

Abstract
Because human energy metabolism evolved to favor adiposity over leanness, the availability of palatable, easily attainable, and calorically dense foods has led to unprecedented levels of obesity and its associated metabolic co-morbidities that appear resistant to traditional lifestyle interventions. However, recent progress identifying the molecular signaling pathways through which the brain and the gastrointestinal system communicate to govern energy homeostasis, combined with emerging insights on the molecular mechanisms underlying successful bariatric surgery, gives reason to be optimistic that novel precision medicines that mimic, enhance, and/or modulate gut-brain signaling can have unprecedented potential for stopping the obesity and type 2 diabetes pandemics.
Introduction
Our ancestral environment pressured an intertwined evolution between the human brain and the gastrointestinal (GI) tract (Aiello and Wheeler, 1995). Over the past 4 million years, the hominid brain has grown from an interior cranium volume of ~500–600 cm3 for our early ancestors to ~1,300–1,400 cm3 for modern humans. In order to maintain a relatively stable metabolic rate, this massive brain expansion was associated with corresponding adjustments in gut-size. Increasing availability of easily digestible energy and nutrient-rich food sources might have been the key prerequisite for this gut-brain coevolution (Leonard et al., 2007), which was paralleled by an analogous evolution of reciprocally coordinated metabolic cues between peripheral tissues and the CNS. Yet, whether or not genetic pre-disposition to storing excess energy continues to be an adaptive advantage selected by natural selection remains a topic of debate.
Over the last three decades, considerable progress has been made in understanding how diet-induced peripheral signals are integrated by neurobiological circuitries that govern energy metabolism and feeding behavior. Nutrient intake and content are immediately sensed and converted into gut-derived humoral and neural signals. The CNS receives and integrates this information, and in conjunction with environmental cues including visual, taste, and olfactory stimuli as well as with internal cues related to adiposity, stress, past experience, and many others, generates appropriate behavioral, autonomic, and endocrine output completing the energy homeostatic loop. Ironically, and despite our exponentially growing insight into the molecular control of metabolism and systems biology, modern humans have (mis)used the legacy of superior brain-power to engineer a dietary environment that supersedes peripherally derived satiation and adiposity signals, exploits the limbic system, is “unnaturally” energy-dense and hyper-palatable, and comes in virtually unlimited quantities. This “evolution” of our environment has, in less than a century, overwritten millions of years of biological optimization and has rapidly transformed Homo sapiens into an obese species (Figure 1). Even worse, this transformation may already be impacting future generations via deleterious epigenetic programming (Huypens et al., 2016).
Figure 1. Contribution of Genetic Heritage and Modern Lifestyle to Body Weight.
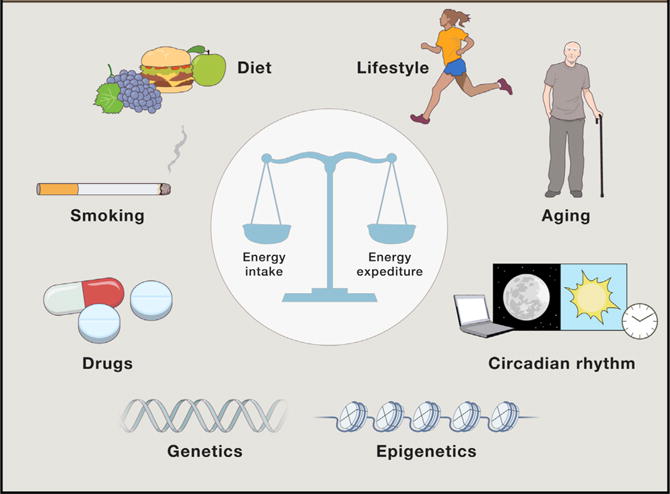
Body weight and metabolic health are determined by the interaction of genetic susceptibility and environmental influences. Major contributors to human adiposity include dietary quality and palatability, exercise habits, smoking, alcohol consumption, age, sleep, and pharmacology. These factors both directly and indirectly impact energy intake and/or energy expenditure to govern energy homeostasis.
Reversing the pathological processes underlying obesity and metabolic co-morbidities represents an essential task for 21st century biomedical research. Thus far, the most effective treatment for severe obesity and diabetes is surgical re-organization of the gastrointestinal (GI) system. Bariatric surgeries, such as gastric bypass, result in a multitude of metabolic benefits that for a majority of patients lead to a remission of their lifestyle-induced complications including obesity, type 2 diabetes, and cardiovascular disease (Mingrone et al., 2015; Schauer et al., 2014). Importantly, the physiological and molecular underpinnings of the metabolic benefits of the bariatric surgeries are beginning to emerge (Seeley et al., 2015), offering insights for inventing more effective pharmacotherapies that could replace these invasive and irreversible surgical interventions.
In this review, we highlight recent advances in unraveling essential cross-talk between the GI system and the brain circuits that govern homeostatic and hedonic components of energy balance. Notable advances include insights resulting from bariatric surgery as a key model of understanding human metabolic physiology, insights that allow differentiating essential from redundant gut-brain communication pathways. Importantly, these insights have now enriched and accelerated drug development programs, including advanced polypharmacy concepts as well as the first metabolic-precision medicines (Finan et al., 2016a; Kühnen et al., 2016; Oral et al., 2002).
Brain Control of Energy Homeostasis
In the middle of the 20th century, a series of elegant brain-lesion studies revealed the existence of distinct hypothalamic areas that foster increased food intake and weight gain (lateral hypothalamic area [LHA]) and reduced food intake and weight loss (ventromedial hypothalamic nucleus [VMH]), respectively (Brobeck, 1946). In parallel, it was speculated that an adipose tissue-derived factor communicates the availability of peripherally stored energy to these same hypothalamic areas (Kennedy, 1953, 1966). Inventive parabiosis experiments confirmed this idea. Experiments utilizing VMH-lesioned rats and mouse strains harboring mutations in obese (ob) or diabetic (db) alleles demonstrated that both a circulating factor and a CNS sensor for this factor are fundamental for the regulation of body fat (Coleman, 1973; Coleman and Hummel, 1969; Hervey, 1959). Decades later, the responsible mutations were genetically identified leading to the discovery of leptin (Zhang et al., 1994) and the leptin receptor (Lepr) (Tartaglia et al., 1995). Leptin was soon after shown to target the brain (Maffei et al., 1995) and to act in the brain to decrease body weight (Green et al., 1995). While the discovery of leptin did not immediately translate into a cure for human obesity, it ignited keen interest of scientists worldwide in uncovering other signals and circuits governing metabolic processes in health and disease. Insights from bariatric surgery, breakthrough advancements in research on molecular metabolism, technical revolutions transforming neuroscience, and a wealth of information generated by Genome-Wide Association Studies (GWAS), corroborated the important role of the CNS for the pathogenesis of obesity and are now positioning gutbrain communication as a prime target for developing superior therapeutics for metabolic diseases.
Hypothalamic Integration of Metabolic Status
The hypothalamic arcuate nucleus (ARC) is located so as to directly receive circulating hormonal and nutrient signals, and ARC cells accordingly express a multitude of hormone receptors and nutrient sensors. The exceptional position for the ARC to serve as a sensing and orchestrating conduit is based in part on its proximity to the neighboring median eminence, which as a circumventricular organ, is not fully protected by a functional blood-brain barrier (Schwartz et al., 2000). One area where particularly significant progress has been made in the last two decades concerns the physiological and molecular roles of sub-populations of ARC neurons that have distinct neuropeptide expression and consequently functional profiles, including those that express the orexigenic Agouti-related protein (Agrp) and the anorexigenic pro-opiomelanocortin (POMC), respectively, neuropeptides that are prototypical regulators of energy metabolism. These neurons are highly sensitive to metabolic status and adjust energy intake through bimodal melanocortin-4 receptor (MC4R) modulation. Another potent orexigenic peptide, neuropeptide Y (NPY), is co-expressed with AGRP. AGRP/NPY neurons project directly to POMC neurons and upon stimulation inhibit POMC firing through inhibitory Y1 and GABA receptors among likely multiple other levels of intercellular communication (reviewed in Waterson and Horvath, 2015).
Coordinated activity of POMC relative to AGRP and NPY is a major determinant of feeding behavior and body weight regulation. Identifying the significance of MC4Rs located in the nearby hypothalamic paraventricular nucleus (PVN) to integrate this information into a larger neural feeding circuitry represents a hallmark success of modern obesity research: loss-of-function mutations in the MC4R gene lead to severe obesity in both rodents (Huszar et al., 1997) and humans (Farooqi et al., 2003; Yeo et al., 1998). Consequently, several drug development initiatives have been initiated to advance efficacious MC4R agonists (Fani et al., 2014). Despite these efforts and the recognized role of MC4R in energy homeostasis, the exact neuronal circuitry downstream of MC4R remains remarkably uncharted (Krashes et al., 2016).
In addition to receiving input from the ARC, the PVN also integrates information from the LHA, the VMH, and the dorsomedial nucleus (DMN). The intercommunication among neurons in these distinct hypothalamic nuclei is multidirectional, and through systematic and persistent efforts, researchers are currently working to delineate the complex intrahypothalamic wiring governing energy homeostasis and systemic metabolism (Branco et al., 2016; García-Cáceres et al., 2016; Koch et al., 2015; Stanley et al., 2016; Steculorum et al., 2015; Tan et al., 2016).
Hindbrain Sensing of Gut-Derived Satiation Factors
Whereas the hypothalamus plays an essential role in directly sensing and integrating peripheral inputs denoting nutrients in the blood with hormonal signals indicating the amount of fat stored in the adipose tissue (leptin and insulin), the regulation of energy homeostasis is actually coordinated by a complex neurocircuitry involving multiple brain regions. In particular, the caudal brainstem represents another major integrative gut-brain hub. During the course of a meal, this brain region is flooded with afferent information pertaining to the composition and quantity of nutrients being ingested (Grill and Hayes, 2012). The area postrema (AP), a circumventricular organ analogous to the hypothalamic ARC, is located on the caudal brainstem and ideally situated to receive and integrate circulating metabolic signals including leptin, amylin, cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1), peptide YY (PYY), ghrelin, and others, each of which can influence satiation, or how much food will be consumed in the ongoing meal. The AP projects to the adjacent nucleus tractus solitarius (NTS) that in turn relays visceral information via monoamine-expressing neuronal projections to other brain areas including several hypothalamic nuclei (Grill and Hayes, 2012).
As discussed in detail in the next section, the NTS receives a continuous flow of information from the gut, in part from vagal afferent fibers that innervate large parts of the intestinal wall and gastric mucosa and that convey the volume and composition of food being ingested by secreting peptide hormones such as CCK and GLP-1. These then act in a paracrine fashion on their receptors located on the surface of local vagal afferent fibers in the intestine that project to the NTS; these vagal neurons thus convert nutrient-dependent changes in the gut-hormonal milieu into electro-chemical information that is relayed to the NTS (Berthoud and Morrison, 2008). Neurons in the NTS integrate the incoming vagal information with other neuroendocrine signals, including locally synthesized GLP-1 and CCK, such that the magnitude of the incoming responses to the gut-derived signals can be enhanced or suppressed before being relayed from the NTS to other brain areas (Barrera et al., 2011; D’Agostino et al., 2016; Herbert and Saper, 1990; Larsen et al., 1997). Thus, multiple layers of perpetual gut-derived information on nutrient availability and metabolic balance highlights that mammals, including Homo sapiens, have evolved a highly complex metabolic control system.
Role of Hedonic Eating and Food Reward in Energy Homeostasis
Around 80 AD, the philosopher Musonius Rufus observed that: “pleasure associated with eating a gourmet cuisine creates a desire for overeating and humans will choose food that is pleasant over food that is nutritious” (Cynthia, 2010). Today, we refer to this as hedonic food intake involving “food reward,” a phenomenon describing the consumption of palatable foods beyond the need-based energy requirements of the organism (Kenny, 2011). Animals and humans given a choice between rewarding and non-rewarding foods disproportionally over-consume the palatable rewarding food, often in quantities beyond their energetic needs (Rising et al., 1992; Sclafani and Springer, 1976). Intriguingly, despite having free access to nutritious but less appealing food, animals willingly endure considerable discomfort such as cold to obtain rewarding and palatable foods (Cabanac and Johnson, 1983; Oswald et al., 2011), underlining the potent impact of the reward system and its temporal uncoupling from energy need-based circuits.
Ample evidence supports the concept that neurons located in the midbrain that project anteriorly to the nucleus accumbens (i.e., the mesoaccumbens dopamine system) play an essential role for integrating and incorporating the rewarding properties of foods into ingestive behavior. Notably, the prototypical endocrine regulators of energy homeostasis, such as leptin, insulin, ghrelin, and GLP-1, besides acting in the hypothalamus and NTS, also directly act upon these dopaminergic neurons in the midbrain to influence hedonic feeding (Dickson et al., 2012; Farooqi et al., 2007; Figlewicz, 2016; Fulton et al., 2006; Skibicka et al., 2012). This information appears to be intertwined with emotion-related information arising from the limbic system and energy-need cues coming from the LHA, and all of this information is relayed to cortical regions where it processed to execute an appropriate behavioral response such as eating or satiation.
