H435Q (c.C1305G) | Neutral | – |
CLIN_SIG: clinical significance predicted by ClinVar
Identification of clinically actionable mutations
Due to the poor understanding of ESC genomic profiles, currently there are no targeted therapies approved for these patients. In this study, patients were subjected to surgical desection and/or chemo/radiotherapies and their survival time was ranged from 1 month to exceeding 71 months (alive at the time of testing) after diagnosis (Fig. 1c), suggesting that their genetic background could possibly be related to the extraordinary differences of their prognosis. In this study, clinically actionable mutations were defined as mutations showing sensitivity to existing targeted therapies or drugs in clinical trials, or having been approved to influence the outcomes of targeted therapies, regardless of the cancer types in the original trials [23]. Of 15 ESC patients, 9 have been identified with at least one clinical actionable mutation based on the results of preclinical or clinical trials (Table 3) [24–32]. Taking TP53 for example, supportive evidences include: 1) patients with carcinomas demonstrated better responses to bevacizumab treatment if carrying TP53 mutations [24]; 2) patients with sarcomas showed better responses to pazopanib treatment [25]. In addition, the PI3-kinase pathway was activated in 6 patients by altering AKT1, PIK3CA or PTEN functions, and it can be targeted by multiple PI3K/mTOR/AKT or MEK inhibitors that are currently in active clinical trials [26, 27].
Table 3.
Clinically actionable gene alterations
| Gene | Alterations | Patient ID | Significances in treatment and prognosis |
|---|---|---|---|
| AKT1 | Amplification | 3 | Response to mTOR inhibitors, AKT inhibitor MK2206 [28] |
| CREBBP | p.Q540X (c.C1618T) | 7 | Response to HDAC inhibitors (active clinical trial) [29] |
| HNF1A | p.288 fs (c.864_865insC) | 3 | Response to mTOR inhibitors in in-vitro experiments [30] |
| IDH1 | p.R132H (c.G395A) | 8 | Response to IDH1 and pan-IDH inhibitors (active clinical trial) [31] |
| MET | Amplification | 7 | Response to c-MET inhibitors |
| NF1 | c.A3975-2 T | 6 | Possibly increased sensitivity to MEK inhibitors [32] |
| PIK3CA | p.E545K (c.G1633A) | 12 | Response to PI3K/AKT/mTOR inhibitors [26, 27] |
| p.M1043 V (c.A3127G) | 2 | ||
| Amplification | 13, 7 | ||
| PTEN | p.R142fs (c.425delG) | 6 | Response to PI3K/AKT/mTOR inhibitors [27] |
| TP53 | p.Y234C (c.A701G) | 13 | Better response to bevacizumab [24]; Better response to pazopanib in advanced sarcoma [25] |
| p.R273C (c.C817T) | 9 | ||
| Nonsense | 2, 7 | ||
| Frameshift | 2, 5, 12 |
Discussion
In this study, we analyzed the genomic profiles of 15 ESC patients by targeted sequencing of pan-cancer genes in archived primary FFPE tissue samples. For the first time, we obtained an overall picture of the genomic alterations for this rare disease that could be inspirational for developing and selecting targeted therapies in the future. Despite the differences of cellular morphology between sarcoma and carcinoma components in the tumor, they seem to share the majority of the mutations according to a study of uterine carcinosarcoma [4]. By comparing the mutational profiles of these ESC patients to a previous study on Chinese ESCC patients, we found similarities but also differences of genomic alterations between these two diseases. Typically, TP53 was mutated in all ESC cases, which is comparable to 93% in ESCC [11]. Besides, histone modifier genes show a high mutation rate in both ESC (40%) and ESCC (63%), and EP300 and its homolog CREBBP are the most commonly mutated genes. EP300 mutations were associated with shorter overall survival (OS) time (median survival time around 20 months) in ESCC [11]. In the ESC patients we tested, the OS time of 3 cases with EP300 alterations are > 10 months, > 12 months (both alive at the time of the return visit) and 27 months. A longer observation time is required to compare the survival rates of ESC patients with or without EP300 mutations.
