

Abstract
Background
Although arachidonic acid metabolites, cysteinyl leukotrienes (cys-LTs; leukotriene [LT] C4, LTD4, and LTE4), and prostaglandin (PG) E2 are generated at the site of inflammation, it is not known whether crosstalk exists between these 2 classes of inflammatory mediators.
Objective
We sought to determine the role of LTD4-PGE2 crosstalk in inducing vascular inflammation in vivo, identify effector cells, and ascertain specific receptors and pathways involved in vitro.
Methods
Vascular (ear) inflammation was assessed by injecting agonists into mouse ears, followed by measuring ear thickness and histology, calcium influx with Fura-2, phosphorylation and expression of signaling molecules by means of immunoblotting, PGD2 and macrophage inflammatory protein 1β generation by using ELISA, and expression of transcripts by using RT-PCR. Candidate receptors and signaling molecules were identified by using antagonists and inhibitors and confirmed by using small interfering RNA.
Results
LTD4 plus PGE2 potentiated vascular permeability and edema, gearing the system toward proinflammation in wild-type mice but not in KitW-sh mice. Furthermore, LTD4 plus PGE2, through cysteinyl leukotriene receptor 1 (CysLT1R) and E-prostanoid receptor (EP) 3, enhanced extracellular signal-regulated kinase (Erk) and c-fos phosphorylation, inflammatory gene expression, macrophage inflammatory protein 1β secretion, COX-2 upregulation, and PGD2 generation in mast cells. Additionally, we uncovered that this synergism is mediated through Gi, protein kinase G, and Erk signaling. LTD4 plus PGE2–potentiated effects are partially sensitive to CysLT1R or EP3 antagonists but completely abolished by simultaneous treatment both in vitro and in vivo.
Conclusions
Our results unravel a unique LTD4-PGE2 interaction affecting mast cells through CysLT1R and EP3 involving Gi, protein kinase G, and Erk and contributing to vascular inflammation in vivo. Furthermore, current results also suggest an advantage of targeting both CysLT1R and EP3 in attenuating inflammation.
Keywords: Mast cells, prostaglandin E2, leukotriene D4, CysLT1R, E-prostanoid receptor 3, prostaglandin D2, c-fos, protein kinase G, extracellular signal-regulated kinase, macrophage inflammatory protein 1β
Mast cells (MCs) are recognized as critical components of our immune system. They are vital in the initiation and amplification of acute inflammatory responses and play an important role in triggering asthma exacerbations through the elaboration of several soluble inflammatory mediators.1,2 MCs reside in connective tissues and are located in close proximity to the blood vessels. Activation of MCs stimulate the formation of leukotrienes (LTs) and prostaglandins (PGs), both of which initiate vascular changes.3 KitW-sh/W-sh mice have the W-sash (Wsh) inversion mutation and remarkable deficiency in MCs, providing a great model system to analyze MC function in vivo.4 Cysteinyl leukotrienes (cys-LTs; LTC4, LTD4, and LTE4) are arachidonic acid derivatives generated by MCs, eosinophils, basophils, and macrophages5 through the action of 5-lipoxygenase enzyme. All PGs are derived from PGH2 and generated through arachidonic acid through the action of PGH synthase (also known as COX). MCs express both COX-1 and COX-2. COX-2 is upregulated by inflammatory stimuli driving PGD2 generation under inflammatory conditions. In MCs PGH2 derived from both COX-1 and COX-2 is converted to PGD2 by a terminal hematopoietic PGD2 synthase.6 Although not a product of MCs, PGE2, a metabolite of PGH2 through the action of PGE2 synthase,7 is the most ubiquitous PG, with prominent and complex functions in inflammation, asthma, and allergic diseases. Remarkably, MCs not only generate cys-LTs but also express corresponding receptors and respond to them.8 Two known G protein–coupled receptors, termed cysteinyl leukotriene receptor 1 (CysLT1R) and cysteinyl leukotriene receptor 2 (CysLT2R), specifically recognize cys-LTs and mediate their biologic functions. CysLT1R binds LTD4 with higher affinity than LTC4, whereas CysLT2R has equal affinity for LTD4 and LTC4.5 GPR17, another cys-LT receptor, has been identified and is expressed primarily in the brain,9 and GPR99 has been recently identified as a cys-LT receptor with a preference for LTE4.10 Mice lacking LTC4 synthase have reduced numbers of MCs in the airway mucosa after sensitization and challenge by allergen,11 suggesting the prominence of cys-LTs in MC function. We have previously demonstrated that stimulation of human cord blood–derived mast cells (hMCs), LAD2 cells, or both with LTD4 potently induces calcium flux and cytokine generation8 through CysLT1R. Additionally, MC proliferative and inflammatory responses are modulated by LTD4 and stem cell factor (SCF) signaling interactions.12 PGs have also been shown to elicit vasodilation and an increase in blood flow. Among PGs, PGE2 is the most abundantly synthesized PG at the inflammation site and is regarded as an important regulator of inflammation.13 The decisive effect of PGE2 is the outcome of specific E-prostanoid receptor (EP) 1 to 4 activation through which the signal is transduced.14 EP1 is coupled to intracellular calcium mobilization through Gq; however, EP2 and EP4 are coupled to stimulation of adenylyl cyclase through Gs, and EP3 is coupled to the inhibition of adenylyl cyclase through Gi. Different splice variants are generated by means of alternative splicing of the C-terminal tail of the EP3 receptor and can couple to different signal transduction pathways. Eight human EP3 isoforms are known thus far, which are identical, except for their carboxyl termini.14
The interactions among various mediator systems that participate in inflammatory responses are complex, and it is difficult to define the unique contribution of any single element. In the current study we show that LTD4 and PGE2 synergistically potentiate peripheral inflammation in vivo and MC activation in vitro through CysLT1R-, EP3-, Gi-, protein kinase (PK) G–, and extracellular signal-regulated kinase (Erk)–dependent pathways. Furthermore, our results indicate that blocking EP3 together with CysLT1R could be a better therapeutic target to control inflammation.
METHODS
Animals
Six- to 8-week-old BALB/c mice, C57BL/6 mice, and KitW-sh mice (W-sh) were obtained from Jackson Laboratories and maintained at the Comparative Medicine Unit, Northeast Ohio Medical University. All animal experiments were done in accordance with standard guidelines, as approved by the Animal Care and Use Committee of Northeast Ohio Medical University.
Reagents
LTD4, PGE2, MK571, BayCysLT2, iloprost, butaprost, sulprostone, L-798, ONO-871, L-161, and PGD2 ELISA kits were purchased from Cayman Chemicals (Ann Arbor, Mich). KT5823, PD98059, pertussis toxin (PTX), H7, GF109203X, Rp-cAMPS, and H89 inhibitors were from Tocris Bioscience (Minneapolis, Minn). Fura-2 AM was from Molecular Probes (Eugene, Ore), phospho-specific antibodies were from Cell Signaling Technology (Danvers, Mass), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was from Fitzgerald (Acton, Mass). All secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, Pa). Nonspecific small interfering RNA (siRNA) and isoform-specific siRNAs for CysLT1R, EP3, and PKG were obtained from Dharmacon (Lafayette, Colo), and the macrophage inflammatory protein 1β (MIP-1β) ELISA kit was from R&D Systems (Minneapolis, Minn). Cytokines for hMC cultures were obtained from PeproTech (Rocky Hill, NJ).