The Overfed Brain: Neuronal Perturbations Leading to Obesity
For the first time in history, we have reached a situation where our planet harbors more obese than underweight humans (Di Angelantonio et al., 2016). The whole-body energy homeostasis model implies that biological processes have emerged to maintain stable fuel availability independent of environmental conditions (Schwartz et al., 2000). Despite the fact that many individuals maintain a relatively constant body weight without heeding energy intake or expenditure, the doubling of the prevalence of obesity in just 30 years arguably implies that human “energy homeostasis” may be an oxymoron. Instead, humans may have evolved a regulatory system biased toward defending body fat over leanness (Chakravarthy and Booth, 2004; Schwartz et al., 2003). The drifty gene hypothesis suggests that weight gain was released from selection pressure when our ancestors evolved from the risk of predation around 2 million years ago (Speakman, 2014). Consistent with this view, the neuronal circuitry defending body weight arguably predominates the circuitry defending against weight gain (Henry et al., 2015; Wu et al., 2012). Further, the majority of the genetic polymorphisms linked to obesity relate to proteins either expressed in the brain or exerting their most important functions in the CNS to control feeding behavior or relevant components of systemic metabolism (Locke et al., 2015).
Although researchers are struggling to comprehend the full scope of the genetic and epigenetic factors, patterns, and processes driving the obesity pandemic, our knowledge of the immediate neuronal response following exposure to an energy-rich palatable diet has grown exponentially over the last 20 years. However, interpretive complications and unforeseen complexities arise as rapidly as new information comes in. For example, whereas a decrease of leptin, the adipose tissue-derived hormone secreted in direct proportion to body fat, potently initiates overfeeding and weight gain, excess leptin generates only moderate physiological responses, limiting leptin’s therapeutic potential as a weight-loss agent (Heymsfield et al., 1999; Zelissen et al., 2005). As another example, in addition to influencing satiation and energy homeostasis via their energetic value, individual nutrients are increasingly acknowledged as signaling molecules that increase brain (including hypothalamic) inflammatory responses that secondarily interfere with the regulation of energy balance (Valdearcos et al., 2015). Finally, there is a disconnect between the long-held concept that body fat is homeostatically defended, sometimes referred to as the set-point model of adiposity, and the reality that palatable diets, stress, experience, and a multitude of other factors determine the actual amount of fat that is achieved and defended (Ramsay and Woods, 2014). Despite the nuances of these complexities, it is increasingly clear that reward, hedonic, and energy need-based circuits are closely interconnected and reciprocally influence one another (Figure 2).
Figure 2. Gut-Brain Cross-Talk in Eating Behavior.
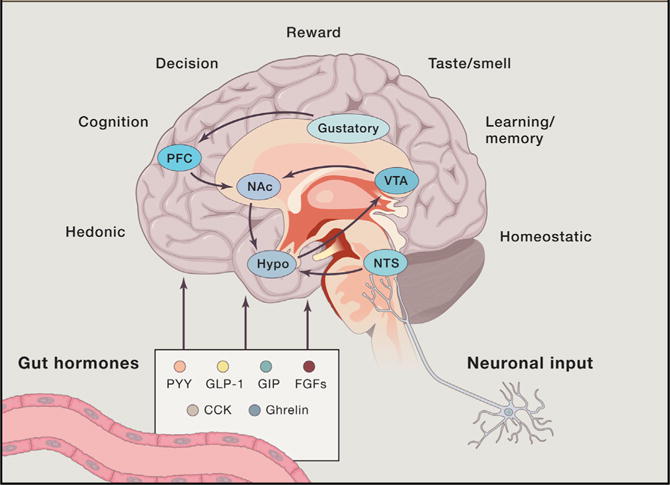
Gut hormones and afferent neurons are key signals in gut-brain communication and human energy metabolism. Brain regions, traditionally divided into homeostatic (hypothalamus and NTS) and hedonic (VTA – Nac), sense diverse humoral factors and generate signals in cooperation with cognitive processes and information arising from visual, gustatory and olfactory stimuli to influence feeding behavior. PFC, prefrontal cortex; NAc, nucleus accumbens; VTA, ventral tegmental area; Hypo, hypothalamus; NTS, nucleus tractus solitaries.
Gastrointestinal Control of Energy Homeostasis
The GI tract is involved in several aspects of energy homeostasis. First, the alimentary canal is essential for nutrient absorption and the delivery of fuels to the organism. Like other vital functions, such as breathing, nutrient processing and absorption are largely automatic, relying on elaborate hormonal and neural feedback systems that have evolved over millions of years. The alimentary tract even has its own “enteric” brain containing as many neurons as the spinal cord and capable of executing all basic functions in the absence of input from the brain and other organs (Furness, 2012). Second, the gastrointestinal tract utilizes a significant portion of energy for itself, and this is due to its considerable mass and the constant turnover of epithelial cells that, in humans, have the surface area of a tennis court (Madara, 2011). Third, nutrient absorption and subsequent metabolic processing determines the thermic effect of food, a small but significant portion of total energy expenditure (de Jonge and Bray, 1997).
Enteroendocrine Cells Are Primary Nutrient Sensors
The regulation of energy balance by the brain requires accurate information regarding the influx of energy. This is accomplished by nutrient sensor mechanisms distributed along the entire alimentary canal, starting with the gustatory system in the mouth and terminating with sensors in the hepatic portal vein (Berthoud, 2008a). Throughout the GI tract, specialized endothelial cells called enteroendocrine cells sense the luminal content and trans-epithelial flux of nutrients and consequently release hormones and paracrine factors (Gribble and Reimann, 2016) informing the brain and other organs either directly through the circulation or by affecting sensory neurons projecting to the brain.
More than 100 years ago, secretin was the first GI hormone discovered, and the list is now quite long and includes the incretins GIP and GLP-1 as well as the satiation peptides CCK and ghrelin (Gribble, 2012; Rehfeld, 1998). All are secreted from enteroendocrine cells interspersed among the enterocytes lining the gut lumen. The existence of different combinations of G protein-coupled receptors and transporters in these enteroendocrine cells, and their differential distribution along the gut, enable them to sense all major nutrients (Reimann et al., 2012).
Intrinsic and Extrinsic Innervation of the Gut
The GI tract is innervated by both the sympathetic and parasympathetic nervous systems as well as by primary sensory nerves. The densest and most widespread innervation comes from vagal afferent neurons located in the nodose ganglia (Berthoud and Neuhuber, 2000; Berthoud et al., 1997). These bipolar neurons innervate all layers of the gut wall from esophagus to colon and their proximal extensions terminate in the NTS. Sensory terminals innervating the external muscle layers and the myenteric plexus of the gut act as mechanical stretch and tension sensors, respectively (Berthoud and Powley, 1992; Zagorodnyuk et al., 2001) while terminals in the mucosa are thought to be mainly chemosensors (Berthoud et al., 1995; Berthoud and Patterson, 1996; Blackshaw and Grundy, 1990; Powley et al., 2011).
Functions of the GI tract, pancreas, and liver are all also modulated by efferent vagal (parasympathetic) and sympathetic motor outflow, with vagal input generally accelerating and sympathetic input generally slowing intestinal transit and absorptive capacity. These autonomic inputs indirectly control food intake and energy balance by changing sensory and hormonal signaling to the brain (Berthoud, 2008b).
Gut Nutrient Sensing Can Be Both Satiating and Appetizing
There is no doubt that gut and brain intimately communicate with each other. What has been less clear is how much this communication contributes to the controls of food intake and energy expenditure and to the regulation of energy homeostasis. While early views focused almost exclusively on the satiating action of post-oral nutrients (Gibbs et al., 1973; Smith, 1996), it is now recognized that nutrients in the gut, and even in the circulation, also act to stimulate appetite and food intake (for review see Sclafani and Ackroff, 2012). This is based on studies using the “electronic esophagus,” wherein rodents can choose to lick from either of two bottles containing different flavored non-nutritive solutions, with each lick triggering intragastric or intraduodenal infusions of glucose, fat, or saline. In many conditions, licking particular solutions leads to a learned preference for the flavor associated with the nutrient (Lucas and Sclafani, 1989; Sclafani, 2004). In distinction to the feed-back mechanisms of satiation, the term “appetition” was coined for this feed-forward phenomenon (Sclafani, 2013). Using this paradigm, it was demonstrated that the sodium-glucose transporter-1 (SGLT1) is required for post-oral glucose (Sclafani et al., 2016), and the fatty acid sensors GPR40 and GPR120 for post-oral fat (Sclafani et al., 2013), to mediate the development of preferences for flavors associated with glucose or fats, respectively. Interestingly, in contrast to the satiating post-oral effects of carbohydrates and fats, neither vagal afferents nor sensory fibers in the splanchnic nerve are essential for these “appetition” effects (Sclafani et al., 2003). Further, ghrelin signaling, which is known to stimulate appetite when administered systemically, is not necessary for sugar or fat to condition flavor preferences (Sclafani et al., 2015).
More recently, sugar sensing in the gut has been linked to the brain’s dopamine/positive hedonic circuits that drive food intake. Specifically, infusion of glucose into the gut, but not into the mouth, triggered dopamine release in the dorsal striatum, a forebrain reward area, whereas glucose or non-caloric sweeteners infused into the mouth triggered dopamine release only in the ventral striatum (nucleus accumbens and ventral pallidum) (Tellez et al., 2016). Importantly, the dorsal striatal dopaminergic circuit sensitive to the nutritional value of glucose in the gut was able to override any inhibitory signals generated by the mouth. If confirmed, this logic may explain why, over the long term, energy content trumps taste when it comes to food choice. The same group of researchers demonstrated that surgically bypassing the duodenum and jejunum in mice abolished the dopamine-stimulating and appetite-enhancing effects of intragastric glucose (Han et al., 2016), suggesting this as a critical mechanism underlying the reduced sweet craving and carbohydrate intake after bariatric surgery in rodents (Shin and Berthoud, 2011; Tichansky et al., 2011; Wilson-Pérez et al., 2013b).
The Overfed Gut: Desensitization of Nutrient-Sensing Pathways
Chronic exposure to high-fat diets (HFDs) can lead to structural and functional changes in the gut nutrient-sensing pathways extending from the mucosa to the brain (for recent reviews see Brandsma et al., 2015; Teixeira et al., 2012). HFD-induced changes generally increase as a function of time on the diet, with changes after 1 week being fully reversible, but with changes after several weeks not being reversible and correlating with the onset of obesity (Hamilton et al., 2015). Importantly, offspring of HFD-induced obese mothers also have impaired gut barrier functions and increased oxidative stress markers (Xue et al., 2014). Impairment of intestinal barrier function activates the innate immune system and ultimately leads to obesity, insulin resistance, and hepatic steatosis. Reduced secretion of gut hormones responsible for the incretin effect and satiation may mediate these effects. In addition, reduced sensitivity of vagal afferent neurons to gut nutrient signaling may enhance hyperphagia and impair glycemic control (de Lartigue et al., 2014). However, because vagal deafferentation with capsaicin at the beginning of a HFD regimen in rats led to a modest decrease, rather than an increase, in visceral fat accumulation (Stearns et al., 2012), the contribution of vagal afferents to the pathogenesis of obesity remains unclear.
Attempts to capitalize on extrinsic and intrinsic enteric nervous system innervation of the gut as targets for neuromodulation strategies in the prevention or reversal of obesity and metabolic diseases have had relatively little success. The recently approved V-Bloc device, which uses high-frequency stimulation of the abdominal vagal trunks to block all (sensory and motor) vagal communication between the gut and the brain, has only modest effects on body weight (Ikramuddin et al., 2014). Similarly, gastric pacing through implanted serosal electrodes that likely affect enteric nervous system activity has not brought the expected results on body weight control (Hasler, 2009). These rather disappointing outcomes are likely due to the non-specificity of the nerve manipulations. Autonomic and sensory innervation of peripheral organs is not an all-or-none phenomenon and rather is functionally highly specific. Until we understand this functional specificity, such crude manipulation attempts are premature and ill-conceived. The modern genetics-based neural manipulation techniques that revolutionized central nervous circuit mapping (Sternson and Roth, 2014; Wouterlood et al., 2014) are now ready to be applied to the peripheral and enteric nervous systems. Early reports with deletion or stimulation of selective populations of vagal afferents demonstrate the potential of this strategy (Udit and Gautron, 2013). This should lead to more informed and specific neuromodulation strategies for more profound and sustained effects on body weight regulation.
In sum, the brain and the gut are each endowed with a rich neural and hormonal intrinsic control system, the two governing separate but overlapping aspects of energy balance. While these two organ systems can function independently of one another, there is normally a highly complex exchange of information between the two that is both neurally and hormonally mediated. Identifying the key signaling mechanisms underlying the normal functioning of all parts of the gut-brain axis, and devising ways to intervene with them, is a major goal and challenge for tackling metabolic disorders.
Bariatric Surgeries: Toward Understanding Gut-Brain Signaling and Cooperation
Some of the most compelling evidence for a role of the gut-brain axis to regulate multiple aspects of metabolism comes from the clinical effects of bariatric surgery. In fact, nothing currently available in the clinic is nearly as effective as bariatric procedures to produce large and sustained weight loss. Interestingly, the dramatic efficacy of these procedures was not the result of careful hypothesizing about cross-talk between the gut and other organ systems. Rather, the effects of procedures such as the Roux-en-Y gastric bypass (RYGB) were secondary effects observed when they were used to treat ulcers. Observant surgeons noticed distinct weight loss and improved metabolic endpoints in patients that received a RYGB (Celio and Pories, 2016).