In addition, 5 out of 15 (33%) patients have PIK3CA alterations, much higher than what was observed in ESCC (5%) (p = 0.0002) [11]. All 5 cases with PIK3CA alterations were male patients at the stage I-II diseases with smoking history. The OS of these patients show tremendous differences, ranging from 1 to 47 months. Due to the limit sample size, we are not be able to link PIK3CA alterations to the possibility of early diagnosis or poor prognosis, but PIK3CA alterations are now clinically actionable with potential responses to PI3K/AKT/mTOR inhibitors [33, 34]. Patients would be subjected to targeted treatment and their responses and disease recurrence can be monitored by regular inspection of this alteration.
This study uncovered the diversity of tumor genetic background in ESC patients. Despite 5 patients carried mutations on MMR and proofreading genes, half of them were predicted to be benign. Therefore, it is not surprised that these 5 patients did not show a significant higher TMB comparing to other patients. The reason is possibly the functional overlapping of MMR proteins that could compensate the functional loss of each other [35], which is also explanatory in patient #2, as he had more than one MMR genes altered and his TMB level is relatively higher comparing to others.
Conclusions
To our knowledge, this is the very first study specifically investigated the genomic alterations in ESC patients. Patients carrying these mutations could be potentially treated with targeted drugs to improve their long-term prognosis. The observations of high mutation rates in PIK3CA and histone modifier genes need to be validated in a larger sample size in the future if used to define clinical strategies for personalized therapy for this disease.
Additional files
Table S1. The gene list of the gene panel used for the targeted sequencing. It includes 416 cancer-relevant genes and introns of 16 fusions genes (bold text). (XLSX 13 kb)
Table S2. Gene alterations in all patients and their functional and clinical significance annotation. All mutations were annotated with SIFT, CLINVAR and COSMIC in ANNOVAR software. (XLSX 21 kb)
Acknowledgements
Not applicable.
Funding
This work was supported by Talent Training Program of Zhejiang Cancer Hospital (ID: 1022). The funding was mainly used to support the experiments of DNA sample preparation, next generation sequencing and bioinformatics data analysis.
Availability of data and materials
The datasets generate and analyzed in this study are not publicly available for the reason of protecting patients’ privacy, but are available from the corresponding authors on reasonable request.
Abbreviations
- ESC
Esophageal sarcomatoid carcinoma
- ESCC
Esophageal squamous cell carcinoma
- FFPE
Formalin-fixed paraffin-embedded
- HMT
Histone methyltransferase complex
- MAF
Mutation allele frequency
- MMR
Mismatch repair
- NGS
Next generation sequencing
- RTK
Receptor tyrosine kinase
- TMB
Tumor mutation burden
Authors’ contributions
HL, YS and WM were mainly in charge of the study design, data review and manuscript preparation. SY and HZ contributed to data analysis and figure preparation. XT and XW were involved in experimental design and performance of DNA library preparation, quality control and sample loading for NGS. FX, JQ, NH and YF screened clinical records and assembled patient information. They also prepared the FFPE samples, determined the tumor purity that qualified for testing and conducted the DNA extraction from these samples. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of Zhejiang Cancer Hospital. All included subjects have provided written informed consent.
Consent for publication
Not applicable.
Competing interests
Yang W. Shao, Xiaoling Tong and Xue Wu are the shareholders or employees of Geneseeq Technology Inc.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4159-2) contains supplementary material, which is available to authorized users.
Contributor Information
Hongyang Lu, Email: luhy@zjcc.org.cn.
Shifeng Yang, Email: yangsf@zjcc.org.cn.
Huineng Zhu, Email: zhn00@126.com.
Xiaoling Tong, Email: xiaoling.tong@geneseeq.com.
Fajun Xie, Email: fjxie@foxmail.com.
Jing Qin, Email: qinjing@zjcc.org.cn.
Na Han, Email: hanna@zjcc.org.cn.
Xue Wu, Email: xue.wu@geneseeq.com.
Yun Fan, Email: fanyun@zjcc.org.cn.