Intradermal injection of agonists and assessment of ear edema
Mice anesthetized with ketamine/xylazine received intradermal injections of 0.5 μmol/L LTD4, PGE2, and LTD4 plus PGE2 (in a 10-μL volume) in the right ear and 10 μL of saline in the left ear in the presence or absence of MK571, L-798, or both. At 0, 30, 60, 120, 240, and 300 minutes after the intradermal injection, ear thickness was measured with a caliper. Mice were killed 60 minutes after the indicated treatment, ear tissues were fixed in 4% paraformaldehyde and embedded in paraffin, and 4-μm-thick sections were cut and stained for hematoxylin and eosin and toluidine blue (to detect MCs). Total (toluidine blue–positive cells that are compact) and degranulated MCs (toluidine-positive cells with no clearly defined cell membrane and diffuse) in the toluidine blue–stained sections were visualized at ×60 magnification and presented in Fig 1, B; counted in each section by a blinded observer; and expressed as the number of MCs per millimeter. Representative images of intact and degranulated MCs were shown in Fig 1, B.
FIG 1.
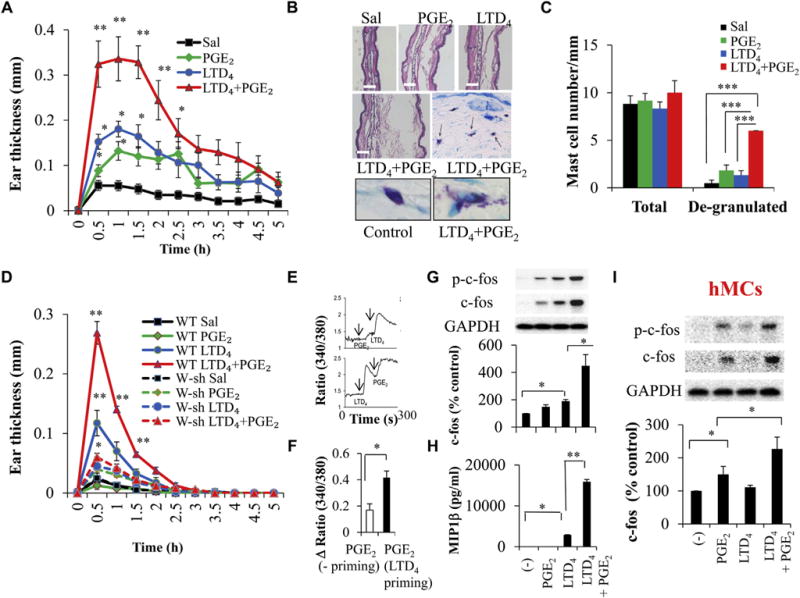
Effect of LTD4 and PGE2 on ear edema in mice in vivo and MC activation in vitro. Wild-type (WT) BALB/c mice were treated with saline (Sal), 0.5 μmol/L LTD4, 0.5 μmol/L PGE2, or LTD4 plus PGE2. A, Ear thickness. B, Hematoxylin and eosin staining and toluidine blue staining. C, Quantification (blind analysis) of MCs per millimeter. D, Ear thickness in C57BL/6 and W-sh mice treated with 0.5 μmol/L LTD4, PGE2, and LTD4 plus PGE2. Results are means ± SEMs from 4 to 6 mice per group per experiment and 3 experiments performed. E–I, LAD2 cells (Fig 1, E–H) and hMCs (Fig 1, I) were stimulated with 0.5 μmol/L LTD4, 0.5 μmol/L PGE2, or both, and calcium flux (Fig 1, E and F), c-fos (Fig 1, G and I), and MIP-1β (Fig 1, H) were analyzed. *P < .05, **P < .01, and ***P < .001.
Cell culture
The LAD2 MC leukemia line15 was a kind gift from Dr Arnold Kirshenbaum (National Institutes of Health) and cultured as described previously.8 Primary hMCs were derived from cord blood mononuclear cells cultured for 6 to 9 weeks in RPMI supplemented with SCF, IL-6, and IL-10.16
Calcium flux
LAD2 cells (0.5 to 1 × 106/sample) were washed and labeled with Fura 2-AM for 30 minutes at 37°C. Cells were stimulated with PGE2 (0.5 μmol/L) with or without LTD4 (0.5 μmol/L) priming, and the changes in intracellular calcium levels measured by using excitation at 340 and 380 nm and emission at 510 nm were recorded in a fluorescence spectrophotometer (Hitachi F-4500).8
Cell activation
LAD2 cells were stimulated with 0.5 μmol/L of LTD4, PGE2, or both for 15 minutes for the phosphorylation of Erk or 1 hour for expression of c-fos, 2 hours for expression of inflammatory gene transcripts, 3 hours for COX-2 protein expression, and 6 hours for measurement of cytokine and PGD2 levels. LTD4 responses were dose dependently inhibited by MK571 (with maximum inhibition at 1 μmol/L), and PGE2 responses were attenuated by L-798 (in a dose-dependent manner, with maximum inhibition at 100 nmol/L). Therefore 1 μmol/L MK571 and 100 nmol/L L-798 were used in all the subsequent experiments. Transfection of isoform-specific siRNA smart pool constructs from Dharmacon (10 nmol/L) were carried out with siLentFect transfection reagent (Bio-Rad Laboratories, Hercules, Calif) for 48 hours, according to the manufacturer’s protocol.
Cell lysates and Western blotting
After stimulation with the respective agonists, LAD2 cells, hMCs, or both (0.5 × 106) were lysed with lysis buffer (BD Biosciences, San Jose, Calif) supplemented with protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail (Pierce, Rockford, Ill). Immunoblotting was performed, as described previously.17 Western blots were incubated with ECL, and the bands were visualized with an imager (ProteinSimple, San Jose, Calif) and quantified by using Alpha View SA (ProteinSimple). The blots were stripped and reprobed with GAPDH antibody.
Real-time quantitative PCR
Expressions of CysLT1R, CysLT2R, EP1, EP2, EP3, EP4, MIP-1β, TNF-α, IL-8, COX-1, COX-2, and PKG transcripts were determined with real-time PCR performed on the LightCycler 480 (Roche). Total RNA was isolated from LAD2 cells after respective treatment with an RNeasy Mini Kit (Qiagen, Hilden, Germany), and cDNA was synthesized with the cDNA synthesis kit from Quanta BioSciences (Gaithersburg, Md). Real-time PCR was performed by using primers mentioned in Table E1 in this article’s Online Repository at www.jacionline.org, the levels of respective genes relative to GAPDH were analyzed, and the ΔΔ cycle threshold values relative to control were expressed as the fold change.
