

Abstract
The sonic hedgehog (SHH) signaling pathway has been shown to play important roles in embryogenesis, cell proliferation as well as in cell differentiation. It is aberrantly activated in various common cancers in adults, but also in pediatric neoplasms, such as rhabdomyosarcoma (RMS) and atypical teratoid/rhabdoid tumors (AT/RTs). Dysregulation and germline mutation in PATCHED1 (PTCH1), a receptor for SHH, is responsible for the Gorlin Syndrome, a familial cancer predisposing syndrome including RMS. Here, we report a newborn diagnosed with congenital embryonal RMS. Whole-exome sequencing (WES) identified the presence of two heterozygous germline mutations in two target genes of the SHH signaling pathway. The PTCH1 mutation p.(Gly38Glu) is inherited from the mother, whereas the PTCH2 p.(His622Tyr) mutation is transmitted from the father. Quantitative RT-PCR expression analysis of GLI and SMO, key players of the SHH pathway, showed significantly increase in the tumor tissue of the patient and also enrichment in the germline sample in comparison to the parents indicating activation of the SHH pathway in the patient. These findings demonstrate that SHH pathway activity seems to play a role in eRMS as evidenced by high expression levels of GLI1 RNA transcripts. We speculate that PTCH2 modulates tumorigenesis linked to the PTCH1 mutation and is likely associated with the congenital onset of the RMS observed in our patient.
Introduction
Congenital cancers rarely occur in pediatric oncology and neonatology counting approximately 2% of childhood cancers with an estimated prevalence of 3.65 per 100,000 live births. Neonatal solid tumors are a heterogeneous group including neuroblastoma and teratoma, followed by Wilms tumor, brain tumor, leukemia, retinoblastoma, and rhabdomyosarcoma (RMS) [1]. These may be caused either by intrauterine exposure to mutagenic factors or point towards an underlying genetic cancer predisposition syndrome (CPS).
In general, embryogenesis is regulated by a variety of complex signaling cascades that are critical for normal development. One of these central cascades is the sonic hedgehog (SHH) signaling pathway, which plays a vital role in human development, cell proliferation, and differentiation [2]. The SHH signaling pathway is complex and depends on several factors including the type of responding cell, the dose of hedgehog (HH) protein received, and the time of exposure to HH [3]. It is activated by binding of HH protein to patched receptor (PTCH) 1 or 2, which suspends the inhibitory effect of PTCH on its signaling partner smoothened (SMO), which releases the suppressor of fused (SUFU) and induces the expression of target genes such as the GLI transcription factors GLI1, GLI2, and GLI3 [2,4–6]. The HH gradient itself is shaped by several proteins and the mechanism of cellular response incorporates multiple feedback loops [3].
Activation of the pathway is widely associated in pediatric and adult malignancies along with inactivation of the HH regulator PTCH1. Inherited or de novo, heterozygous germline mutations in the PTCH1 gene are responsible for the Gorlin syndrome (GS), a familial CPS [7, 8]. GS is characterized by developmental defects, neurological, genitourinary, and dental symptoms as well as an increased risk of tumor development such as basal cell carcinoma, medulloblastoma, meningioma, and RMS [9].
RMS accounts for 4–8% of all malignancies in childhood [10]. It occurs during all age groups with a peak between 2 to 6 years, however, it is extremely rare in the perinatal period (0.4–2%) [1].
Case report
We report the case of a newborn diagnosed with congenital embryonal rhabdomyosarcoma (eRMS). The non-consanguineous parents are of Turkish origin. The family history is unremarkable without a cancer diagnosis in the three preceding generations (Fig. 1a).
Fig. 1.

a Pedigree of the family; circles, females; squares, males; half symbol, heterozygous mutation; black symbol, PTCH1 mutation; blue symbol, PTCH2 mutation. b Chest X-ray (upper row) and photos (bottom row) of the new born boy at the age of 3 days and 7 months. c MRI scans, sagittal axis, show the very large paravertebral tumor at diagnosis until the end of treatment. d Hematoxylin and Eosin staining of the tumour at diagnosis
Pregnancy was uneventful until 36 weeks of gestation, when a large paravertebral tumor was prenatally detected by routine ultrasound. Elective cesarean section was performed at 37 + 2 weeks of gestational age due to breech presentation. Postnatal adaptation of the 2490 g and 45 cm male small for gestational age neonate was unremarkable.