Yang W. Shao, Email: yang.shao@geneseeq.com
Weimin Mao, Email: weiminmao@163.com.
References
- 1.Hatch GF, 3rd, Wertheimer-Hatch L, Hatch KF, Davis GB, Blanchard DK, Foster RS, Jr, Skandalakis JE. Tumors of the esophagus. World J Surg. 2000;24(4):401–411. doi: 10.1007/s002689910065. [DOI] [PubMed] [Google Scholar]
- 2.Iyomasa S, Kato H, Tachimori Y, Watanabe H, Yamaguchi H, Itabashi M. Carcinosarcoma of the esophagus: a twenty-case study. Jpn J Clin Oncol. 1990;20(1):99–106. [PubMed] [Google Scholar]
- 3.Jin Z, Ogata S, Tamura G, Katayama Y, Fukase M, Yajima M, Motoyama T. Carcinosarcomas (malignant mullerian mixed tumors) of the uterus and ovary: a genetic study with special reference to histogenesis. Int J Gynecol Pathol. 2003;22(4):368–373. doi: 10.1097/01.pgp.0000092134.88121.56. [DOI] [PubMed] [Google Scholar]
- 4.McConechy MK, Hoang LN, Chui MH, Senz J, Yang W, Rozenberg N, Mackenzie R, McAlpine JN, Huntsman DG, Clarke BA, et al. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J Pathol Clin Res. 2015;1(3):173–185. doi: 10.1002/cjp2.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong AB, Wang M, Eble JN, MacLennan GT, Montironi R, Tan PH, Lopez-Beltran A, Zhang S, Baldridge LA, Spartz H, et al. TP53 mutational analysis supports monoclonal origin of biphasic sarcomatoid urothelial carcinoma (carcinosarcoma) of the urinary bladder. Mod Pathol. 2009;22(1):113–118. doi: 10.1038/modpathol.2008.176. [DOI] [PubMed] [Google Scholar]
- 6.Sano A, Sakurai S, Kato H, Sakai M, Tanaka N, Inose T, Saito K, Sohda M, Nakajima M, Sakamoto K, et al. Clinicopathological and immunohistochemical characteristics of esophageal carcinosarcoma. Anticancer Res. 2009;29(8):3375–3380. [PubMed] [Google Scholar]
- 7.Wang L, Lin Y, Long H, Liu H, Rao H, He Y, Rong T, Liang Y. Esophageal carcinosarcoma: a unique entity with better prognosis. Ann Surg Oncol. 2013;20(3):997–1004. doi: 10.1245/s10434-012-2658-y. [DOI] [PubMed] [Google Scholar]
- 8.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46(10):1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 12.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104(13):5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102(3):802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D, Angiuoli SV, Sausen M, Kann L, Shukla M, et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun. 2014;5:5006. doi: 10.1038/ncomms6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawada G, Niida A, Uchi R, Hirata H, Shimamura T, Suzuki Y, Shiraishi Y, Chiba K, Imoto S, Takahashi Y, et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology. 2016;150(5):1171–1182. doi: 10.1053/j.gastro.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 16.Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, Hempen PM, Hilgers W, Yeo CJ, Hruban RH, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003;163(4):1255–1260. doi: 10.1016/S0002-9440(10)63485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wael H, Yoshida R, Kudoh S, Hasegawa K, Niimori-Kita K, Ito T. Notch1 signaling controls cell proliferation, apoptosis and differentiation in lung carcinoma. Lung Cancer. 2014;85(2):131–140. doi: 10.1016/j.lungcan.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Rossi D, Gaidano G. Molecular genetics of high-risk chronic lymphocytic leukemia. Expert Rev Hematol. 2012;5(6):593–602. doi: 10.1586/ehm.12.58. [DOI] [PubMed] [Google Scholar]
- 19.Kourtis N, Strikoudis A, Aifantis I. Emerging roles for the FBXW7 ubiquitin ligase in leukemia and beyond. Curr Opin Cell Biol. 2015;37:28–34. doi: 10.1016/j.ceb.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014;26(4):455–464. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 22.Henninger EE, Pursell ZF. DNA polymerase epsilon and its roles in genome stability. IUBMB Life. 2014;66(5):339–351. doi: 10.1002/iub.1276. [DOI] [PubMed] [Google Scholar]