ELISA
Concentrations of PGD2 and MIP-1β in the supernatants were assayed by using the PGD2-MOX and MIP-1β ELISA kits purchased from Cayman Chemicals (Ann Arbor, Mich) and R&D Systems, respectively, according to the manufacturer’s instructions. PGD2 secreted into medium was first converted into PGD2-MOX and then assessed by using the PGD2-MOX ELISA kit.
Statistical analysis
Blots presented are representative of 3 experiments performed, and data are expressed as means ± SEMs from at least 3 experiments, except where otherwise indicated. Significance was determined by using the Student t test, as well as 1-way ANOVA, followed by Tukey post hoc analysis.
RESULTS
Combined treatment with PGE2 and LTD4 synergistically potentiates peripheral vascular inflammation in mice
To elucidate the possible interaction between LTD4 and PGE2, we first evaluated the increase in vascular permeability and edema in the mouse ear, a widely used model to assess peripheral vascular inflammation,18,19 in response to LTD4 plus PGE2 compared with agonists alone. LTD4 or PGE2 and LTD4 plus PGE2 (0.5 μmol/L, a dose at which both the agonists generated maximum response; data not shown) were injected into the ears of BALB/c mice, and tissue edema was assessed. The patterns of ear edema induced by LTD4, PGE2, and LTD4 plus PGE2 were similar, peaking at 30 minutes and returning to baseline at 300 minutes. However, we observed a significant enhancement in the magnitude of ear edema with LTD4 plus PGE2 compared with LTD4 or PGE2 alone (Fig 1, A). The increase in ear thickness with LTD4 plus PGE2 was rapid, transient, and approximately 6-fold higher compared with control values (0.32 ± 0.05 vs 0.05 ± 0.01) in the first half hour. We also observed an increase in ear thickness in response to both LTD4 (approximately 3-fold, 0.15 ± 0.01) and PGE2 (approximately 1.6-fold, 0.08 ± 0.01) individually during the first 30 minutes compared with control values. Histologic analysis of ear tissues revealed expansion of the extracellular space correlating with ear thickness (Fig 1, B). Interestingly, we found a robust increase in the number of degranulating MCs with LTD4 plus PGE2 treatment (Fig 1, B, middle and bottom right panel, arrows point to degranulating MCs, and Fig 1, C) compared with other groups, suggesting that LTD4 plus PGE2 can synergistically potentiate peripheral inflammation through their action on MCs. To elucidate the role of MCs, we repeated the above experiment in C57BL/6 and W-sh mice (on C57BL/6 background). Although we observed a similar pattern of ear inflammation in C57BL/6 wild-type mice as in BALB/c mice with all the agonists, the response was more transient and smaller (Fig 1, D). LTD4 plus PGE2 synergistically enhanced the inflammation in C57BL/6 wild-type mice, which is significantly attenuated in W-sh mice (Fig 1, D).
LTD4 primes PGE2-dependent calcium flux and potentiates c-fos phosphorylation and MIP-1β production in MCs
Next, we investigated the molecular mechanisms through which LTD4 and PGE2 activate MCs and potentiate inflammation. The expression pattern of cys-LT receptor transcript in LAD2 cells is similar to that in hMCs, with high levels of CysLT1R compared with CysLT2R (see Fig E1, C, in this article’s Online Repository at www.jacionline.org). Additionally, LAD2 cells express all 4 EPs (EP3 > EP2 > EP4 > EP1; see Fig E1, C). Furthermore, Western blot analysis of LAD2 cells and hMCs confirmed the expression of CysLT1R, CysLT2R, and EP1-4 (see Fig E1, D). hMCs expressed higher CysLT1R levels compared with LAD2 cells, and the expression pattern of EP3 is comparable.
First, we analyzed the ability of LAD2 cells to flux calcium in response to PGE2 and LTD4. We observed that PGE2 induced modest calcium flux (Fig 1, E, top panel, and Fig 1, F) and LTD4 priming before PGE2 stimulation led to an approximately 2.5-fold increase (0.42 ± 0.05 vs 0.16 ± 0.04; Fig 1, E, bottom panel, and Fig 1, F). Furthermore, LTD4 and PGE2 together increased phosphorylation and expression of c-fos (Fig 1, G) and secretion of MIP-1β (Fig 1, H). Importantly, in hMCs also, LTD4 plus PGE2 treatment significantly enhanced c-fos phosphorylation and expression (Fig 1, I). Stimulating LAD2 cells with a constant dose of LTD4 (0.5 μmol/L) and varying concentrations of PGE2 (0.001, 0.01, 0.1, and 0.5 μmol/L), we observed dose-dependent c-fos phosphorylation and expression and MIP-1β secretion (see Fig E1, A and B).
CysLT1R inhibition partially attenuates LTD4-primed PGE2 responses
We then sought to identify the receptors involved in mediating this synergy by using MK571 (CysLT1R antagonist8) and BayCysLT2 (CysLT2R antagonist20). MK571 pretreatment abolished LTD4-induced calcium influx but attenuated LTD4-primed, PGE2-generated calcium influx by 30% (Fig 2, A). LTD4 plus PGE2–stimulated c-fos phosphorylation and expression and MIP-1β secretion were reduced with MK571 preincubation (Fig 2, B-D), whereas BayCysLT2 had no effect (Fig 2).
FIG 2.