These observations led some surgeons to advocate for greater use of bypass procedures in obese patients, as well as to hypothesize how it is that surgical rearrangement of the GI tract can result in such profound effects. Not surprisingly, these surgeons focused their hypotheses on the mechanical aspects of the surgery itself. RYGB involves creating a small gastric pouch just below the distal end of the esophagus, thereby “restricting” patients from overeating by limiting the size of individual bouts of ingestion. RYGB also reroutes calories because the jejunum becomes linked directly to the small gastric pouch, the alimentary canal now bypassing the rest of the stomach and upper duodenum. With the absorptive capacity of the duodenum lost, the procedure was also thought to result in “malabsorption,” with some otherwise absorbable calories lost in the feces (Celio and Pories, 2016).
These mechanical hypotheses about bariatric surgery have generated a powerful narrative that remains the dominant paradigm for explaining surgical effects, including to patients. It also happens to be almost entirely false. In fact, a basic understanding of how body weight is regulated presents conundrums for the mechanical explanations. Body weight loss is typically counter-acted by powerful responses that include increased hunger in response to falling leptin and insulin levels (Schwartz et al., 2000). If surgery made it harder to ingest or absorb calories, patients would become dramatically hungrier. What is observed, however, is that despite pronounced weight loss, patients report being less hungry (le Roux and Bueter, 2014).
Other evidence comes from comparing surgical procedures, which result in relatively comparable and profound efficacy. In vertical sleeve gastrectomy (VSG), 80% of the stomach is removed along the greater curvature. This is often considered a “restrictive” procedure due to the greatly reduced size of the remaining stomach; however, with no rerouted calories, caloric absorption should be normal. Relative to RYGB, VSG creates only a portion of the restriction (i.e., the remaining stomach is much larger than RYGB pouch) and it does not result in malabsorption. Further, in some reports, VSG has comparable efficacy as RYGB in terms of weight loss and resolution of type 2 diabetes (Colquitt et al., 2014).
Further evidence that mechanical explanations do not adequately explain the effects of these procedures comes from rodent models that mimic the weight loss and improved metabolism observed in humans. First, following either RYGB or VSG, rodents resume eating the same number of calories as the sham-surgery controls within 2 months (Stefater et al., 2010). They maintain the weight loss but do so with normal food intake despite no evidence of dilation of the restrictive elements of the surgery that might undermine its efficacy from a mechanical perspective (Chambers et al., 2011). Moreover, rodents defend this new lower body weight in the same manner as unoperated control animals. When further food restriction is forced upon the animal, it loses weight as expected. However, once ad lib food is returned, it overeats and recovers the lost weight, thus defending body weight, and this occurs after both RYGB and VSG (Stefater et al., 2010). If the lost weight were primarily the result of restriction, animals should be unable to further increase their intake because they were already “underweight” compared to their sham-operated controls. As there is nothing physically preventing them from consuming additional calories, the key point is that animals choose not to overeat after the surgery, presumably because of altered signals from the gut indicating that this new reduced weight is appropriate.
Lastly, other biological circumstances that increase food intake in unoperated controls also do so in animals with surgical interventions. For example, female rats with a VSG gain weight during pregnancy in a manner identical to that of sham-operated controls (Grayson et al., 2013). Moreover, once the litter arrives and the dam now has to feed 10–12 hungry mouths through lactation, VSG females can double their caloric intake to keep up with the increased caloric demand just as happens in sham-operated controls.
The important point is that while it may seem obvious that surgery results in a mechanical alteration of the gastrointestinal tract, it is less obvious that this mechanical alteration (restriction and/or malabsorption) actually underlies its effect. The alternative is to hypothesize that bariatric surgery’s effects are the result of altered gut signals that act in the brain and other organs to alter the physiological system that regulates body weight and other important aspects of metabolism. Changes in the relative levels of various gut hormones represent one such potential signal and have received considerable attention as potential mediators of the effects of bariatric surgery (Makaronidis and Batterham, 2016). The evidence for an important role of classic gut hormones such as GLP-1, PYY, or ghrelin are decidedly mixed. In genetic mouse models, loss of function of the GLP-1 receptor or of ghrelin does not impair the effects of either VSG or RYGB (Chambers et al., 2013; Wilson-Pérez et al., 2013a). In humans, however, acute blockade of the GLP-1R is at least partly efficacious at reversing the impact of RYGB on classical incretin effects, including effects to increase insulin secretion (Salehi et al., 2011). A causal role of altered gut hormones remains an area of great controversy and a full explication of that controversy is beyond the scope of the present review. Nevertheless, the mouse data point to the possibility that there are mechanisms beyond these classic hormone systems that are important contributors to surgical effects on weight and metabolism (Figure 3).
Figure 3. Gut-Brain Adjustments following Bariatric Surgery.
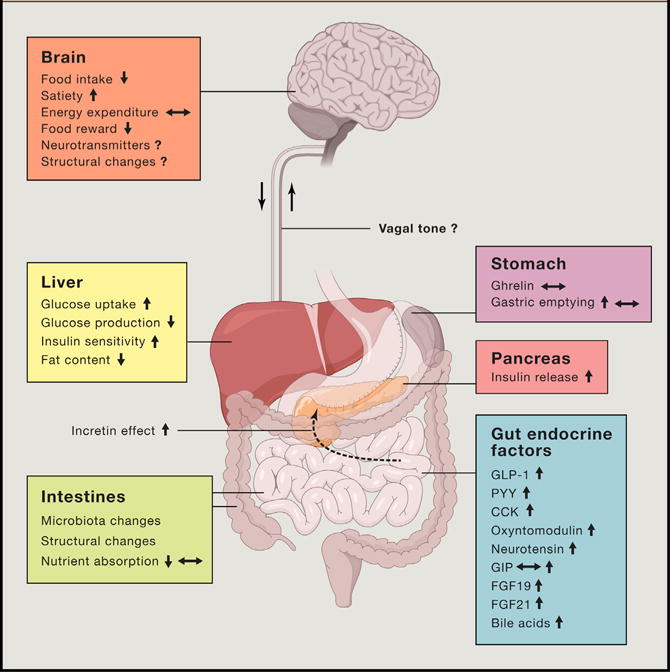
Cartoon overview of the biological adjustments in the gut-brain axis following bariatric surgery in obese subjects. In the gut, meal-induced peptides and microbiota are significantly altered following bariatric surgery. These adjustments contribute to improved glycemic control and to numerous behavioral adjustments that collectively contribute to reduced caloric intake following the surgery.
Role of Bile Acids to the Metabolic Benefits of Bariatric Surgery
Bile acids are made in the liver and, particularly in response to the ingestion of fatty foods, are secreted into the duodenum where they are essential for lipid absorption. Bile acids can be absorbed into the blood from the gut and then function as hormones by acting on nuclear receptors such as FXR and cell-surface receptors such as TGR5 (Kawamata et al., 2003; Wang et al., 1999). Both VSG and RYGB increase the circulating levels of bile acids, as well as alter their composition, in both rodents and humans (Kohli et al., 2013; Patti et al., 2009), suggesting the intriguing possibility that altered bile acid dynamics may underlie some of the effects of these procedures. Whole-body deletion of FXR results in mice that gain less weight on a HFD but nevertheless have reduced responses to VSG for long-term weight loss, reductions in food intake, and improvements in glucose tolerance (Ryan et al., 2014). Mice with no TGR5 signaling have normal body weight loss following VSG, but have less improvements in glucose regulation (McGavigan et al., 2017). The point is that molecular targeting of bile acid signaling provides evidence that alterations in bile acids are an important physiological mechanism by which surgical intervention can alter energy balance and metabolism.
A key question is how bariatric surgery alters these physiological systems, including gut hormone secretion and bile acid dynamics, to enable such enduring improvements in body weight and glycemic control (Figure 3). The most obvious possibility in the case of RYGB is the presentation of under-digested chyme into the distal GI tract. Such chyme is then hypothesized to activate nutrient signaling on entroendocrine cells to drive increased secretion of gut hormones such as GLP-1 and PYY. However, intestinal rerouting cannot be the only mechanism by which the GI tract alters signals to brain and other organs. For example, greatly enhanced secretion of GLP-1 is observed in VSG even though there is no rerouting of the chyme (Chambers et al., 2011; Steinert et al., 2013). Thus, a critical issue is just how the gut adapts to its new surgically altered reality and whether there are their commonalities among successful procedures that may suggest common underlying mechanisms for their effects.
Brain Adaptations following Bariatric Surgery
The earliest observations suggested that following RYGB, patients changed their hedonic and emotional rapport with food—they were less preoccupied with thinking about food and began to prefer low-calorie foods such as fruits and vegetables over high-calorie foods such as French fries and ice cream (Ernst et al., 2009; Schultes et al., 2010). More vigorously controlled human studies confirmed a reduced hedonic impact of palatable foods (Ullrich et al., 2013), and rodent studies found reduced “liking” and “wanting” of sweet and oily foods (Shin et al., 2011) as well as a shift in preference away from fat and to carbohydrate (Wilson-Pérez et al., 2013b).
Neural correlates of these hedonic shifts after RYGB were identified in fMRI studies demonstrating weaker responses to the sight of high-calorie foods and stronger responses to pictures of low-calorie foods in corticolimbic and other brain structures compared to responses in obese subjects (Frank et al., 2014; Ochner et al., 2012; Scholtz et al., 2014). In addition, one PET study reported decreased dopamine receptor-2 availability in the dorsal striatum 6 weeks after RYGB surgery in female patients (Dunn et al., 2010) (see Steele et al. [2010] for opposite outcome). However, and importantly, all of these studies were observational and associative. No study has directly tested the importance of such changes in brain activity for the beneficial effects of bariatric surgeries.
Potential Role for Gut Microbiota in Influencing Gut-Brain Signaling and Energy Balance
The human gut is inhabited by trillions of microbes. Understanding the symbiotic relationship between the gut microbiota and the human host—and the possible functional contributions of this ecosystem to human physiology and health—is currently being actively pursued. The gut microbiome co-evolved with our human ancestors, and gut bacteria have likely played a vital complementary role for extracting energy and nutrients from dissimilar food sources throughout our evolutionary history (Schnorr et al., 2014) and they likely interact with gut-brain signaling. For example, in response to a HFD, increased microbiota acetate production has been suggested to drive a pathological feedback loop of overeating involving direct stimulation of the parasympathetic nervous system and ghrelin secretion (Perry et al., 2016). Conversely, prebiotic-induced microbiota modulation may amplify GLP-1 and PYY secretion, exemplifying that cross talk between enteroendocrine anorectic hormones and gut bacteria might be exploited to benefit metabolic control (Cani and Delzenne, 2009). Finally, insights into a gut-brain circuitry involving microbiota and bile acid signaling suggest potential opportunities for innovative therapeutic intervention points (Degirolamo et al., 2014; Sayin et al., 2013). However, most reports find metabolic changes to be either rather modest in size, linked merely by association, or still awaiting replication, and massive alterations of gut microbiota in clinical studies have recently been documented not to impact energy homeostasis or systemic metabolism in humans (Mikkelsen et al., 2015; Reijnders et al., 2016).
Therapeutic Applications: Emerging Treatment Avenues for Metabolic Diseases Based on Gut-Brain Signaling
Converging evidence suggests that the metabolic benefits of bariatric surgery are consequential to complex gastrointestinal adjustments that generate signals “educating” the brain on the new gut-environment, and these altered gut-brain signals somehow lower the defended level of body fat and restore glycemic control. Identifying and mimicking these altered signals should be a major focus of research aimed to thwart the obesity/diabetes pandemic. Here, we provide an overview of a few prominent pre-clinical avenues and highlight opportunities—and knowledge-gaps—in the ongoing enterprise of transforming our heightened understanding of the gut-brain axis into effective and safe pharmacotherapies.
Mimicking Bariatric Surgical Enteroendocrine Responses with Polypharmacy
Successful bariatric surgeries are associated with altered circulating levels of numerous gut-derived factors including GLP-1 (Korner et al., 2007), CCK (Dirksen et al., 2013), PYY (Chan et al., 2006), GIP (Näslund et al., 1998), bile acids (Patti et al., 2009), ghrelin (Cummings et al., 2002), neurotensin (Ratner et al., 2016), FGF19, and FGF21 (Jansen et al., 2011). Because each of these factors influences appetite and substrate metabolism, mapping out the alterations in gut hormone secretion following bariatric surgery has received sizeable attention over the last decade (Madsbad et al., 2014). In context, this endeavor is considered an appropriate starting point for designing novel pharmacotherapies that could mimic the metabolic benefits of the surgeries.
Gut Peptide-Based Combinatorial Strategies
Perhaps the most outstanding endocrine adjustment following bariatric surgery is amplification of postprandial GLP-1 secretion (Falkén et al., 2011; Romero et al., 2012). GLP-1 targets the CNS to promote satiation and the pancreas to intensify insulin secretion, and through laborious pharmacological optimization, GLP-1-based therapies have now obtained pharmacological pole-position for the dual treatment of obesity and diabetes (Clemmensen et al., 2016). As a stand-alone therapy for type 2 diabetes, GLP-1R agonists provide relevant yet insufficient metabolic benefits that are limited by a dose-dependent increase in adverse GI events. Thus, combining GLP-1R agonism with suitable endocrine partners aiming for additive or synergistic metabolic benefits without compromising safety may provide an optimal path toward an incretin-based cure for obesity and its sequelae (Finan et al., 2015a).