- 23.Shu Y, Wu X, Tong X, Wang X, Chang Z, Mao Y, Chen X, Sun J, Wang Z, Hong Z, et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep. 2017;7(1):583. doi: 10.1038/s41598-017-00520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Said R, Hong DS, Warneke CL, Lee JJ, Wheler JJ, Janku F, Naing A, Falchook GS, Fu S, Piha-Paul S, et al. P53 mutations in advanced cancers: clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget. 2013;4(5):705–714. doi: 10.18632/oncotarget.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koehler K, Liebner D, Chen JL. TP53 mutational status is predictive of pazopanib response in advanced sarcomas. Ann Oncol. 2016;27(3):539–543. doi: 10.1093/annonc/mdv598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung KS, Lee J, Park SH, Park JO, Park YS, Lim HY, Kang WK, Kim ST. Pilot study of sirolimus in patients with PIK3CA mutant/amplified refractory solid cancer. Mol Clin Oncol. 2017;7(1):27–31. doi: 10.3892/mco.2017.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R, et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203–210. doi: 10.2147/OTT.S89967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, Fenga C, Spandidos D, Libra M, Tsatsakis AM. Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use (review) Int J Oncol. 2016;48(3):869–885. doi: 10.3892/ijo.2015.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hahn NM, Picus J, Bambury RM, Pal SK, Hart LL, Grivas P, Milowsky MI, Alva AS, Sonpavde G, Mortazavi A, et al. A phase 2 study of the histone deacetylase (HDAC) inhibitor mocetinostat in patients with urothelial carcinoma (UC) and inactivating alterations of acetyltransferase genes. J Clin Oncol. 2015;33(15_suppl):TPS4575. [Google Scholar]
- 30.Pelletier L, Rebouissou S, Paris A, Rathahao-Paris E, Perdu E, Bioulac-Sage P, Imbeaud S, Zucman-Rossi J. Loss of hepatocyte nuclear factor 1alpha function in human hepatocellular adenomas leads to aberrant activation of signaling pathways involved in tumorigenesis. Hepatology. 2010;51(2):557–566. doi: 10.1002/hep.23362. [DOI] [PubMed] [Google Scholar]
- 31.Garrett-Bakelman FE, Melnick AM. Mutant IDH: a targetable driver of leukemic phenotypes linking metabolism, epigenetics and transcriptional regulation. Epigenomics. 2016;8(7):945–957. doi: 10.2217/epi-2016-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE, Rizvi TA, et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med. 2016;375(26):2550–2560. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JJ, Loh K, Yap YS. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol Med. 2015;12(4):342–354. doi: 10.7497/j.issn.2095-3941.2015.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baselga J, Im S-A, Iwata H, Clemons M, Ito Y, Awada A, Chia S, Jagiello-Gruszfeld A, Pistilli B, Tseng L-M, et al. Abstract S6-01: PIK3CA status in circulating tumor DNA (ctDNA) predicts efficacy of buparlisib (BUP) plus fulvestrant (FULV) in postmenopausal women with endocrine-resistant HR+/HER2– advanced breast cancer (BC): first results from the randomized, phase III BELLE-2 trial. Cancer Res. 2016;76(4 Supplement):S6-01-S06-01. [Google Scholar]
- 35.Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics. 1998;148(4):1637–1646. doi: 10.1093/genetics/148.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The gene list of the gene panel used for the targeted sequencing. It includes 416 cancer-relevant genes and introns of 16 fusions genes (bold text). (XLSX 13 kb)
Table S2. Gene alterations in all patients and their functional and clinical significance annotation. All mutations were annotated with SIFT, CLINVAR and COSMIC in ANNOVAR software. (XLSX 21 kb)
Data Availability Statement
The datasets generate and analyzed in this study are not publicly available for the reason of protecting patients’ privacy, but are available from the corresponding authors on reasonable request.