The boy was referred to our department 2 days later. Initial laboratory tests including various tumor markers yielded exclusively normal results. MRI revealed a very large (11.5 cm craniocaudal extension), paravertebral tumor with intraspinal extension and epidural compression, massive secondary thinning of several ribs, pedicles of vertebral arch, and vertebral body, and multiple liver metastases (Fig. 1b, c). Abdominal ultrasound confirmed multiple lesions with low echogenicity on the rim and hyperechoic center in all liver segments, clearly increasing in size within the next 2 weeks until treatment start.
Biopsy of the paravertebral tumor was taken and pathological examination revealed eRMS with positive staining for desmin, myoD1, and CD56 and negative staining for S100 protein, synaptophysin, and smooth muscle actin (Fig. 1d).
Chemotherapy was initiated according to soft tissue sarcoma study group guidance for stage IV patients with metastatic disease (carboplatine, epirubicin, vincristine, actinomycin D, ifosfamide, etoposide) with drug doses calculated by body weight, additionally 1/3 dose reduction and substitution of ifosfamide by cyclophosphamide. Treatment was well tolerated under supportive care.
Response reassessment after three courses of chemotherapy revealed discrepant findings with partial response of the primary tumor and stable disease of the liver lesions. After nine courses of chemotherapy, at the end of treatment, second look biopsy of both, the tumor and liver lesions, was performed. Pathology evaluation showed completely necrotic tumor tissue, whereas the liver lesions showed no skeletal muscle differentiation, thus, the lesions were re-classified as non-malignant, most likely of fibromatous origin. Treatment was terminated without local therapy as tumor resection was deemed not feasible.
Since then, the patient is closely monitored by regular follow-up investigations including MRI and ultrasound. Until now, 30 months after the diagnosis and 22 months after the end of treatment, the patient is well and relapse-free without further signs of GS.
Results
Trio whole-exome sequencing analysis identifies two heterozygous germline variants in PTCH1 and PTCH2
Whole-exome sequencing (WES) of peripheral blood-derived DNA of the parent-child trio was performed. Details on data analysis are given in the Supplemental material [11]. Taken together, we identified 461 inherited heterozygous sequence variations of probable consequences including missense (444), frameshift (9), inframe insertion and deletions (6), and start/stop-codons (2), occurring with an allele frequency of less than 5% (MAF ≤ 0.05). Out of these single-nucleotide variants (SNVs), we identified two concomitant monoallelic germline mutations in SHH pathway target genes, PTCH1 and PTCH2. The PTCH1 p.(Gly38Glu) missense variant was in silico predicted by SIFT and PolyPhen as tolerated and probably damaging, respectively. According to ClinVar the variant has uncertain clinical significance (VUS) and a CADD score (Combined Annotation Dependent Depletion) [12], of 7.8 was calculated.
The identified PTCH2 variant p.(His622Tyr) was in silico predicted to be deleterious and benign according to SIFT and PolyPhen, respectively, while a CADD score of 14.82 was calculated (Fig. 2). Both variants are heterozygous in the patient, the PTCH1 variant is transmitted from the mother, while the PTCH2 variant is inherited from the father. Furthermore, the SNVs were validated by conventional Sanger sequencing (Supplementary Fig. 1). Additionally, tumor tissue was also analyzed by WES, but showed no loss of heterozygosity (LOH) in PTCH1 and PTCH2.
Fig. 2.