Secretion of both CCK and PYY are augmented by bariatric surgery and each has thus been suggested to contribute to the satiating effects resulting from the procedure. Whereas administration of CCK together with GLP-1 has been reported to bestow no additional benefits on satiation (Brennan et al., 2005; Gutzwiller et al., 2004), co-infusion studies using GLP-1 and PYY report additive lowering of food intake in animals (Neary et al., 2005) and humans (De Silva et al., 2011), and this is further potentiated by the addition of neurotensin (Grunddal et al., 2016). Nonetheless, co-administration of GLP-1 and PYY (with or without an appropriate third agent) has yet to be tested in a controlled clinical setting. The fact that both GLP-1 and PYY dose-dependently increase nausea limits therapeutic utility of the combination.
A major obstacle to the development of efficacious weight loss drugs is counter-regulatory biological adjustments to pharmacotherapy-induced perturbations in energy balance. Targeting anorectic pathways decreases energy expenditure and, conversely, targeting energy expenditure stimulates appetite. Thus, in order to sustain energy expenditure in the weight-reduced state, administration of a thermogenic agent may be useful. This would also imply that polypharmacy solely targeting anorectic mechanisms (such as PYY and GLP-1 co-administration) might not offer sufficiently broad joint action profiles to successfully treat obesity.
Glucagon agonism has recently been discovered to offer the previously missing potential for safe and chemically achievable addition of energy expenditure on top of incretin biology. Indeed, mounting evidence based on innovative chemistry and extensive biological studies now positions glucagon as a relevant candidate to uphold energy expenditure in pharmacotherapy-induced weight loss (Day et al., 2009; Habegger et al., 2010). Novel single-molecule glucagon/GLP-1 co-agonists cause superior weight reduction relative to GLP-1 mono-agonism (Day et al., 2009). In pre-clinical models, these dual-compounds achieved benefits similar to bariatric surgery but without apparent side effects. Follow-up studies confirmed this phenomenon in rodent studies (Pocai et al., 2009) as well as by using co-infusion of GLP-1 and glucagon in humans (Cegla et al., 2014; Tan et al., 2013). Consequently, numerous clinical trials based on GLP-1R and glucagon-receptor co-agonism are now ongoing (Tschöp et al., 2016), and translational efficacy is based on promising data from non-human primate studies (Tschöp et al., 2016).
Another thermogenic agent that has gained momentum as a gut-derived drug candidate is the fibroblast growth factor-21 (FGF21). FGF21 is predominantly derived from the liver to stimulate energy utilization (Xu et al., 2009), and pharmacological FGF21 is currently being evaluated in clinical trials for treating obesity and diabetes (Kharitonenkov and DiMarchi, 2015). Relevantly, glucagon is a powerful FGF21 secretagogue, and the pre-clinical success of GLP-1/glucagon co-agonism may partially be based on the involvement of a glucagon-FGF21 axis (Habegger et al., 2013). Albeit causality is uncharted, both glucagon and FGF-21 are increased following bariatric surgery (Jacobsen et al., 2012; Jansen et al., 2011). Interestingly, recent data demonstrate immune-metabolic dependency between GLP-1 and FGF21 (Lynch et al., 2016), and the metabolic efficacy of pharmacologically inhibiting FGF21 degradation is blunted in GLP-1R-deficient mice (Sánchez-Garrido et al., 2016), emphasizing the hormonal interplay between GLP-1 and FGF21.
While many obese patients might benefit from glucagon/GLP1 dual agonism, patients with severe comorbid type 2 diabetes might profit more from the combination of GLP-1 with the incretin GIP. Whereas GLP-1 pharmacology offers to offset undesirable effects of glucagon on glycemic control, combining GLP-1 with companion GIP delivers superior glycemic control relative to mono-incretin treatment (Finan et al., 2016b). The first so-called “twincretin” molecule, an intermixed peptide GLP-1/GIP hybrid with balanced potency at each cognate receptor, was generated in 2013 and found to have superior anti-diabetic efficacy and, perhaps unexpectedly, also to achieve superior body weight loss in obesity, relative to GLP-1R mono-agonism (Finan et al., 2013). This triggered a broad series of follow-up studies that are now re-characterizing previously underappreciated GIP as an important gut hormone offering metabolic benefits beyond the action profile of GLP-1 (Finan et al., 2016b). Noteworthy, the glycemic benefits of GLP-1/GIP dual activation translate into primates including humans (Finan et al., 2013). Unraveling the exact mechanistic underpinnings of how GIP amplifies the anorectic efficacy of GLP-1, and vice versa, is an imperative task that will provide relevant novel insight to be exploited pharmacologically.
The enhanced performance of GLP-1/glucagon and GLP-1/GIP co-agonism to reverse obesity and diabetes, respectively, and the structural similarity of these members of the same peptide family, facilitated the engineering of the first single-molecule triagonist (Finan et al., 2015b). In rodent models of obesity and diabetes, the first unimolecular GcgR/GLP-1R/GIPR triple-agonist produced unprecedented reversal of metabolic complications. In fact, the efficacy of the tri-agonist to correct obesity and diabetes rivals what can be achieved with bariatric surgery. Clinical studies are now evaluating if this pre-clinical success, which includes validation in primates, can be replicated in obese diabetic patients (Tschöp et al., 2016).
Studies looking at other possible polypharmacological strategies implicating GLP-1 are currently being explored. Co-administration GLP-1 and gastrin, and a GLP-1-gastrin dual agonist, appears to offer promising effects to prevent progressive deterioration of beta cell functional mass in rodent models of diabetes (Fosgerau et al., 2013; Suarez-Pinzon et al., 2008; Tamaki et al., 2010). A heightened understanding of the involvement of gut microbiota in metabolic diseases has revived interest in exploiting the gut barrier-protecting effects of GLP-2. However, the first pre-clinical studies with mixed GLP-1/GLP-2 co-agonists have revealed limited efficacy to improve metabolic parameters (Finan et al., 2015a). Nonetheless, it is paramount to continue to explore different combinations of gut-hormone mixtures for ameliorating metabolic diseases. History has taught this research community not to discard endocrine factors based on their (lack of) efficacy as single agents. It is indisputable that the prandial endocrine gut response has evolved to work in a coordinated, concerted fashion, and parsing this in further detail may continue to unlock superior blueprints for pharmacological mimicry.
Mimicking Bariatric Surgery-Induced Bile Acid Adjustments with Pharmacology
As discussed above, the discovery that bile acids are substantially altered following both RYGB (Patti et al., 2009) and VSG (Myronovych et al., 2014) has positioned bile acids and bile-acid transducers as critical instruments coordinating the enteroendocrine orchestra governing energy homeostasis. In particular, the bile acid receptors FXR and TGR5 continue to be pursued as potential drug targets that might offer ways to mimic the metabolic benefits of bariatric surgery (Penney et al., 2015). Consistent with that approach, the relatively gut-selective dual TGR5 and FXR agonist fexaramine substantially increases energy expenditure and reverses the metabolic syndrome in obese mice (Fang et al., 2015). Conversely, however, the small molecule Gly-MCA, that inhibits intestinal FXR signaling, reverses diet-induced and genetic obesity in mice, leaving the optimal strategy to target gut FXR unsettled (Jiang et al., 2015). The bile acid chenodeoxycholic acid (CDCA) increases brown adipose tissue (BAT) activity and whole-body energy expenditure in humans (Broeders et al., 2015). Mechanistic analysis points to BAT-TGR5 signaling as the mediating entity, which might in turn influence other metabolic pathways including thyroid hormone action (Watanabe et al., 2006), thereby emphasizing the widespread potential of targeting bile acid receptors pharmacologically.
Of note, ileal-FXR activation leads to increased production and release of the gut hormone FGF19 (mouse ortholog FGF15). FGF19 is increased following bariatric surgery (Gerhard et al., 2013; Jansen et al., 2011) and acts as a negative feedback signal to suppress hepatic bile acid synthesis (Holt et al., 2003; Inagaki et al., 2005). In addition, via activating hypothalamic FGFR1, FGF19 constitutes a key gut-brain axis signal that has been reported to contribute to the regulation of energy and glucose homeostasis (Morton et al., 2013; Ryan et al., 2013). Therefore, an increasing rationale for incorporating selective targeting of bile acid metabolism-related receptors such as FGFRs, FXR, and/or TGR5 in next-generation metabolic pharmacology is emerging. To mention one more example, activation of the FGF1R-βKlotho complex, together with GLP-1-based multi-agonists, appears to provide metabolic benefits that approximate the post-surgical endocrine milieu.
Gut-Hormone Potentiation of the Brain’s Melanocortin System
Despite the fact that leptin treatment has a negligible effect on appetite and body weight in diet-induced obesity, growing evidence supports the concept that specific enteroendocrine hormones may offer potential to act as central leptin sensitizers. Accordingly, gut-hormone-based pharmacotherapies are increasingly being evaluated for their ability to restore or enhance central leptin actions (Quarta et al., 2016). Obese mice shifted from a high-fat, high sugar diet to a low-fat chow diet and treated with a GLP-1R agonist (Exendin-4) have restored exogenous leptin responsiveness (Müller et al., 2012). Amylin, which is co-secreted from beta cells along with insulin, has also been established as a powerful leptin sensitizer (Trevaskis et al., 2010), and reports of central interactions between leptin and other gut-hormones including PYY and CCK are indicative of combinatorial potential (Unniappan and Kieffer, 2008; Wang et al., 2000). Recently, GLP-1R/GcgR co-agonism was found to elicit unprecedented sensitization of leptin actions (Clemmensen et al., 2014). Four weeks of combined GLP-1/glucagon and leptin treatment resulted in a 45% reduction in body weight in obese mice with free access to a hyper-palatable and energy-dense diet. Such weight loss potency is equivalent to what can be achieved with bariatric surgery and only rivaled by a few other pre-clinical agents (Finan et al., 2015b; Liu et al., 2015).
Considerable effort has been invested in therapeutic modulation of the second-order neuronal integrator of hypothalamic leptin action, MC4R. Unfortunately, most attempts have failed due to safety issues, particularly a negative impact on the cardiovascular system (Fani et al., 2014). Encouragingly, however, a recently developed selective MC4R agonist (setmelanotide) was reported to reverse obesity in monkeys and humans without compromising cardiovascular safety (Chen et al., 2015; Kievit et al., 2013). This same compound was recently employed to successfully treat human POMC deficiency (Kühnen et al., 2016). Finally, corroborating the emerging paradigm that co-targeting anorectic and thermogenic pathways is a superior medicinal strategy, dual MC4R and GLP-1R agonism administered to HFD-induced obese mice caused a greater improvement in reversing obesity and dyslipidemia as well as restoring glycemic control relative to each mono-agonist (Clemmensen et al., 2015). Yet, contrasting the obvious benefits of combining drugs that target diverse non-redundant metabolic pathways, the ability of gut-factors to potentiate melanocortin-linked satiation or centrally-regulated thermogenic circuits, remains comparatively understudied.
Pharmacological Targeting of Hedonic Feeding and Food Reward Circuits
The potential of targeting behavioral feeding circuitries is evident by the capacity of selective cannabinoid receptor type-1 (CB1) inhibition to ameliorate rodent (Ravinet Trillou et al., 2004) and human (Pi-Sunyer et al., 2006; Ravinet Trillou et al., 2004) obesity. Numerous previous and current anti-obesity drugs directly act on neural pathways that influence eating behavior (Gautron et al., 2015). Accordingly, targeting of food hedonics in combination with the archetypical anorectic and thermogenic pathways is likely to yield hitherto unparalleled pharmacological efficacy to reverse human obesity and to maintain a leaner phenotype. However, indirect impact, via modulating endogenous gut-brain signals, on behavioral neuro-circuits likely offers superior specificity with lower risk for side effects and toxicity.
Several gut-hormones including GLP-1, PYY, and ghrelin directly affect mesolimbic dopaminergic tone and modulate food reward (Abizaid et al., 2006; Batterham et al., 2007; Dossat et al., 2011; Fulton et al., 2006). Yet, it remains to be determined if, for example, the therapeutic benefits of GLP-1R agonism are in part attributable to effects on behavioral circuits. Indeed, there is an unmet demand for exploring how gut-derived peptides influence relevant neuronal intervention points beyond the caudal brainstem and the hypothalamus to modulate energy homeostasis. Such insights may facilitate the design of refined polypharmacy that relevantly targets food hedonics without severe off-target effects that are currently frequently linked to behavioral pharmacology.
Therapeutic Challenges and Opportunities: A Look to the Future
We are currently witnessing important progress in transforming basic research into novel therapeutics for obesity and diabetes. However, if granted spotlight exposure, several less traveled avenues might provide additional opportunities (Figure 4). Perhaps surprisingly, the most challenging obstacle in obesity treatment may not be losing excess adipose mass, but rather maintaining a lower body weight once it has been re-established. Thus, weight loss and weight loss maintenance may be un-coupled biological challenges and require different pharmacology. For example, calorie-restriction-induced weight loss disproportionally reduces metabolic rate, which contributes to the powerful counter-regulatory “homeostatic” mechanisms driving weight regain (Bray, 1969; Leibel et al., 1995). Probing innovative strategies that efficaciously dampen CNS-perceived “under-weight” seems critical to maintain a healthy level of body fat in spite of a history of obesity.
Figure 4. Understanding Gut-Brain Cross-Talk: Therapeutic Targets for Treating Metabolic Disease.