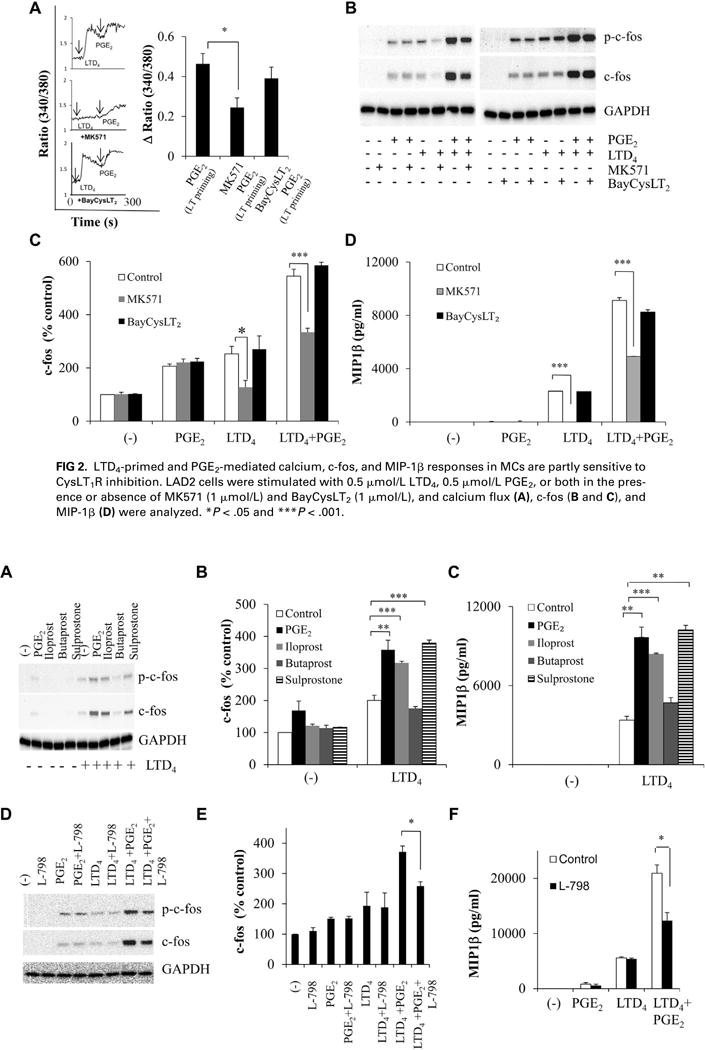
LTD4-primed and PGE2-mediated calcium, c-fos, and MIP-1β responses in MCs are partly sensitive to CysLT1R inhibition. LAD2 cells were stimulated with 0.5 μmol/L LTD4, 0.5 μmol/L PGE2, or both in the presence or absence of MK571 (1 μmol/L) and BayCysLT2 (1 μmol/L), and calcium flux (A), c-fos (B and C), and MIP-1β (D) were analyzed. *P < .05 and ***P < .001.
Effects of LTD4 plus PGE2 are partially mediated through EP3
We next investigated which of the putative EPs were interacting with CysLT1R and enhancing PGE2 responses. Stimulation of LAD2 cells with iloprost (EP1/EP3 agonist) and sulprostone (EP3 agonist) in the presence of LTD4 enhanced calcium flux (data not shown), c-fos (Fig 3, A and B), and MIP-1β expression (Fig 3, C), which is similar to what is seen with LTD4 plus PGE2, suggesting the involvement of EP3 and a possible involvement of EP1 in this synergism. The EP2 agonist butaprost had no effect (Fig 3, A-C). Because EP1 and EP3 act dominantly through Gq- and Gi-mediated signaling pathways, respectively,14 we used PTX, a Gαi inhibitor, and evaluated LTD4 plus PGE2 synergy. Pretreatment of LAD2 cells with PTX (100 ng/mL for 18 hours) completely abolished the enhanced activation of c-fos and MIP-1β by LTD4 and PGE2 (see Fig E2, A-C, in this article’s Online Repository at www.jacionline.org), suggesting the involvement of EP3. Furthermore, we found that the synergistic responses by LTD4 plus PGE2 were partially sensitive to the inhibition of EP3 by L-798 (EP3 antagonist, 100 nmol/L; Fig 3, D-F), which is similar to what is seen with CysLT1R inhibition. Neither ONO-8711 (EP1 antagonist, 2 nmol/L) nor L-161 (EP4 antagonist, 100 nmol/L) had any effect on LTD4 plus PGE2–mediated c-fos expression/phosphorylation or MIP-1β generation (see Fig E2, D and E).
FIG 3.
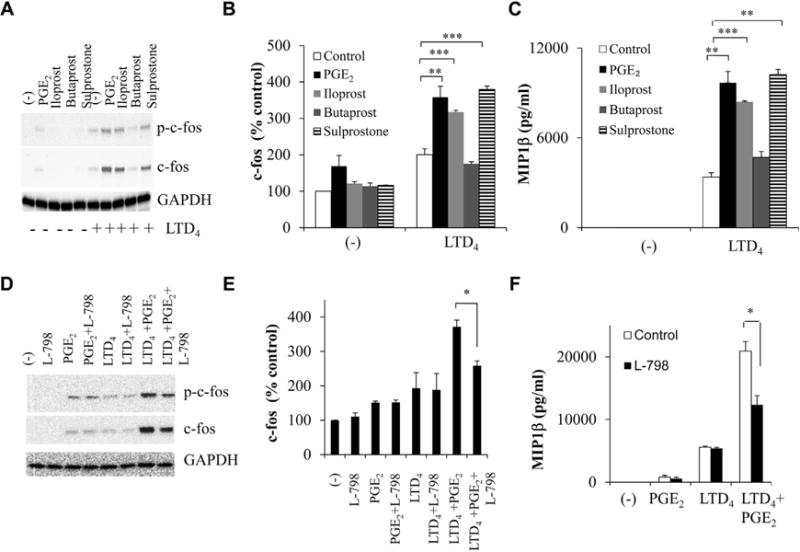
EP3 relays synergistic activation of c-fos and MIP-1β in response to PGE2 and LTD4. LAD2 cells were treated with LTD4 (0.5 μmol/L) ± PGE2 (0.5 μmol/L) or iloprost (10 μmol/L), butaprost (5 μmol/L), or sulprostone (100 nmol/L) ± L-798 (100 nmol/L), and c-fos (A, B, D, and E) and MIP-1β (C and F) were analyzed. *P < .05, **P < .01, and ***P < .001.
Synergistic responses to LTD4 and PGE2 were completely attenuated by blocking both CysLT1R and EP3 simultaneously
Next, we speculated that blocking both CysLT1R and EP3 simultaneously might completely block the synergistic responses. We observed that although the effects mediated by LTD4 and PGE2 are partially blocked by CysLT1R and EP3 antagonists alone (40% and 35% for c-fos and 58% and 57% for MIP-1β, respectively), combined treatment with MK571 and L-798 completely blocked the effects of LTD4 plus PGE2 (78% for c-fos phosphorylation and expression and 92% for MIP-1β; Fig 4, A-C). The combination of MK571 and L-798 also attenuated augmentation of c-fos and MIP-1β generation by LTD4 plus sulprostone (data not shown). Importantly, in hMCs MK571 plus L-798 totally inhibited c-fos phosphorylation and expression (Fig 4, D and E), suggesting a functional relevance for this interaction and associated signaling.
FIG 4.