a WES reveals a heterozygous variant in PTCH1 (c.113G > A, p.(Gly38Glu)), and in PTCH2 (c.1864C > T, p.(His622Tyr)) in the patient´s peripheral blood. Both variants are inherited by the parents. b The PTCH1 variant is located in exon 1 and the PTCH2 variant is located in exon 14
Quantitative reverse transcription PCR (qRT-PCR) analysis of the SHH pathway activity
qRT-PCR was performed to determine the SHH pathway activity by mRNA expression levels of four different SHH target genes comprising PTCH1, PTCH2, SMO, and GLI1 in the tumor as well as in the germline samples. We could show high GLI1 expression levels in the peripheral blood-derived germline sample from the patient with a tenfold significant increase in the tumor sample indicating an activation of the SHH pathway, while the parents showed a sixfold lower GLI1 expression compared to the patients’ germline, revealing low activity of the SHH pathway. Furthermore, SMO was also significantly increased in the tumor tissue, whereas all three germline samples showed low expression levels. The patient showed a twofold decrease of PTCH1 expression levels in the germline and tumor tissue in comparison to the peripheral blood-derived cDNA from the parents. The PTCH2 expression level was threefold increased in the tumor sample in comparison to the germline of the patient and his parents (Fig. 3). Therefore, the variant in PTCH2 might play a decisive role and contributes to both the activation of the SHH pathway and the extremely rare clinical presentation with congenital RMS.
Fig. 3.
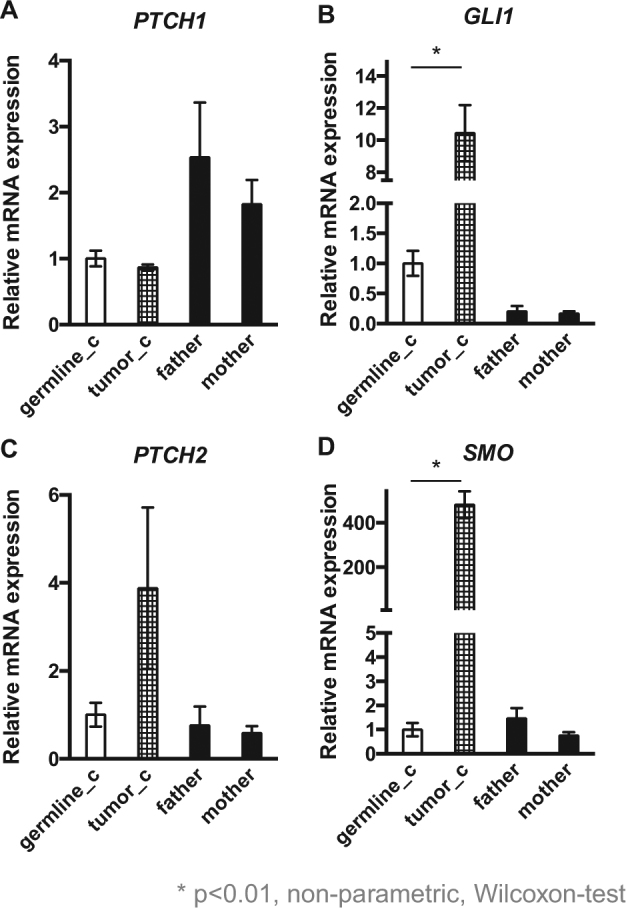
a Quantitative RT-PCR (qRT-PCR) showed a twofold decrease of PTCH1 expression levels in the germline and tumor tissue in comparison to the peripheral blood-derived cDNA from the parents and b a tenfold significant increased GLI1 expression level in the tumor sample and a sixfold lower GLI1 expression in the parents compared to the patients’ germline. c The PTCH2 expression level in the tumor sample was threefold increased in comparison to the germline of the patient and his parents. d SMO was 500-fold increased in the tumor tissue, whereas all three germline samples showed low expression levels
Discussion
In this case report, we describe the co-occurrence of two concomitant heterozygous germline mutations in SHH pathway genes in a newborn with congenital eRMS. To our knowledge, this is the first report of a patient with eRMS harboring combined heterozygous mutations in PTCH1 and PTCH2.