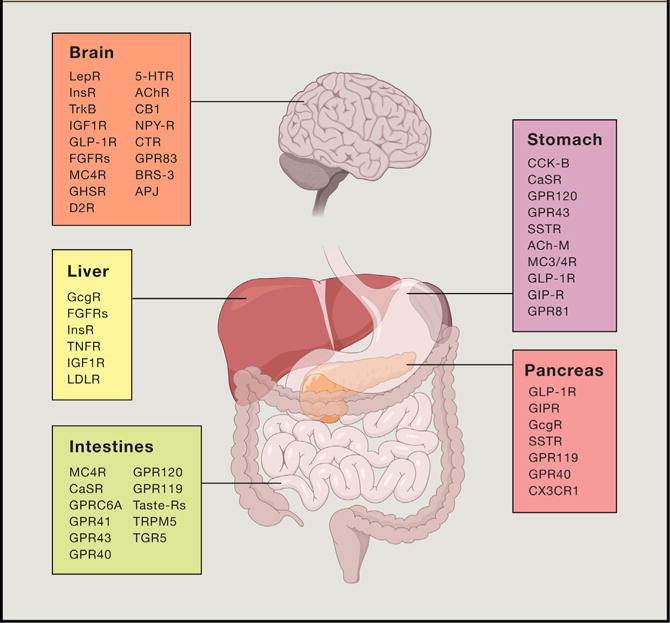
Current and promising therapeutic targets for treating obesity, type 2 diabetes, and associated co-morbidities are summarized. Future efforts hope to identify refined polypharmacy approaches intended to match the efficacy of the bariatric surgeries to cure obesity and diabetes. LepR, leptin receptor; InsR, insulin receptor; TrkB, tyrosine receptor kinase B; IGF1R, insulin-like growth factor 1 receptor; GLP-1R, glucagon-like peptide-1 receptor; FGFRs, fibroblast growth factor receptors; MC4R, melanocortin receptor 4; GHSR, growth hormone secretagogue receptor; D2R, dopamine receptor D2; 5-HTR, serotonin receptors; AChR, acetylcholine receptors; CB1, cannabinoid receptor type 1; NPY-R, neuropeptide Y receptors; CTR, calcitonin receptor; BRS-3, Bombesin receptor subtype 3; AJP, Apelin receptor; GcGR, glucagon receptor; TNFR, tumor necrosis factor receptors; LDLR, low-density lipoprotein receptor; CaSR, calcium sensing receptor; GPR, G protein-coupled receptor; TRPM5, transient receptor potential cation channel subfamily M member 5; CCK-B, cholecystokinin B receptor; SSTR, somatostatin receptor; ACh-M, muscarinic acetylcholine receptors; GIP-R, gastric inhibitory polypeptide receptor; CX3CR1, fractalkine receptor.
While the incorporation of glucagon in weight loss polypharmacy relevantly supplements a thermogenic component, other factors could be (re)-considered for successful pharmacological weight loss maintenance. Inarguably, thyroid hormone plays a central role in human energy metabolism. Thyroid hormone increases ATP turnover and reduces the thermodynamic efficiency of ATP synthesis resulting in heat production (Mullur et al., 2014). Reduced thyroid hormone levels are a hallmark metabolic adaptation to weight loss and may play a fundamental role for “pathological” weight regain (Rosenbaum et al., 2000). Yet, the use of thyroid hormones to treat obesity has, for decades, been largely discarded due to severe adverse effects on the cardiovascular system, bone, and skeletal muscle (Baxter and Webb, 2009). Recent technological advances may permit circumventing the off-target effects of thyroid hormone and enable safely harvesting the metabolic benefits on body weight and lipid metabolism (Tschöp et al., 2016). Thus, via using peptide hormones as shuttles, it is possible to selectively target nuclear hormones to tissues harboring the peptide-receptors (Finan et al., 2012). Recently, glucagon was employed to deliver thyroid hormone to hepatocytes and adipocytes. This pharmacological approach utilizes the metabolic benefits of both hormonal constituents to reverse diet-induced and genetic dyslipidemia, ramp up metabolic futile cellular cycles and consequently reverse obesity (Finan et al., 2016a). Importantly, using glucagon to restrict thyroid hormone actions circumvents adverse cardiovascular events, emphasizing the promising potential of this targetable pharmacological strategy.
The efficacy of bariatric surgery to introduce diabetes remission independent of weight loss has stipulated hope to cure diabetes with pharmacology. Now, baffling pre-clinical results are hinting that this is not science fiction and may be achievable in the not-so-distant future. Scarlett et al. (2016) found that a single central administration of FGF1 reverses diabetes in rodent models. Consistent with this, peripheral FGF1 administration delivers admirable effects on glycemic control in rodent models (Suh et al., 2014). Understanding the molecular details and the potential pharmacological utility of FGF1 in energy and glucose metabolism is pertinent and warrants further investigation.
Conclusions and Future Directions
Our species is on the verge of transcending biologically determined limits. Natural selection has provided humans with a disproportionally large playing field, and with the rise of sophisticated gene-editing techniques and other biotechnological advances, we are standing at a crossroads. Evident by the escalating obesity and diabetes epidemics as well as the multitude of associated co-morbidities, we have, from the perspective of optimal health, inarguably over-engineered our lives. Consequently, the demand for solutions is intensifying.
In a counterproductive manner, overweight and obese people are still frequently being accused of having self-inflicted complications, mirroring their (poor) choice of lifestyle. This is not the case. As with most major diseases, a complex interaction between environmental factors and genetic predisposition, rather than personal responsibility, is driving the disease etiologies. Whereas the genetic components of obesity point to CNS circuits as the major culprit of the pathogenesis, the gut has emerged as a significant therapeutic intervention point. In particular, the capacity of bariatric surgeries to effectively reverse obesity and diabetes has placed a spotlight on the gut-brain axis as a holistic, multifaceted, bidirectional control system governing energy metabolism.
In recent developments, new clinical guidelines for the treatment of type 2 diabetes recommend that bariatric surgery should be included as a standard treatment option for eligible candidates (Rubino et al., 2016). This radical amendment is following clinical trials consistently reporting that surgery improves glycemic control more effectively than lifestyle or pharmacological interventions—and in many cases produces long-term disease remission. However, risks and costs of surgery, and not yet fully understood long-term complications caused by the intervention, required specialized medical units and the necessity for trained surgeons and medical personnel needs to be added to the equation. The math just does not add up, and offering bariatric surgery to hundreds of millions of patients is not the solution to the growing epidemics of obesity and diabetes. Likely, there will be specific indication to perform surgery on defined small subsets of patients as a part of metabolic-precision medicine in the future, but the majority of obese and diabetic subjects will have to be cured—or ideally prevented—by novel medicines.
Tremendous progress in delineating the perpetual bimodal gut-brain cross talk is ongoing, and if the current momentum toward unraveling the molecular underpinnings of these signals can be maintained across academia, biotech companies, and the pharmaceutical industry, efficacious and safe precision medicines for obesity and type 2 diabetes may soon become a reality.
Acknowledgments
We would like to acknowledge and thank the numerous colleagues and collaborators with whom we have interacted over the years. C.C. received support from the Alfred Benzon Foundation and the Lundbeck Foundation. T.D.M. and M.H.T. received support from Helmholtz Alliance ICEMED, the Helmholtz Initiative on Personalized Medicine iMed, and the Helmholtz cross-program topic “Metabolic Dysfunction”. M.H.T. received support from the Alexander von Humboldt Foundation, the German Research Foundation DFG (SFB1123) and the European Research Council ERC (AdG HypoFlam no. 695054). H.R.B. was supported by NIH grant R01 DK047348. R.J.S. was supported by NIH grant R01 DK107652 and is a paid consultant for Ethicon Surgical Care, Novo Nordisk, Novartis, Daiichi Sankyo, Janssen/Johnson & Johnson, Zafgen, Paul Hastings Law Firm, Takeda, Boehringer-Ingelheim, and Sanofi.
Footnotes
AUTHOR CONTRIBUTIONS
C.C. conceptualized the review, generated substantial text, and provided edits for all sections. T.D.M generated substantial text, and provided edits for all sections. S.C.W. generated substantial text, and provided edits for all sections. H.R.B. generated substantial text, and provided edits for all sections. R.J.S. generated substantial text, and provided edits for all sections. M.H.T co-conceptualized the review, generated substantial text, and provided edits for all sections.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello L, Wheeler P. The expensive-tissue hypothesis: the brain and the digestive system in human and primate evolution. Curr Anthropol. 1995;36:199–221. [Google Scholar]
- Barrera JG, Sandoval DA, D’Alessio DA, Seeley RJ. GLP-1 and energy balance: an integrated model of short-term and long-term control. Nat Rev Endocrinol. 2011;7:507–516. doi: 10.1038/nrendo.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterham RL, ffytche DH, Rosenthal JM, Zelaya FO, Barker GJ, Withers DJ, Williams SC. PYY modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans. Nature. 2007;450:106–109. doi: 10.1038/nature06212. [DOI] [PubMed] [Google Scholar]
- Baxter JD, Webb P. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug Discov. 2009;8:308–320. doi: 10.1038/nrd2830. [DOI] [PubMed] [Google Scholar]
- Berthoud HR. Vagal and hormonal gut-brain communication: from satiation to satisfaction. Neurogastroenterol Motil. 2008a;20(Suppl 1):64–72. doi: 10.1111/j.1365-2982.2008.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR. The vagus nerve, food intake and obesity. Regul Pept. 2008b;149:15–25. doi: 10.1016/j.regpep.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR, Morrison C. The brain, appetite, and obesity. Annu Rev Psychol. 2008;59:55–92. doi: 10.1146/annurev.psych.59.103006.093551. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci. 2000;85:1–17. doi: 10.1016/S1566-0702(00)00215-0. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Patterson LM. Anatomical relationship between vagal afferent fibers and CCK-immunoreactive entero-endocrine cells in the rat small intestinal mucosa. Acta Anat (Basel) 1996;156:123–131. doi: 10.1159/000147837. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Powley TL. Vagal afferent innervation of the rat fundic stomach: morphological characterization of the gastric tension receptor. J Comp Neurol. 1992;319:261–276. doi: 10.1002/cne.903190206. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Kressel M, Raybould HE, Neuhuber WL. Vagal sensors in the rat duodenal mucosa: distribution and structure as revealed by in vivo DiI-tracing. Anat Embryol (Berl) 1995;191:203–212. doi: 10.1007/BF00187819. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Patterson LM, Neumann F, Neuhuber WL. Distribution and structure of vagal afferent intraganglionic laminar endings (IGLEs) in the rat gastrointestinal tract. Anat Embryol (Berl) 1997;195:183–191. doi: 10.1007/s004290050037. [DOI] [PubMed] [Google Scholar]
- Blackshaw LA, Grundy D. Effects of cholecystokinin (CCK-8) on two classes of gastroduodenal vagal afferent fibre. J Auton Nerv Syst. 1990;31:191–201. doi: 10.1016/0165-1838(90)90185-l. [DOI] [PubMed] [Google Scholar]
- Branco T, Tozer A, Magnus CJ, Sugino K, Tanaka S, Lee AK, Wood JN, Sternson SM. Near-Perfect Synaptic Integration by Nav1.7 in Hypothalamic Neurons Regulates Body Weight. Cell. 2016;165:1749–1761. doi: 10.1016/j.cell.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsma E, Houben T, Fu J, Shiri-Sverdlov R, Hofker MH. The immunity-diet-microbiota axis in the development of metabolic syndrome. Curr Opin Lipidol. 2015;26:73–81. doi: 10.1097/MOL.0000000000000154. [DOI] [PubMed] [Google Scholar]
- Bray GA. Effect of caloric restriction on energy expenditure in obese patients. Lancet. 1969;2:397–398. doi: 10.1016/s0140-6736(69)90109-3. [DOI] [PubMed] [Google Scholar]
- Brennan IM, Feltrin KL, Horowitz M, Smout AJ, Meyer JH, Wishart J, Feinle-Bisset C. Evaluation of interactions between CCK and GLP-1 in their effects on appetite, energy intake, and antropyloroduodenal motility in healthy men. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1477–R1485. doi: 10.1152/ajpregu.00732.2004. [DOI] [PubMed] [Google Scholar]
- Brobeck JR. Mechanism of the development of obesity in animals with hypothalamic lesions. Physiol Rev. 1946;26:541–559. doi: 10.1152/physrev.1946.26.4.541. [DOI] [PubMed] [Google Scholar]
- Broeders EP, Nascimento EB, Havekes B, Brans B, Roumans KH, Tailleux A, Schaart G, Kouach M, Charton J, Deprez B, et al. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab. 2015;22:418–426. doi: 10.1016/j.cmet.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Cabanac M, Johnson KG. Analysis of a conflict between palatability and cold exposure in rats. Physiol Behav. 1983;31:249–253. doi: 10.1016/0031-9384(83)90128-2. [DOI] [PubMed] [Google Scholar]
- Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des. 2009;15:1546–1558. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]
- Cegla J, Troke RC, Jones B, Tharakan G, Kenkre J, McCullough KA, Lim CT, Parvizi N, Hussein M, Chambers ES, et al. Coinfusion of low-dose GLP-1 and glucagon in man results in a reduction in food intake. Diabetes. 2014;63:3711–3720. doi: 10.2337/db14-0242. [DOI] [PubMed] [Google Scholar]
- Celio AC, Pories WJ. A History of Bariatric Surgery: The Maturation of a Medical Discipline. Surg Clin North Am. 2016;96:655–667. doi: 10.1016/j.suc.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Chakravarthy MV, Booth FW. Eating, exercise, and “thrifty” genotypes: connecting the dots toward an evolutionary understanding of modern chronic diseases. J Appl Physiol. 2004;96:3–10. doi: 10.1152/japplphysiol.00757.2003. [DOI] [PubMed] [Google Scholar]