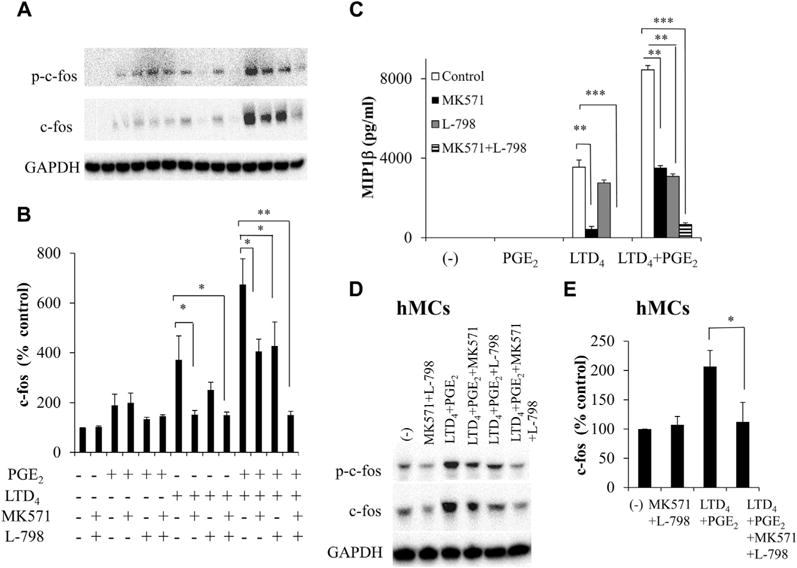
Combined effect of CysLT1R antagonist and EP3 antagonist on synergistic responses to LTD4 and PGE2. LAD2 cells (A–C) and hMCs (D and E) were preincubated with MK571 (1 μmol/L) and L-798 (100 nmol/L) separately or in combination and treated with LTD4 (0.5 μmol/L) ± PGE2 (0.5 μmol/L), and c-fos (Fig 4, A, B, D, and E) and MIP-1β (Fig 4, C) were analyzed. *P < .05, **P < .01, and ***P < .001.
LTD4 and PGE2 treatment upregulated inflammatory genes, PGD2 generation, and Erk phosphorylation in LAD2 cells
We then investigated whether stimulation of MCs with LTD4 plus PGE2 would upregulate any other inflammatory chemokines and cytokines. LTD4 treatment upregulated MIP-1β, TNF-α, IL-8, and COX-2, whereas PGE2 upregulated COX-2 alone. LTD4 plus PGE2 treatment significantly upregulated MIP-1β (145 ± 30; Fig 5, A), TNF-α (90 ± 11; Fig 5, B), IL-8 (72 ± 20; Fig 5, C), and COX-2 (60 ± 10; Fig 5, D) transcripts, and the expression of COX-1 remained unchanged (Fig 5, E). Consistently, we observed a significant 3-fold upregulation of COX-2 protein with LTD4 plus PGE2 treatment (Fig 5, F). Furthermore, LTD4 plus PGE2 treatment upregulated PGD2 secretion compared with control values (1630 ± 51 vs 1000 ± 75 pg/mL), and combined treatment with both MK571 and L-798 inhibited this secretion (Fig 6, A). Then we investigated the ability of LTD4, PGE2, and a combination of both in inducing Erk phosphorylation. PGE2 and LTD4 stimulation enhanced phosphorylation of Erk, and LTD4 plus PGE2 further potentiated this effect, which is sensitive to the MEK inhibitor PD989059 (50 μmol/L; Fig 6, B).
FIG 5.
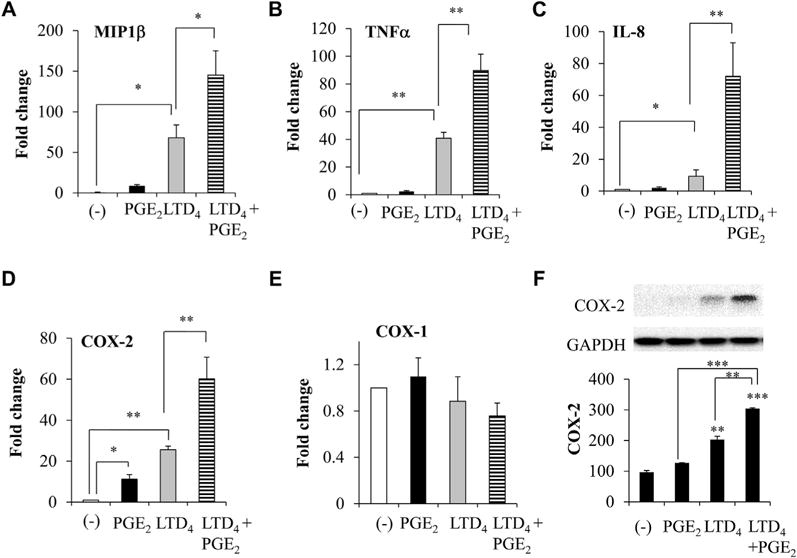
Inflammatory gene induction and COX-2 protein by LTD4 and PGE2 in LAD2 cells. LAD2 cells were stimulated with 0.5 μmol/L LTD4, PGE2, or both; transcript levels of MIP-1β, TNF-α, IL-8, COX-2, and COX-1 were analyzed by means of real-time PCR at 2 hours of stimulation (A–E); and COX-2 protein levels were determined (F) in cell lysates. *P < .05, **P < .01, and ***P < .001.
FIG 6.
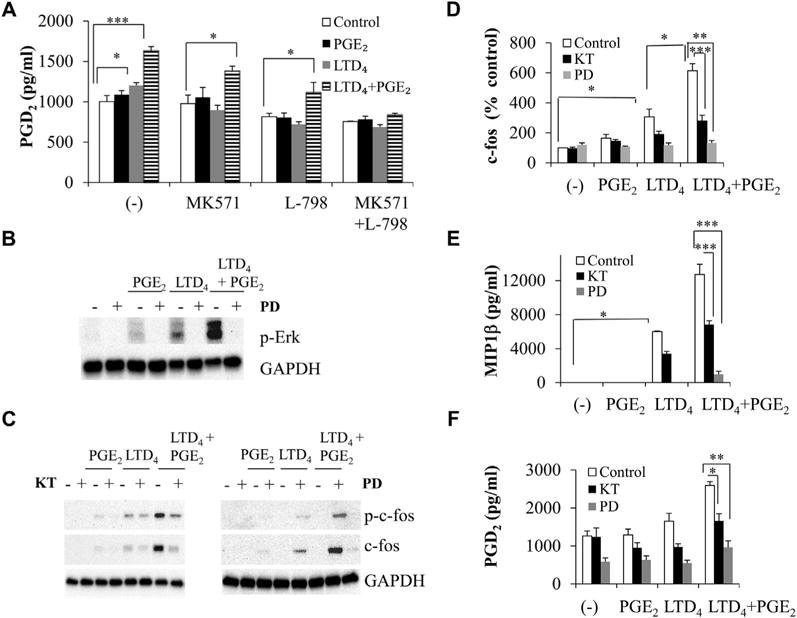
LTD4 and PGE2 crosstalk causes a synergistic increase in PGD2 secretion and Erk phosphorylation and is mediated through PKG and Erk. LAD2 cells were stimulated with 0.5 μmol/L LTD4, 0.5 μmol/L PGE2, or both in the presence or absence of MK571 (1 μmol/L), L-798 (100 nmol/L), KT5823 (KT; 5 μmol/L), or PD98059 (PD; 50 μmol/L) before treatment, and PGD2 secretion (A and F), Erk phosphorylation (B), c-fos (C and D), and MIP-1β generation (E) were analyzed. *P < .05, **P < .01, and ***P < .001.