Usually, in tumor suppressor genes linked hereditary syndromes such as GS, the tumor develops a deletion of the wild-type allele resulting in LOH [13]. However, we excluded causative LOH in PTCH1 and PTCH2 in the tumor tissue of our patient. This is in line with findings in PTCH1 heterozygous mice, in which retained wild-type allele in medulloblastomas and RMS-like tumors have been shown, indicating that haploinsufficiency of PTCH1 may already be sufficient for tumor development [14, 15]. These mice develop many features of GS and, on a CD1 background, RMS-like tumors at a frequency of 10–15% [16–18]. As a consequence of non-functional PTCH1, the negative feedback loop is disrupted and PTCH1, SMO, GLI1, and GLI3 mRNA accumulate in the cell leading to a constitutive activation of the SHH signaling that leads to cellular proliferation at the expense of maturation of the target cell into a post-mitotic state [19, 20]. Therefore, a subsequent defect in the DNA damage response caused by PTCH1 haploinsufficiency may be relevant as a mechanism contributing to tumor formation. According to Pressey et al. [7], tumors with high GLI1 and low PTCH1 expression could exert a more active SHH signaling due to silencing of PTCH. This is in line with the expression profile in our patient showing high levels of GLI1 and SMO. Upregulation of GLI1 provides the potential for downstream activation of the SHH signaling pathway and has been shown to play a role in the development and pathogenesis of a subset of eRMS tumors [7].
Inhibition of SMO prevents downstream activation of GLI transcription factors, which leads to suppression of those genes that are associated with cancer growth and progression. Therefore, SMO has been the primary target for the development of SHH pathway inhibitors. Until now, the US Food and Drug Administration (FDA) approved two SHH signaling inhibitors, vismodegib and sonidegib, for the treatment of advanced basal cell carcinoma (BCC). Sonidegib and vismodegib bind to SMO and act as an inhibitor preventing downstream activation of GLI1 and blocking the biological activity of the SHH pathway [21, 22].
In a mouse model reported by Lee et al. [8], it was shown that PTCH1 and PTCH2 compound mutants—as identified in our patient—have an increased tumor susceptibility and tumorigenesis [8]. Concomitant loss of PTCH2 led to an increase in RMS frequency and affected tumor formation in combination with PTCH1 haploinsufficiency. Therefore, these animals showed a higher incidence of tumors and an increase of different tumor types including mostly sarcomas, which was not previously observed in PTCH1 haploinsufficient mice. Consequently, we speculate that PTCH2 modulates tumorigenesis linked to the PTCH1 mutation and likely is associated with the congenital onset of RMS observed in our patient.
The phenomenon of double heterozygosity for mutations in two different genes in the same pathway has previously been described in breast cancer patients [23]. It remains to be determined whether this functional perturbation of key cancer pathways by inherited heterozygous mutations from each parent is a more widespread genetic phenomenon in the germline of children with cancer.
Electronic supplementary material
Acknowledgements
This work was supported by the Elterninitiative Kinderkrebsklinik e.V. Duesseldorf and by the BMBF (to SF). We wish to thank Mrs. S. Furlan and Mrs. K. Alemazkour for technical assistance, Prof. Dr. I. Leuschner for pathologic analyses, Prof. Dr. M. Dugas and C. Walter for WES data analyses and establishment of the bioinformatics pipeline as well as Dr. J.I. Hoell for critical reading of the manuscript.
Author contributions
JT and MK drafted the manuscript. JT performed the experiments with help of NQ and JB. TB obtained informed consent, asked for the family history, and cared for the child. JS performed the radiological diagnostic. SG was responsible for the internal SQL database. JF contributed to pathology analysis. SF contributed to the design of the experiments and data analysis. CV performed pathologic analyses. AB and MK designed and supervised the project. AB critically revised the manuscript for important intellectual content. All authors approved the final manuscript as submitted.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the ethics committee of the Heinrich-Heine-University Duesseldorf, Germany.
Informed consent
Written informed consent was obtained from both the parents.
Footnotes
Arndt Borkhardt and Michaela Kuhlen contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1038/s41431-017-0048-4) contains supplementary material, which is available to authorized users.