- Chambers AP, Jessen L, Ryan KK, Sisley S, Wilson-Pérez HE, Stefater MA, Gaitonde SG, Sorrell JE, Toure M, Berger J, et al. Weight-independent changes in blood glucose homeostasis after gastric bypass or vertical sleeve gastrectomy in rats. Gastroenterology. 2011;141:950–958. doi: 10.1053/j.gastro.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AP, Kirchner H, Wilson-Perez HE, Willency JA, Hale JE, Gaylinn BD, Thorner MO, Pfluger PT, Gutierrez JA, Tschop MH, et al. The effects of vertical sleeve gastrectomy in rodents are ghrelin independent. Gastroenterology. 2013;144:50–52. doi: 10.1053/j.gastro.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JL, Mun EC, Stoyneva V, Mantzoros CS, Goldfine AB. Peptide YY levels are elevated after gastric bypass surgery. Obesity (Silver Spring) 2006;14:194–198. doi: 10.1038/oby.2006.25. [DOI] [PubMed] [Google Scholar]
- Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, et al. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015;100:1639–1645. doi: 10.1210/jc.2014-4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmensen C, Chabenne J, Finan B, Sullivan L, Fischer K, Küchler D, Sehrer L, Ograjsek T, Hofmann SM, Schriever SC, et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes. 2014;63:1422–1427. doi: 10.2337/db13-1609. [DOI] [PubMed] [Google Scholar]
- Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, et al. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015;7:288–298. doi: 10.15252/emmm.201404508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmensen C, Müller TD, Finan B, Tschöp MH, DiMarchi R. Current and Emerging Treatment Options in Diabetes Care. Handb Exp Pharmacol. 2016;233:437–459. doi: 10.1007/164_2015_7. [DOI] [PubMed] [Google Scholar]
- Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9:294–298. doi: 10.1007/BF01221857. [DOI] [PubMed] [Google Scholar]
- Coleman DL, Hummel KP. Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol. 1969;217:1298–1304. doi: 10.1152/ajplegacy.1969.217.5.1298. [DOI] [PubMed] [Google Scholar]
- Colquitt JL, Pickett K, Loveman E, Frampton GK. Surgery for weight loss in adults. Cochrane Database Syst Rev. 2014;(8):CD003641. doi: 10.1002/14651858.CD003641.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DE, Weigle DS, Frayo RS, Breen PA, Ma MK, Dellinger EP, Purnell JQ. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. 2002;346:1623–1630. doi: 10.1056/NEJMoa012908. [DOI] [PubMed] [Google Scholar]
- Cynthia K. Musonius Rufus (Lectures and Sayings. CreateSpace Independent Publishing Platform) 2010 [Google Scholar]
- D’Agostino G, Lyons DJ, Cristiano C, Burke LK, Madara JC, Campbell JN, Garcia AP, Land BB, Lowell BB, Dileone RJ, Heisler LK. Appetite controlled by a cholecystokinin nucleus of the solitary tract to hypothalamus neurocircuit. eLife. 2016;5:e12225. doi: 10.7554/eLife.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, Findeisen H, Bruemmer D, Drucker DJ, Chaudhary N, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- de Jonge L, Bray GA. The thermic effect of food and obesity: a critical review. Obes Res. 1997;5:622–631. doi: 10.1002/j.1550-8528.1997.tb00584.x. [DOI] [PubMed] [Google Scholar]
- de Lartigue G, Ronveaux CC, Raybould HE. Vagal plasticity the key to obesity. Mol Metab. 2014;3:855–856. doi: 10.1016/j.molmet.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva A, Salem V, Long CJ, Makwana A, Newbould RD, Rabiner EA, Ghatei MA, Bloom SR, Matthews PM, Beaver JD, Dhillo WS. The gut hormones PYY 3-36 and GLP-1 7-36 amide reduce food intake and modulate brain activity in appetite centers in humans. Cell Metab. 2011;14:700–706. doi: 10.1016/j.cmet.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degirolamo C, Rainaldi S, Bovenga F, Murzilli S, Moschetta A. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep. 2014;7:12–18. doi: 10.1016/j.celrep.2014.02.032. [DOI] [PubMed] [Google Scholar]
- Di Angelantonio E, Bhupathiraju ShN, Wormser D, Gao P, Kaptoge S, Berrington de Gonzalez A, Cairns BJ, Huxley R, Jackson ChL, Joshy G, et al. Global BMI Mortality Collaboration Body-mass index and all-cause mortality: individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet. 2016;388:776–786. doi: 10.1016/S0140-6736(16)30175-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson SL, Shirazi RH, Hansson C, Bergquist F, Nissbrandt H, Skibicka KP. The glucagon-like peptide 1 (GLP-1) analogue, exendin-4, decreases the rewarding value of food: a new role for mesolimbic GLP-1 receptors. J Neurosci. 2012;32:4812–4820. doi: 10.1523/JNEUROSCI.6326-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen C, Jørgensen NB, Bojsen-Møller KN, Kielgast U, Jacobsen SH, Clausen TR, Worm D, Hartmann B, Rehfeld JF, Damgaard M, et al. Gut hormones, early dumping and resting energy expenditure in patients with good and poor weight loss response after Roux-en-Y gastric bypass. Int J Obes. 2013;37:1452–1459. doi: 10.1038/ijo.2013.15. [DOI] [PubMed] [Google Scholar]
- Dossat AM, Lilly N, Kay K, Williams DL. Glucagon-like peptide 1 receptors in nucleus accumbens affect food intake. J Neurosci. 2011;31:14453–14457. doi: 10.1523/JNEUROSCI.3262-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JP, Cowan RL, Volkow ND, Feurer ID, Li R, Williams DB, Kessler RM, Abumrad NN. Decreased dopamine type 2 receptor availability after bariatric surgery: preliminary findings. Brain Res. 2010;1350:123–130. doi: 10.1016/j.brainres.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst B, Thurnheer M, Wilms B, Schultes B. Differential changes in dietary habits after gastric bypass versus gastric banding operations. Obes Surg. 2009;19:274–280. doi: 10.1007/s11695-008-9769-3. [DOI] [PubMed] [Google Scholar]
- Falkén Y, Hellström PM, Holst JJ, Näslund E. Changes in glucose homeostasis after Roux-en-Y gastric bypass surgery for obesity at day three, two months, and one year after surgery: role of gut peptides. J Clin Endocrinol Metab. 2011;96:2227–2235. doi: 10.1210/jc.2010-2876. [DOI] [PubMed] [Google Scholar]
- Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21:159–165. doi: 10.1038/nm.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fani L, Bak S, Delhanty P, van Rossum EF, van den Akker EL. The melanocortin-4 receptor as target for obesity treatment: a systematic review of emerging pharmacological therapeutic options. Int J Obes. 2014;38:163–169. doi: 10.1038/ijo.2013.80. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat: Historical perspective. Brain Res. 2016;1645:68–70. doi: 10.1016/j.brainres.2015.12.041. [DOI] [PubMed] [Google Scholar]
- Finan B, Yang B, Ottaway N, Stemmer K, Müller TD, Yi CX, Habegger K, Schriever SC, García-Cáceres C, Kabra DG, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med. 2012;18:1847–1856. doi: 10.1038/nm.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan B, Ma T, Ottaway N, Müller TD, Habegger KM, Heppner KM, Kirchner H, Holland J, Hembree J, Raver C, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5:209ra151. doi: 10.1126/scitranslmed.3007218. [DOI] [PubMed] [Google Scholar]
- Finan B, Clemmensen C, Müller TD. Emerging opportunities for the treatment of metabolic diseases: Glucagon-like peptide-1 based multi-agonists. Mol Cell Endocrinol. 2015a;418:42–54. doi: 10.1016/j.mce.2015.07.003. [DOI] [PubMed] [Google Scholar]
- Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, Chabenne J, Zhang L, Habegger KM, Fischer K, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015b;21:27–36. doi: 10.1038/nm.3761. [DOI] [PubMed] [Google Scholar]
- Finan B, Clemmensen C, Zhu Z, Stemmer K, Gauthier K, Muller L, De Angelis M, Moreth K, Neff F, Perez-Tilve D, et al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell. 2016a;167:843–857. doi: 10.1016/j.cell.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Finan B, Müller TD, Clemmensen C, Perez-Tilve D, DiMarchi RD, Tschöp MH. Reappraisal of GIP pharmacology for metabolic diseases. Trends Mol Med. 2016b;22:359–376. doi: 10.1016/j.molmed.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Fosgerau K, Jessen L, Lind Tolborg J, Østerlund T, Schæffer Larsen K, Rolsted K, Brorson M, Jelsing J, Skovlund Ryge Neerup T. The novel GLP-1-gastrin dual agonist, ZP3022, increases β-cell mass and prevents diabetes in db/db mice. Diabetes Obes Metab. 2013;15:62–71. doi: 10.1111/j.1463-1326.2012.01676.x. [DOI] [PubMed] [Google Scholar]
- Frank S, Wilms B, Veit R, Ernst B, Thurnheer M, Kullmann S, Fritsche A, Birbaumer N, Preissl H, Schultes B. Altered brain activity in severely obese women may recover after Roux-en Y gastric bypass surgery. Int J Obes. 2014;38:341–348. doi: 10.1038/ijo.2013.60. [DOI] [PubMed] [Google Scholar]
- Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, Maratos-Flier E, Flier JS. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9:286–294. doi: 10.1038/nrgastro.2012.32. [DOI] [PubMed] [Google Scholar]
- García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell. 2016;166:867–880. doi: 10.1016/j.cell.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautron L, Elmquist JK, Williams KW. Neural control of energy balance: translating circuits to therapies. Cell. 2015;161:133–145. doi: 10.1016/j.cell.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard GS, Styer AM, Wood GC, Roesch SL, Petrick AT, Gabrielsen J, Strodel WE, Still CD, Argyropoulos G. A role for fibroblast growth factor 19 and bile acids in diabetes remission after Roux-en-Y gastric bypass. Diabetes Care. 2013;36:1859–1864. doi: 10.2337/dc12-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs J, Young RC, Smith GP. Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature. 1973;245:323–325. doi: 10.1038/245323a0. [DOI] [PubMed] [Google Scholar]
- Grayson BE, Schneider KM, Woods SC, Seeley RJ. Improved rodent maternal metabolism but reduced intrauterine growth after vertical sleeve gastrectomy. Sci Transl Med. 2013;5:199ra112. doi: 10.1126/scitranslmed.3006505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ED, Maffei M, Braden VV, Proenca R, DeSilva U, Zhang Y, Chua SC, Jr, Leibel RL, Weissenbach J, Friedman JM. The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res. 1995;5:5–12. doi: 10.1101/gr.5.1.5. [DOI] [PubMed] [Google Scholar]
- Gribble FM. The gut endocrine system as a coordinator of postprandial nutrient homoeostasis. Proc Nutr Soc. 2012;71:456–462. doi: 10.1017/S0029665112000705. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol. 2016;78:277–299. doi: 10.1146/annurev-physiol-021115-105439. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. 2012;16:296–309. doi: 10.1016/j.cmet.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunddal KV, Ratner CF, Svendsen B, Sommer F, Engelstoft MS, Madsen AN, Pedersen J, Nøhr MK, Egerod KL, Nawrocki AR, et al. Neurotensin is coexpressed, coreleased, and acts together with GLP-1 and PYY in enteroendocrine control of metabolism. Endocrinology. 2016;157:176–194. doi: 10.1210/en.2015-1600. [DOI] [PubMed] [Google Scholar]
- Gutzwiller JP, Degen L, Matzinger D, Prestin S, Beglinger C. Interaction between GLP-1 and CCK-33 in inhibiting food intake and appetite in men. Am J Physiol Regul Integr Comp Physiol. 2004;287:R562–R567. doi: 10.1152/ajpregu.00599.2003. [DOI] [PubMed] [Google Scholar]
- Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6:689–697. doi: 10.1038/nrendo.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habegger KM, Stemmer K, Cheng C, Müller TD, Heppner KM, Ottaway N, Holland J, Hembree JL, Smiley D, Gelfanov V, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes. 2013;62:1453–1463. doi: 10.2337/db12-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton MK, Boudry G, Lemay DG, Raybould HE. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am J Physiol Gastrointest Liver Physiol. 2015;308:G840–G851. doi: 10.1152/ajpgi.00029.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Tellez LA, Niu J, Medina S, Ferreira TL, Zhang X, Su J, Tong J, Schwartz GJ, van den Pol A, de Araujo IE. Striatal dopamine links gastrointestinal rerouting to altered sweet appetite. Cell Metab. 2016;23:103–112. doi: 10.1016/j.cmet.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler WL. Methods of gastric electrical stimulation and pacing: a review of their benefits and mechanisms of action in gastroparesis and obesity. Neurogastroenterol Motil. 2009;21:229–243. doi: 10.1111/j.1365-2982.2009.01277.x. [DOI] [PubMed] [Google Scholar]
- Henry FE, Sugino K, Tozer A, Branco T, Sternson SM. Cell type-specific transcriptomics of hypothalamic energy-sensing neuron responses to weight-loss. eLife. 2015;4 doi: 10.7554/eLife.09800. http://dx.doi.org/10.7554/eLife.09800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert H, Saper CB. Cholecystokinin-, galanin-, and corticotropin-releasing factor-like immunoreactive projections from the nucleus of the solitary tract to the parabrachial nucleus in the rat. J Comp Neurol. 1990;293:581–598. doi: 10.1002/cne.902930405. [DOI] [PubMed] [Google Scholar]