LTD4 plus PGE2 signals operate through PKG and Erk-dependent pathway
We next examined the signaling involved in LTD4 plus PGE2 synergism downstream of receptor activation. PKs are known to be activated downstream of CysLT1R activation.8,21 A general PK inhibitor, H7 (10 μmol/L), completely blocked LTD4, as well as LTD4 plus PGE2–induced effects (see Fig E3, A, E, and F, in this article’s Online Repository at www.jacionline.org). However, the general PKC inhibitor GF109203X (2 μmol/L) inhibited LTD4 signals but modestly blocked enhanced c-fos and MIP-1β effects by LTD4 plus PGE2 (see Fig E3, B, E, and F). Furthermore, the PKA blockers Rp-cAMPS and H-89 had no effect on synergistic LTD4 plus PGE2 effects (see Fig E3, C-F). Interestingly, pretreatment of LAD2 cells with the PKG inhibitor KT5823 (5 μmol/L) or PD98059 attenuated LTD4 plus PGE2 synergistic responses (Fig 6, C-F). We then knocked down CysLT1R, EP3, both CysLT1R and EP3, and PKG in LAD2 cells by using protein-specific siRNAs (10 nmol/L) and nonspecific siRNAs as a control and analyzed LTD4 plus PGE2 effects. Transfection of MCs with CysLT1R, EP3, and PKG siRNAs significantly and specifically downregulated CysLT1R (55%), EP3 (60%), and PKG (64%) expression (see Fig E4 in this article’s Online Repository at www.jacionline.org). Importantly, downregulation of CysLT1R or EP3 partially inhibited both LTD4- and LTD4 plus PGE2–induced responses, but knockdown of both CysLT1R and EP3 completely inhibited LTD4 plus PGE2–induced synergy (Fig 7, A-C). Also, knockdown of PKG significantly inhibited LTD4 plus PGE2–induced effects (Fig 7, A-C), whereas the control siRNA did not affect LTD4- or PGE2-induced inflammatory responses.
FIG 7.
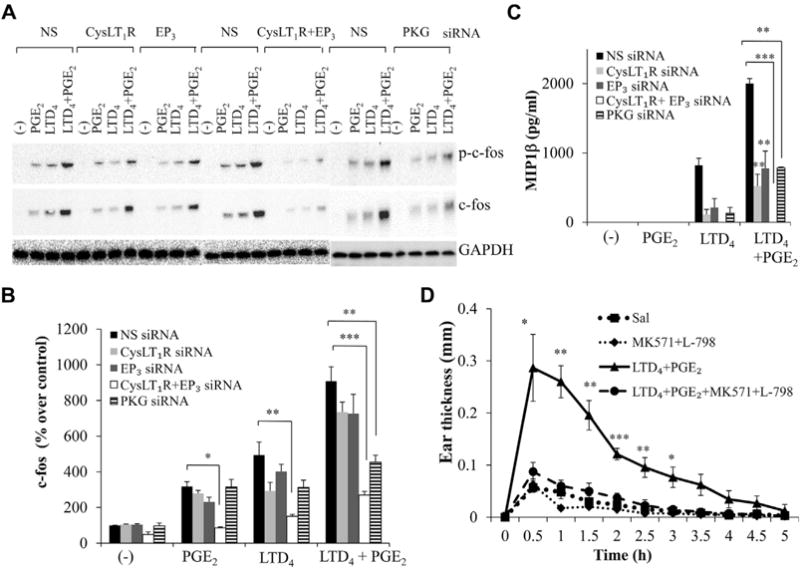
Simultaneous inhibition of CysLT1R and EP3 on LTD4 plus PGE2–mediated MC activation in vitro and ear edema in vivo. CysLT1R, EP3, or both and PKG were knocked down in LAD2 cells by corresponding siR-NAs (10 nmol/L), and c-fos (A and B) and MIP-1β (C) were analyzed with LTD4 plus PGE2 treatment. D, Ear thickness in BALB/c mice treated with 0.5 μmol/L LTD4 plus PGE2 ± MK571 (1 μmol/L) or L-798 (100 nmol/L). Results in Fig 7, D, are means ± SEMs from 4 to 6 mice per group per experiment and 3 experiments performed. *P < .05, **P < .01, and ***P < .001.
Combined treatment with CysLT1R and EP3 antagonists attenuates vascular inflammation induced by LTD4 plus PGE2
Next, we determined the effect of blocking both CysLT1R and EP3 simultaneously in evoking vascular inflammation. The synergistic ear edema response caused by combined treatment with PGE2 and LTD4 was substantially attenuated with MK571 plus L-798 treatment (Fig 7, D).
DISCUSSION
Edema formation is a prominent feature of the inflammatory response and serves an important function in local host defense and tissue repair. Inflammatory responses result in the movement of fluid and plasma proteins into the extracellular space from leaky blood vessels in response to various chemical mediators.22 In the current study, for the first time, we demonstrate that 2 major eicosanoids, LTD4 and PGE2, both derived through alternate pathways from arachidonic acid, synergistically enhanced vascular inflammation in vivo (edema formation) and MC activation in vitro. Although W-sh mice have been well characterized as MC deficient, they have additional defects beyond just MC deficiency, and hence there exists a possibility that the in vivo vascular permeability attenuated in W-sh mice could be mediated through other cell types in addition to MCs. A number of mediators have been implicated in the regulation of inflammation in an MC-mediated mechanism.23 Although MCs are primarily known to be activated in an antigen-dependent manner, evidence also suggests a role for them in antigen-independent activation.19,22,24 They are regulatory cells throughout the course of acute inflammation, from its initiation to resolution,25 and they contribute to the development of allergic disease.26 MC activation in patients with various kinds of inflammatory diseases is significant from a clinical perspective. Translational implications of LTD4 plus PGE2 crosstalk provoked us to examine the candidate molecules and signaling mechanisms involved. We identified that LTD4-PGE2 synergism was mediated through CysLT1R and EP3. It is known that G protein–coupled receptors interact with one another, modulating each other’s function both positively and negatively.27,28 Also, PGE2 relays differential responses depending on the receptor subtype, receptor coupling, and the cell type. For instance, PGE2 blocks FcεRI-mediated exocytosis of rat peritoneal MCs29 and human lung MCs30 in vitro through an increase in cyclic AMP levels (EP2, EP4, or both) but activates murine bone marrow-derived mast cells through EP3 in vitro.31 On the contrary, PGE2 in hMCs did not suppress FcεRI-dependent exocytosis but caused exocytosis on its own when the cells were primed with IL-4.16 It is likely that the suppressive effects of PGE2 on MC activation largely reflects EP2 signaling, and in agreement with this, EP2 protein expression is downregulated in MCs in the nasal polyp mucosa of patients with aspirin-exacerbated respiratory disease.32 PGE2 has been shown to stimulate chemotaxis and adhesion of MCs through EP.33,34 In a recent report Morimoto et al19 demonstrated that EP3 signaling in MCs generated PGE2-induced vasodilatation and subsequent edema formation and speculated that EP3-mediated MC activation might be involved in antigen-independent innate immune reactions. Interestingly, we identified that the LTD4 plus PGE2 synergism is also EP3 dependent, strengthening the idea that EP3 activation might contribute to a proinflammatory role of PGE2.