References
- 1.Orbach D, Sarnacki S, Brisse HJ, et al. Neonatal cancer. Lancet Oncol. 2013;14:e609–20. doi: 10.1016/S1470-2045(13)70236-5. [DOI] [PubMed] [Google Scholar]
- 2.Hahn H, Wojnowski L, Miller G, Zimmer A. The patched signaling pathway in tumorigenesis and development: lessons from animal models. J Mol Med. 1999;77:459–68. doi: 10.1007/s001099900018. [DOI] [PubMed] [Google Scholar]
- 3.Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29:469–81. doi: 10.1038/onc.2009.392. [DOI] [PubMed] [Google Scholar]
- 4.Nilsson M, Unden AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci USA. 2000;97:3438–43. doi: 10.1073/pnas.97.7.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–17. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 6.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–9. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 7.Pressey JG, Anderson JR, Crossman DK, Lynch JC, Barr FG. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2011;57:930–8. doi: 10.1002/pbc.23174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee Y, Miller HL, Russell HR, Boyd K, Curran T, McKinnon PJ. Patched2 modulates tumorigenesis in patched1 heterozygous mice. Cancer Res. 2006;66:6964–71. doi: 10.1158/0008-5472.CAN-06-0505. [DOI] [PubMed] [Google Scholar]
- 9.Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. N Engl J Med. 1960;262:908–12. doi: 10.1056/NEJM196005052621803. [DOI] [PubMed] [Google Scholar]
- 10.Pappo AS. Rhabdomyosarcoma and other soft tissue sarcomas of childhood. Curr Opin Oncol. 1995;7:361–6. doi: 10.1097/00001622-199507000-00012. [DOI] [PubMed] [Google Scholar]
- 11.Brozou T, Taeubner J, Velleuer E, et al. Genetic predisposition in children with cancer—affected families’ acceptance of Trio-WES. Eur J Pediatr. (e-pub ahead of print 19 September 2017; 10.1007/s00431-017-2997-6).
- 12.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pino LC, Balassiano LK, Sessim M, et al. Basal cell nevus syndrome: clinical and molecular review and case report. Int J Dermatol. 2015;55:365–75. doi: 10.1111/ijd.12993. [DOI] [PubMed] [Google Scholar]
- 14.Wetmore C, Eberhart DE, Curran T. The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ-line mutation of patched. Cancer Res. 2000;60:2239–46. [PubMed] [Google Scholar]
- 15.Calzada-Wack J, Kappler R, Schnitzbauer U, et al. Unbalanced overexpression of the mutant allele in murine Patched mutants. Carcinogenesis. 2002;23:727–33. doi: 10.1093/carcin/23.5.727. [DOI] [PubMed] [Google Scholar]
- 16.Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619–22. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- 17.Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH, Jr., Scott MP. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–21. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 18.Fan H, Oro AE, Scott MP, Khavari PA. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat Med. 1997;3:788–92. doi: 10.1038/nm0797-788. [DOI] [PubMed] [Google Scholar]
- 19.Toftgard R. Hedgehog signalling in cancer. Cell Mol life Sci: CMLS. 2000;57:1720–31. doi: 10.1007/PL00000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Malley S, Weitman D, Olding M, Sekhar L. Multiple neoplasms following craniospinal irradiation for medulloblastoma in a patient with nevoid basal cell carcinoma syndrome. J Neurosurg. 1997;86:286–8. doi: 10.3171/jns.1997.86.2.0286. [DOI] [PubMed] [Google Scholar]
- 21.Wahid M, Jawed A, Mandal RK, et al. Vismodegib, itraconazole and sonidegib as hedgehog pathway inhibitors and their relative competencies in the treatment of basal cell carcinomas. Crit Rev Oncol Hematol. 2016;98:235–41. doi: 10.1016/j.critrevonc.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Jain S, Song R, Xie J. Sonidegib: mechanism of action, pharmacology, and clinical utility for advanced basal cell carcinomas. OncoTargets Ther. 2017;10:1645–53. doi: 10.2147/OTT.S130910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heidemann S, Fischer C, Engel C, et al. Double heterozygosity for mutations in BRCA1 and BRCA2 in German breast cancer patients: implications on test strategies and clinical management. Breast Cancer Res Treat. 2012;134:1229–39. doi: 10.1007/s10549-012-2050-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.