- Hervey GR. The effects of lesions in the hypothalamus in parabiotic rats. J Physiol. 1959;145:336–352. doi: 10.1113/jphysiol.1959.sp006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCamish M. Re-combinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Huypens P, Sass S, Wu M, Dyckhoff D, Tschöp M, Theis F, Marschall S, Hrabě de Angelis M, Beckers J. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat Genet. 2016;48:497–499. doi: 10.1038/ng.3527. [DOI] [PubMed] [Google Scholar]
- Ikramuddin S, Blackstone RP, Brancatisano A, Toouli J, Shah SN, Wolfe BM, Fujioka K, Maher JW, Swain J, Que FG, et al. Effect of reversible intermittent intra-abdominal vagal nerve blockade on morbid obesity: the ReCharge randomized clinical trial. JAMA. 2014;312:915–922. doi: 10.1001/jama.2014.10540. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Jacobsen SH, Olesen SC, Dirksen C, Jørgensen NB, Bojsen-Møller KN, Kielgast U, Worm D, Almdal T, Naver LS, Hvolris LE, et al. Changes in gastrointestinal hormone responses, insulin sensitivity, and beta-cell function within 2 weeks after gastric bypass in non-diabetic subjects. Obes Surg. 2012;22:1084–1096. doi: 10.1007/s11695-012-0621-4. [DOI] [PubMed] [Google Scholar]
- Jansen PL, van Werven J, Aarts E, Berends F, Janssen I, Stoker J, Schaap FG. Alterations of hormonally active fibroblast growth factors after Roux-en-Y gastric bypass surgery. Dig Dis. 2011;29:48–51. doi: 10.1159/000324128. [DOI] [PubMed] [Google Scholar]
- Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun. 2015;6:10166. doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- Kennedy GC. The role of depot fat in the hypothalamic control of food intake in the rat. Proc R Soc Lond B Biol Sci. 1953;140:578–596. doi: 10.1098/rspb.1953.0009. [DOI] [PubMed] [Google Scholar]
- Kennedy GC. Food intake, energy balance and growth. Br Med Bull. 1966;22:216–220. doi: 10.1093/oxfordjournals.bmb.a070476. [DOI] [PubMed] [Google Scholar]
- Kenny PJ. Reward mechanisms in obesity: new insights and future directions. Neuron. 2011;69:664–679. doi: 10.1016/j.neuron.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A, DiMarchi R. FGF21 Revolutions: Recent Advances Illuminating FGF21 Biology and Medicinal Properties. Trends Endocrinol Metab. 2015;26:608–617. doi: 10.1016/j.tem.2015.09.007. [DOI] [PubMed] [Google Scholar]
- Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013;62:490–497. doi: 10.2337/db12-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519:45–50. doi: 10.1038/nature14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R, Setchell KD, Kirby M, Myronovych A, Ryan KK, Ibrahim SH, Berger J, Smith K, Toure M, Woods SC, Seeley RJ. A surgical model in male obese rats uncovers protective effects of bile acids post-bariatric surgery. Endocrinology. 2013;154:2341–2351. doi: 10.1210/en.2012-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner J, Bessler M, Inabnet W, Taveras C, Holst JJ. Exaggerated glucagon-like peptide-1 and blunted glucose-dependent insulinotropic peptide secretion are associated with Roux-en-Y gastric bypass but not adjustable gastric banding. Surg Obes Relat Dis. 2007;3:597–601. doi: 10.1016/j.soard.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Lowell BB, Garfield AS. Melanocortin-4 receptor-regulated energy homeostasis. Nat Neurosci. 2016;19:206–219. doi: 10.1038/nn.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühnen P, Clément K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, Mai K, Blume-Peytavi U, Grüters A, Krude H. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. 2016;375:240–246. doi: 10.1056/NEJMoa1512693. [DOI] [PubMed] [Google Scholar]
- Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77:257–270. doi: 10.1016/s0306-4522(96)00434-4. [DOI] [PubMed] [Google Scholar]
- le Roux CW, Bueter M. The physiology of altered eating behaviour after Roux-en-Y gastric bypass. Exp Physiol. 2014;99:1128–1132. doi: 10.1113/expphysiol.2014.078378. [DOI] [PubMed] [Google Scholar]
- Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. 1995;332:621–628. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- Leonard WR, Snodgrass JJ, Robertson ML. Effects of brain evolution on human nutrition and metabolism. Annu Rev Nutr. 2007;27:311–327. doi: 10.1146/annurev.nutr.27.061406.093659. [DOI] [PubMed] [Google Scholar]
- Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas F, Sclafani A. Flavor preferences conditioned by intragastric fat infusions in rats. Physiol Behav. 1989;46:403–412. doi: 10.1016/0031-9384(89)90011-5. [DOI] [PubMed] [Google Scholar]
- Lynch L, Hogan AE, Duquette D, Lester C, Banks A, LeClair K, Cohen DE, Ghosh A, Lu B, Corrigan M, et al. iNKT cells induce FGF21 for thermogenesis and are required for maximal weight loss in GLP1 therapy. Cell Metab. 2016;24:510–519. doi: 10.1016/j.cmet.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JL. Handbook of Physiology: The Gastrointestinal System, Intestinal Absorption and Secretion. Wiley-Blackwell; 2011. Functional morphology of epithelium of the small intestine. [Google Scholar]
- Madsbad S, Dirksen C, Holst JJ. Mechanisms of changes in glucose metabolism and bodyweight after bariatric surgery. Lancet Diabetes Endocrinol. 2014;2:152–164. doi: 10.1016/S2213-8587(13)70218-3. [DOI] [PubMed] [Google Scholar]
- Maffei M, Fei H, Lee GH, Dani C, Leroy P, Zhang Y, Proenca R, Negrel R, Ailhaud G, Friedman JM. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc Natl Acad Sci USA. 1995;92:6957–6960. doi: 10.1073/pnas.92.15.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makaronidis JM, Batterham RL. Potential mechanisms mediating sustained weight loss following Roux-en-Y gastric bypass and sleeve gastrectomy. Endocrinol Metab Clin North Am. 2016;45:539–552. doi: 10.1016/j.ecl.2016.04.006. [DOI] [PubMed] [Google Scholar]
- McGavigan AK, Garibay D, Henseler ZM, Chen J, Bettaieb A, Haj FG, Ley RE, Chouinard ML, Cummings BP. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut. 2017;66:226–234. doi: 10.1136/gutjnl-2015-309871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen KH, Frost M, Bahl MI, Licht TR, Jensen US, Rosenberg J, Pedersen O, Hansen T, Rehfeld JF, Holst JJ, et al. Effect of antibiotics on gut microbiota, gut hormones and glucose metabolism. PLoS ONE. 2015;10:e0142352. doi: 10.1371/journal.pone.0142352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Nanni G, Castagneto M, Bornstein S, Rubino F. Bariatric-metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomised controlled trial. Lancet. 2015;386:964–973. doi: 10.1016/S0140-6736(15)00075-6. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Matsen ME, Bracy DP, Meek TH, Nguyen HT, Stefanovski D, Bergman RN, Wasserman DH, Schwartz MW. FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest. 2013;123:4799–4808. doi: 10.1172/JCI70710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller TD, Sullivan LM, Habegger K, Yi CX, Kabra D, Grant E, Ottaway N, Krishna R, Holland J, Hembree J, et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. J Pept Sci. 2012;18:383–393. doi: 10.1002/psc.2408. [DOI] [PubMed] [Google Scholar]
- Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355–382. doi: 10.1152/physrev.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myronovych A, Kirby M, Ryan KK, Zhang W, Jha P, Setchell KD, Dexheimer PJ, Aronow B, Seeley RJ, Kohli R. Vertical sleeve gastrectomy reduces hepatic steatosis while increasing serum bile acids in a weight-loss-independent manner. Obesity (Silver Spring) 2014;22:390–400. doi: 10.1002/oby.20548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näslund E, Backman L, Holst JJ, Theodorsson E, Hellström PM. Importance of small bowel peptides for the improved glucose metabolism 20 years after jejunoileal bypass for obesity. Obes Surg. 1998;8:253–260. doi: 10.1381/096089298765554449. [DOI] [PubMed] [Google Scholar]
- Neary NM, Small CJ, Druce MR, Park AJ, Ellis SM, Semjonous NM, Dakin CL, Filipsson K, Wang F, Kent AS, et al. Peptide YY3-36 and glucagon-like peptide-17-36 inhibit food intake additively. Endocrinology. 2005;146:5120–5127. doi: 10.1210/en.2005-0237. [DOI] [PubMed] [Google Scholar]
- Ochner CN, Laferrère B, Afifi L, Atalayer D, Geliebter A, Teixeira J. Neural responsivity to food cues in fasted and fed states pre and post gastric bypass surgery. Neurosci Res. 2012;74:138–143. doi: 10.1016/j.neures.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- Oswald KD, Murdaugh DL, King VL, Boggiano MM. Motivation for palatable food despite consequences in an animal model of binge eating. Int J Eat Disord. 2011;44:203–211. doi: 10.1002/eat.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, Badman MK, Maratos-Flier E, Mun EC, Pihlajamaki J, et al. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 2009;17:1671–1677. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney NC, Kinross J, Newton RC, Purkayastha S. The role of bile acids in reducing the metabolic complications of obesity after bariatric surgery: a systematic review. Int J Obes. 2015;39:1565–1574. doi: 10.1038/ijo.2015.115. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi-Sunyer FX, Aronne LJ, Heshmati HM, Devin J, Rosenstock J, RIO-North America Study Group Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: a randomized controlled trial. JAMA. 2006;295:761–775. doi: 10.1001/jama.295.7.761. [DOI] [PubMed] [Google Scholar]
- Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, Du X, Petrov A, Lassman ME, Jiang G, et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258–2266. doi: 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powley TL, Spaulding RA, Haglof SA. Vagal afferent innervation of the proximal gastrointestinal tract mucosa: chemoreceptor and mechanoreceptor architecture. J Comp Neurol. 2011;519:644–660. doi: 10.1002/cne.22541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarta C, Sánchez-Garrido MA, Tschöp MH, Clemmensen C. Renaissance of leptin for obesity therapy. Diabetologia. 2016;59:920–927. doi: 10.1007/s00125-016-3906-7. [DOI] [PubMed] [Google Scholar]
- Ramsay DS, Woods SC. Clarifying the roles of homeostasis and allostasis in physiological regulation. Psychol Rev. 2014;121:225–247. doi: 10.1037/a0035942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner C, Skov LJ, Raida Z, Bächler T, Bellmann-Sickert K, Le Foll C, Sivertsen B, Dalbøge LS, Hartmann B, Beck-Sickinger AG, et al. Effects of peripheral neurotensin on appetite regulation and its role in gastric bypass surgery. Endocrinology. 2016;157:3482–3492. doi: 10.1210/en.2016-1329. [DOI] [PubMed] [Google Scholar]
- Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrié P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord. 2004;28:640–648. doi: 10.1038/sj.ijo.0802583. [DOI] [PubMed] [Google Scholar]
- Rehfeld JF. The new biology of gastrointestinal hormones. Physiol Rev. 1998;78:1087–1108. doi: 10.1152/physrev.1998.78.4.1087. [DOI] [PubMed] [Google Scholar]
- Reijnders D, Goossens GH, Hermes GD, Neis EP, van der Beek CM, Most J, Holst JJ, Lenaerts K, Kootte RS, Nieuwdorp M, et al. Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: a randomized double-blind placebo-controlled trial. Cell Metab. 2016;24:341. doi: 10.1016/j.cmet.2016.07.008. [DOI] [PubMed] [Google Scholar]
- Reimann F, Tolhurst G, Gribble FM. G-protein-coupled receptors in intestinal chemosensation. Cell Metab. 2012;15:421–431. doi: 10.1016/j.cmet.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Rising R, Alger S, Boyce V, Seagle H, Ferraro R, Fontvieille AM, Ravussin E. Food intake measured by an automated food-selection system: relationship to energy expenditure. Am J Clin Nutr. 1992;55:343–349. doi: 10.1093/ajcn/55.2.343. [DOI] [PubMed] [Google Scholar]
- Romero F, Nicolau J, Flores L, Casamitjana R, Ibarzabal A, Lacy A, Vidal J. Comparable early changes in gastrointestinal hormones after sleeve gastrectomy and Roux-En-Y gastric bypass surgery for morbidly obese type 2 diabetic subjects. Surg Endosc. 2012;26:2231–2239. doi: 10.1007/s00464-012-2166-y. [DOI] [PubMed] [Google Scholar]
- Rosenbaum M, Hirsch J, Murphy E, Leibel RL. Effects of changes in body weight on carbohydrate metabolism, catecholamine excretion, and thyroid function. Am J Clin Nutr. 2000;71:1421–1432. doi: 10.1093/ajcn/71.6.1421. [DOI] [PubMed] [Google Scholar]
- Rubino F, Nathan DM, Eckel RH, Schauer PR, Alberti KG, Zimmet PZ, Del Prato S, Ji L, Sadikot SM, Herman WH, et al. Delegates of the 2nd Diabetes Surgery Summit Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Diabetes Care. 2016;39:861–877. doi: 10.2337/dc16-0236. [DOI] [PubMed] [Google Scholar]