LTD4 plus PGE2 treatment also upregulated transcripts for inflammatory mediators, such as TNF-α and IL-8, both of which are chemoattractants for neutrophils. Interestingly, we observed synergistic COX-2 induction by LTD4 plus PGE2 treatment and PGD2 generation, revealing another major finding that LTD4, in concert with PGE2, can generate proinflammatory metabolite PGD2 and amplify associated signaling. MCs express both COX-1 and COX-2, and COX-2 is upregulated by inflammatory stimuli, suggesting a mechanism that can amplify PGD2 generation under inflammatory conditions. Interestingly, PGD2 has been recently shown to synergize with LTE4 to stimulate diverse TH2 functions and TH2 cell and neutrophil crosstalk.35,36 It is possible that during inflammation, LTD4 plus PGE2 treatment not only activates MCs and initiates inflammation, but also the product of this signaling, PGD2, can potentially perpetuate inflammation through combined action with LTE4. Both LTD4 and PGE2 have been demonstrated to phosphorylate Erk in MCs.8,16 In agreement, we noted phosphorylation of Erk by both agonists and observed enhanced Erk phosphorylation with LTD4 plus PGE2. Furthermore, we identified that LTD4-PGE2 synergy is relayed through PKG and Erk downstream of CysLT1R and EP3. Surprisingly, we found that PKC signals, which are vital in various aspects of CysLT1R,17,21,37 or PKA signals, which mediate EP-induced effects,16 are dispensable for this crosstalk, but it is dependent on PKG and Erk.
We have recently shown that LTD4 can influence SCF responses, potentiating MC proliferation.12 We also found that LTD4 potentiates endothelial cell adhesion mediated by TNF-α,20 suggesting that cys-LTs can modulate inflammation by acting in concert with other inflammatory mediators. Indeed, findings from the present study revealed that LTD4 and PGE2 produced at the inflammation site can synergistically activate MCs and further enhance inflammation. Notably, this enhanced inflammation by LTD4 and PGE2 both in vitro and in vivo is blocked only by the combined treatment of CysLT1R and EP3 antagonists and not by either antagonist alone. This argues for the possibility that the synergy is achieved through the contribution of a specific signaling component from LTD4 and another component from PGE2 signaling, rather than crosstalk at the receptor level. Although we found PKG and Erk are effector molecules in this signal, the convergence of these signaling events are still elusive.
Studies from EP knockout mice suggest that EP3 is mainly responsible for PGE2-induced MC activation and associated proinflammatory signaling pathways.31 High expression of EP3 is observed in the brain, and recent findings suggest an injurious role for the PGE2-EP3 signaling axis in modulating brain injury and inflammation after intracerebral hemorrhage.38 Along similar lines, enhanced EP3 expression and signaling were found in patients with diabetes and have been speculated as a new therapeutic target for β-cell dysfunction in patients with type 2 diabetes.39
All the above studies point to the pathologic role of EP3. Current work alludes to the advantage of blocking CysLT1R along with EP3, and it is tempting to speculate that the combined treatment of EP3 and CysLT1R antagonists might contribute effectively to targeting inflammation compared with the available CysLT1R antagonists alone and could open new avenues in clinical approaches for MC-mediated inflammatory diseases. Although PGE2 alone did not induce significant inflammatory transcripts and MIP-1β generation, it potentiated all the inflammatory readouts in concert with LTD4, suggesting that cys-LTs could modulate PGE2 signaling and blocking LTD4 plus PGE2 could be a potential therapeutic target to combat initiation and progression of inflammation.
Supplementary Material
Key messages.
LTD4 synergizes with PGE2 and activates MCs in vitro and vascular inflammation in vivo through CysLT1R and EP3, which might contribute to MC-mediated allergic inflammation.
LTD4-PGE2 synergism triggers diverse MC inflammatory responses through Gi-, PKG-, and Erk-dependent pathways.
LTD4 plus PGE2 crosstalk is efficiently blocked by simultaneous inhibition of CysLT1R and EP3 but not by either of the agonists alone, suggesting that vascular inflammation can be targeted efficiently by combining currently available CysLT1R antagonists with EP3 antagonists.
Acknowledgments
Supported by National Institutes of Health grant HL098953 and by James Foght Assistant Professor endowed professorship grant (to S.P.).
V. Kondeti and S. Paruchuri have received research support from the National Institutes of Health (HL098953) and the James L. and Martha J. Foght Assistant Professorship.
Abbreviations used
- cys-LT
Cysteinyl leukotriene
- CysLT1R
Cysteinyl leukotriene receptor 1
- CysLT2R
Cysteinyl leukotriene receptor 2
- EP
E-prostanoid receptor
- Erk
Extracellular signal-regulated kinase
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- hMC
Human cord blood–derived mast cell
- LT
leukotriene
- MC
Mast cell
- MIP-1β
Macrophage inflammatory protein 1β
- PG
Prostaglandin
- PK
Protein kinase
- PTX
Pertussis toxin
- SCF
Stem cell factor
- siRNA
Small interfering RNA
Footnotes
Disclosure of potential conflict of interest: The rest of the authors declare that they have no relevant conflicts of interest.