- Ryan KK, Kohli R, Gutierrez-Aguilar R, Gaitonde SG, Woods SC, Seeley RJ. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology. 2013;154:9–15. doi: 10.1210/en.2012-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Pérez HE, Sandoval DA, Kohli R, Bäckhed F, Seeley RJ. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509:183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi M, Prigeon RL, D’Alessio DA. Gastric bypass surgery enhances glucagon-like peptide 1-stimulated postprandial insulin secretion in humans. Diabetes. 2011;60:2308–2314. doi: 10.2337/db11-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Garrido MA, Habegger KM, Clemmensen C, Holleman C, Müller TD, Perez-Tilve D, Li P, Agrawal AS, Finan B, Drucker DJ, et al. Fibroblast activation protein (FAP) as a novel metabolic target. Mol Metab. 2016;5:1015–1024. doi: 10.1016/j.molmet.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Scarlett JM, Rojas JM, Matsen ME, Kaiyala KJ, Stefanovski D, Bergman RN, Nguyen HT, Dorfman MD, Lantier L, Wasserman DH, et al. Central injection of fibroblast growth factor 1 induces sustained remission of diabetic hyperglycemia in rodents. Nat Med. 2016;22:800–806. doi: 10.1038/nm.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Brethauer SA, Navaneethan SD, Aminian A, Pothier CE, Kim ES, Nissen SE, Kashyap SR, STAMPEDE Investigators Bariatric surgery versus intensive medical therapy for diabetes–3-year outcomes. N Engl J Med. 2014;370:2002–2013. doi: 10.1056/NEJMoa1401329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, Turroni S, Biagi E, Peano C, Severgnini M, et al. Gut microbiome of the Hadza hunter-gatherers. Nat Commun. 2014;5:3654. doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtz S, Miras AD, Chhina N, Prechtl CG, Sleeth ML, Daud NM, Ismail NA, Durighel G, Ahmed AR, Olbers T, et al. Obese patients after gastric bypass surgery have lower brain-hedonic responses to food than after gastric banding. Gut. 2014;63:891–902. doi: 10.1136/gutjnl-2013-305008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultes B, Ernst B, Wilms B, Thurnheer M, Hallschmid M. Hedonic hunger is increased in severely obese patients and is reduced after gastric bypass surgery. Am J Clin Nutr. 2010;92:277–283. doi: 10.3945/ajcn.2009.29007. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Seeley RJ, Barsh GS, Baskin DG, Leibel RL. Is the energy homeostasis system inherently biased toward weight gain? Diabetes. 2003;52:232–238. doi: 10.2337/diabetes.52.2.232. [DOI] [PubMed] [Google Scholar]
- Sclafani A. Oral and postoral determinants of food reward. Physiol Behav. 2004;81:773–779. doi: 10.1016/j.physbeh.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Sclafani A. Gut-brain nutrient signaling. Appetition vs satiation Appetite. 2013;71:454–458. doi: 10.1016/j.appet.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclafani A, Ackroff K. Role of gut nutrient sensing in stimulating appetite and conditioning food preferences. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1119–R1133. doi: 10.1152/ajpregu.00038.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclafani A, Springer D. Dietary obesity in adult rats: similarities to hypothalamic and human obesity syndromes. Physiol Behav. 1976;17:461–471. doi: 10.1016/0031-9384(76)90109-8. [DOI] [PubMed] [Google Scholar]
- Sclafani A, Ackroff K, Schwartz GJ. Selective effects of vagal deafferentation and celiac-superior mesenteric ganglionectomy on the reinforcing and satiating action of intestinal nutrients. Physiol Behav. 2003;78:285–294. doi: 10.1016/s0031-9384(02)00968-x. [DOI] [PubMed] [Google Scholar]
- Sclafani A, Zukerman S, Ackroff K. GPR40 and GPR120 fatty acid sensors are critical for postoral but not oral mediation of fat preferences in the mouse. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1490–R1497. doi: 10.1152/ajpregu.00440.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclafani A, Touzani K, Ackroff K. Ghrelin signaling is not essential for sugar or fat conditioned flavor preferences in mice. Physiol Behav. 2015;149:14–22. doi: 10.1016/j.physbeh.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclafani A, Koepsell H, Ackroff K. SGLT1 sugar transporter/sensor is required for post-oral glucose appetition. Am J Physiol Regul Integr Comp Physiol. 2016;310:R631–R639. doi: 10.1152/ajpregu.00432.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley RJ, Chambers AP, Sandoval DA. The role of gut adaptation in the potent effects of multiple bariatric surgeries on obesity and diabetes. Cell Metab. 2015;21:369–378. doi: 10.1016/j.cmet.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin AC, Berthoud HR. Food reward functions as affected by obesity and bariatric surgery. Int J Obes. 2011;35(Suppl 3):S40–S44. doi: 10.1038/ijo.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin AC, Zheng H, Pistell PJ, Berthoud HR. Rouxen-Y gastric bypass surgery changes food reward in rats. Int J Obes. 2011;35:642–651. doi: 10.1038/ijo.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibicka KP, Hansson C, Egecioglu E, Dickson SL. Role of ghrelin in food reward: impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict Biol. 2012;17:95–107. doi: 10.1111/j.1369-1600.2010.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP. The direct and indirect controls of meal size. Neurosci Biobehav Rev. 1996;20:41–46. doi: 10.1016/0149-7634(95)00038-g. [DOI] [PubMed] [Google Scholar]
- Speakman JR. If body fatness is under physiological regulation, then how come we have an obesity epidemic? Physiology (Bethesda) 2014;29:88–98. doi: 10.1152/physiol.00053.2013. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Kelly L, Latcha KN, Schmidt SF, Yu X, Nectow AR, Sauer J, Dyke JP, Dordick JS, Friedman JM. Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature. 2016;531:647–650. doi: 10.1038/nature17183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns AT, Balakrishnan A, Radmanesh A, Ashley SW, Rhoads DB, Tavakkolizadeh A. Relative contributions of afferent vagal fibers to resistance to diet-induced obesity. Dig Dis Sci. 2012;57:1281–1290. doi: 10.1007/s10620-011-1968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steculorum SM, Paeger L, Bremser S, Evers N, Hinze Y, Idzko M, Kloppenburg P, Brüning JC. Hypothalamic UDP increases in obesity and promotes feeding via p2y6-dependent activation of AgRP neurons. Cell. 2015;162:1404–1417. doi: 10.1016/j.cell.2015.08.032. [DOI] [PubMed] [Google Scholar]
- Steele KE, Prokopowicz GP, Schweitzer MA, Magunsuon TH, Lidor AO, Kuwabawa H, Kumar A, Brasic J, Wong DF. Alterations of central dopamine receptors before and after gastric bypass surgery. Obes Surg. 2010;20:369–374. doi: 10.1007/s11695-009-0015-4. [DOI] [PubMed] [Google Scholar]
- Stefater MA, Perez-Tilve D, Chambers AP, Wilson-Perez HE, Sandoval DA, Berger J, Toure M, Tschop M, Woods SC, Seeley RJ. Sleeve gastrectomy induces loss of weight and fat mass in obese rats, but does not affect leptin sensitivity. Gastroenterology. 2010;138:2426–2436. doi: 10.1053/j.gastro.2010.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert RE, Peterli R, Keller S, Meyer-Gerspach AC, Drewe J, Peters T, Beglinger C. Bile acids and gut peptide secretion after bariatric surgery: a 1-year prospective randomized pilot trial. Obesity (Silver Spring) 2013;21:E660–E668. doi: 10.1002/oby.20522. [DOI] [PubMed] [Google Scholar]
- Sternson SM, Roth BL. Chemogenetic tools to interrogate brain functions. Annu Rev Neurosci. 2014;37:387–407. doi: 10.1146/annurev-neuro-071013-014048. [DOI] [PubMed] [Google Scholar]
- Suarez-Pinzon WL, Power RF, Yan Y, Wasserfall C, Atkinson M, Rabinovitch A. Combination therapy with glucagon-like peptide-1 and gastrin restores normoglycemia in diabetic NOD mice. Diabetes. 2008;57:3281–3288. doi: 10.2337/db08-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JM, Jonker JW, Ahmadian M, Goetz R, Lackey D, Osborn O, Huang Z, Liu W, Yoshihara E, van Dijk TH, et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature. 2014;513:436–439. doi: 10.1038/nature13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki M, Fujitani Y, Uchida T, Hirose T, Kawamori R, Watada H. Combination treatment of db/db mice with exendin-4 and gastrin preserves β-cell mass by stimulating β-cell growth and differentiation. J Diabetes Investig. 2010;1:172–183. doi: 10.1111/j.2040-1124.2010.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TM, Field BC, McCullough KA, Troke RC, Chambers ES, Salem V, Gonzalez Maffe J, Baynes KC, De Silva A, Viardot A, et al. Coadministration of glucagon-like peptide-1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes. 2013;62:1131–1138. doi: 10.2337/db12-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CL, Cooke EK, Leib DE, Lin YC, Daly GE, Zimmerman CA, Knight ZA. Warm-sensitive neurons that control body temperature. Cell. 2016;167:47–59. doi: 10.1016/j.cell.2016.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- Teixeira TF, Collado MC, Ferreira CL, Bressan J, Peluzio MdoC. Potential mechanisms for the emerging link between obesity and increased intestinal permeability. Nutr Res. 2012;32:637–647. doi: 10.1016/j.nutres.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Tellez LA, Han W, Zhang X, Ferreira TL, Perez IO, Shammah-Lagnado SJ, van den Pol AN, de Araujo IE. Separate circuitries encode the hedonic and nutritional values of sugar. Nat Neurosci. 2016;19:465–470. doi: 10.1038/nn.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichansky DS, Glatt AR, Madan AK, Harper J, Tokita K, Boughter JD. Decrease in sweet taste in rats after gastric bypass surgery. Surg Endosc. 2011;25:1176–1181. doi: 10.1007/s00464-010-1335-0. [DOI] [PubMed] [Google Scholar]
- Trevaskis JL, Parkes DG, Roth JD. Insights into amylin-leptin synergy. Trends Endocrinol Metab. 2010;21:473–479. doi: 10.1016/j.tem.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Tschöp MH, Finan B, Clemmensen C, Gelfanov V, Perez-Tilve D, Müller TD, DiMarchi RD. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 2016;24:51–62. doi: 10.1016/j.cmet.2016.06.021. [DOI] [PubMed] [Google Scholar]
- Udit S, Gautron L. Molecular anatomy of the gut-brain axis revealed with transgenic technologies: implications in metabolic research. Front Neurosci. 2013;7:134. doi: 10.3389/fnins.2013.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich J, Ernst B, Wilms B, Thurnheer M, Schultes B. Rouxen Y gastric bypass surgery reduces hedonic hunger and improves dietary habits in severely obese subjects. Obes Surg. 2013;23:50–55. doi: 10.1007/s11695-012-0754-5. [DOI] [PubMed] [Google Scholar]
- Unniappan S, Kieffer TJ. Leptin extends the anorectic effects of chronic PYY(3-36) administration in ad libitum-fed rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R51–R58. doi: 10.1152/ajpregu.00234.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdearcos M, Xu AW, Koliwad SK. Hypothalamic inflammation in the control of metabolic function. Annu Rev Physiol. 2015;77:131–160. doi: 10.1146/annurev-physiol-021014-071656. [DOI] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- Wang L, Barachina MD, Martínez V, Wei JY, Taché Y. Synergistic interaction between CCK and leptin to regulate food intake. Regul Pept. 2000;92:79–85. doi: 10.1016/s0167-0115(00)00153-1. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- Waterson MJ, Horvath TL. Neuronal regulation of energy homeostasis: beyond the hypothalamus and feeding. Cell Metab. 2015;22:962–970. doi: 10.1016/j.cmet.2015.09.026. [DOI] [PubMed] [Google Scholar]
- Wilson-Pérez HE, Chambers AP, Ryan KK, Li B, Sandoval DA, Stoffers D, Drucker DJ, Pérez-Tilve D, Seeley RJ. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like Peptide 1 receptor deficiency. Diabetes. 2013a;62:2380–2385. doi: 10.2337/db12-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Pérez HE, Chambers AP, Sandoval DA, Stefater MA, Woods SC, Benoit SC, Seeley RJ. The effect of vertical sleeve gastrectomy on food choice in rats. Int J Obes. 2013b;37:288–295. doi: 10.1038/ijo.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouterlood FG, Bloem B, Mansvelder HD, Luchicchi A, Deisseroth K. A fourth generation of neuroanatomical tracing techniques: exploiting the offspring of genetic engineering. J Neurosci Methods. 2014;235:331–348. doi: 10.1016/j.jneumeth.2014.07.021. [DOI] [PubMed] [Google Scholar]
- Wu Q, Clark MS, Palmiter RD. Deciphering a neuronal circuit that mediates appetite. Nature. 2012;483:594–597. doi: 10.1038/nature10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wang H, Du M, Zhu MJ. Maternal obesity induces gut inflammation and impairs gut epithelial barrier function in nonobese diabetic mice. J Nutr Biochem. 2014;25:758–764. doi: 10.1016/j.jnutbio.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- Zagorodnyuk VP, Chen BN, Brookes SJ. Intraganglionic laminar endings are mechano-transduction sites of vagal tension receptors in the guinea-pig stomach. J Physiol. 2001;534:255–268. doi: 10.1111/j.1469-7793.2001.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelissen PM, Stenlof K, Lean ME, Fogteloo J, Keulen ET, Wilding J, Finer N, Rössner S, Lawrence E, Fletcher C, McCamish M, Author Group Effect of three treatment schedules of recombinant methionyl human leptin on body weight in obese adults: a randomized, placebo-controlled trial. Diabetes Obes Metab. 2005;7:755–761. doi: 10.1111/j.1463-1326.2005.00468.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]