References
- 1.Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–86. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- 2.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–79. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 3.Hart PH. Regulation of the inflammatory response in asthma by mast cell products. Immunol Cell Biol. 2001;79:149–53. doi: 10.1046/j.1440-1711.2001.00983.x. [DOI] [PubMed] [Google Scholar]
- 4.Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167:835–48. doi: 10.1016/S0002-9440(10)62055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanaoka Y, Boyce JA. Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J Immunol. 2004;173:1503–10. doi: 10.4049/jimmunol.173.3.1503. [DOI] [PubMed] [Google Scholar]
- 6.Urade Y, Ujihara M, Horiguchi Y, Igarashi M, Nagata A, Ikai K, et al. Mast cells contain spleen-type prostaglandin D synthetase. J Biol Chem. 1990;265:371–5. [PubMed] [Google Scholar]
- 7.Woodward DF, Jones RL, Narumiya S. International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- 8.Paruchuri S, Jiang Y, Feng C, Francis SA, Plutzky J, Boyce JA. Leukotriene E4 activates peroxisome proliferator-activated receptor gamma and induces prostaglandin D2 generation by human mast cells. J Biol Chem. 2008;283:16477–87. doi: 10.1074/jbc.M705822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006;25:4615–27. doi: 10.1038/sj.emboj.7601341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanaoka Y, Maekawa A, Austen KF. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J Biol Chem. 2013;288:10967–72. doi: 10.1074/jbc.C113.453704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim DC, Hsu FI, Barrett NA, Friend DS, Grenningloh R, Ho IC, et al. Cysteinyl leukotrienes regulate Th2 cell-dependent pulmonary inflammation. J Immunol. 2006;176:4440–8. doi: 10.4049/jimmunol.176.7.4440. [DOI] [PubMed] [Google Scholar]
- 12.Al-Azzam N, Kondeti V, Duah E, Gombedza F, Thodeti CK, Paruchuri S. Modulation of mast cell proliferative and inflammatory responses by leukotriene d4 and stem cell factor signaling interactions. J Cell Physiol. 2015;230:595–602. doi: 10.1002/jcp.24777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stables MJ, Gilroy DW. Old and new generation lipid mediators in acute inflammation and resolution. Prog Lipid Res. 2011;50:35–51. doi: 10.1016/j.plipres.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 15.Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, et al. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk Res. 2003;27:677–82. doi: 10.1016/s0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]
- 16.Feng C, Beller EM, Bagga S, Boyce JA. Human mast cells express multiple EPs for prostaglandin E2 that differentially modulate activation responses. Blood. 2006;107:3243–50. doi: 10.1182/blood-2005-07-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paruchuri S, Hallberg B, Juhas M, Larsson C, Sjolander A. Leukotriene D(4) activates MAPK through a Ras-independent but PKCepsilon-dependent pathway in intestinal epithelial cells. J Cell Sci. 2002;115:1883–93. doi: 10.1242/jcs.115.9.1883. [DOI] [PubMed] [Google Scholar]
- 18.Maekawa A, Balestrieri B, Austen KF, Kanaoka Y. GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc Natl Acad Sci U S A. 2009;106:11685–90. doi: 10.1073/pnas.0905364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morimoto K, Shirata N, Taketomi Y, Tsuchiya S, Segi-Nishida E, Inazumi T, et al. Prostaglandin E2-EP3 signaling induces inflammatory swelling by mast cell activation. J Immunol. 2014;192:1130–7. doi: 10.4049/jimmunol.1300290. [DOI] [PubMed] [Google Scholar]
- 20.Duah E, Adapala RK, Al-Azzam N, Kondeti V, Gombedza F, Thodeti CK, et al. Cysteinyl leukotrienes regulate endothelial cell inflammatory and proliferative signals through CysLT(2) and CysLT(1) receptors. Sci Rep. 2013;3:3274. doi: 10.1038/srep03274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondeti V, Duah E, Al-Azzam N, Thodeti CK, Boyce JA, Paruchuri S. Differential regulation of cysteinyl leukotriene receptor signaling by protein kinase C in human mast cells. PLoS One. 2013;8:e71536. doi: 10.1371/journal.pone.0071536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan DH, Igyarto BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol. 2012;12:114–24. doi: 10.1038/nri3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. 2014;14:478–94. doi: 10.1038/nri3690. [DOI] [PubMed] [Google Scholar]
- 24.Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Kohler A, et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity. 2011;34:973–84. doi: 10.1016/j.immuni.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 25.St John AL, Abraham SN. Innate immunity and its regulation by mast cells. J Immunol. 2013;190:4458–63. doi: 10.4049/jimmunol.1203420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720–45. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Y, Borrelli LA, Kanaoka Y, Bacskai BJ, Boyce JA. CysLT2 receptors interact with CysLT1 receptors and down-modulate cysteinyl leukotriene-dependent mitogenic responses of mast cells. Blood. 2007;110:3263–70. doi: 10.1182/blood-2007-07-100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laidlaw TM, Boyce JA. Cysteinyl leukotriene receptors, old and new; implications for asthma. Clin Exp Allergy. 2012;42:1313–20. doi: 10.1111/j.1365-2222.2012.03982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaliner M, Austen KF. Cyclic AMP, ATP, and reversed anaphylactic histamine release from rat mast cells. J Immunol. 1974;112:664–74. [PubMed] [Google Scholar]
- 30.Peachell PT, MacGlashan DW, Jr, Lichtenstein LM, Schleimer RP. Regulation of human basophil and lung mast cell function by cyclic adenosine monophosphate. J Immunol. 1988;140:571–9. [PubMed] [Google Scholar]
- 31.Nguyen M, Solle M, Audoly LP, Tilley SL, Stock JL, McNeish JD, et al. Receptors and signaling mechanisms required for prostaglandin E2-mediated regulation of mast cell degranulation and IL-6 production. J Immunol. 2002;169:4586–93. doi: 10.4049/jimmunol.169.8.4586. [DOI] [PubMed] [Google Scholar]
- 32.Ying S, Meng Q, Scadding G, Parikh A, Corrigan CJ, Lee TH. Aspirin-sensitive rhinosinusitis is associated with reduced E-prostanoid 2 receptor expression on nasal mucosal inflammatory cells. J Allergy Clin Immunol. 2006;117:312–8. doi: 10.1016/j.jaci.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 33.Sakanaka M, Tanaka S, Sugimoto Y, Ichikawa A. Essential role of EP3 subtype in prostaglandin E2-induced adhesion of mouse cultured and peritoneal mast cells to the Arg-Gly-Asp-enriched matrix. Am J Physiol Cell Physiol. 2008;295:C1427–33. doi: 10.1152/ajpcell.00218.2008. [DOI] [PubMed] [Google Scholar]
- 34.Weller CL, Collington SJ, Hartnell A, Conroy DM, Kaise T, Barker JE, et al. Chemotactic action of prostaglandin E2 on mouse mast cells acting via the PGE2 receptor 3. Proc Natl Acad Sci U S A. 2007;104:11712–7. doi: 10.1073/pnas.0701700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue L, Fergusson J, Salimi M, Panse I, Ussher JE, Hegazy AN, et al. Prostaglandin D2 and leukotriene E4 synergize to stimulate diverse TH2 functions and TH2 cell/neutrophil crosstalk. J Allergy Clin Immunol. 2015;135:1358–66. e1–11. doi: 10.1016/j.jaci.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue L, Gyles SL, Wettey FR, Gazi L, Townsend E, Hunter MG, et al. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J Immunol. 2005;175:6531–6. doi: 10.4049/jimmunol.175.10.6531. [DOI] [PubMed] [Google Scholar]
- 37.Paruchuri S, Sjolander A. Leukotriene D4 mediates survival and proliferation via separate but parallel pathways in the human intestinal epithelial cell line Int 407. J Biol Chem. 2003;278:45577–85. doi: 10.1074/jbc.M302881200. [DOI] [PubMed] [Google Scholar]
- 38.Leclerc JL, Lampert AS, Diller MA, Dore S. Genetic deletion of the prostaglandin E prostanoid receptor subtype 3 improves anatomical and functional outcomes after intracerebral hemorrhage. Eur J Neurosci. 2015;41:1381–91. doi: 10.1111/ejn.12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimple ME, Keller MP, Rabaglia MR, Pasker RL, Neuman JC, Truchan NA, et al. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes. 2013;62:1904–12. doi: 10.2337/db12-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.