

Summary
Objective
Ketogenic parenteral nutrition (kPN) is indicated when enteral intake is temporarily limited or impossible, but evidence‐based prescriptions are lacking. Objective was to evaluate the efficacy and safety of kPN in children with epileptic encephalopathies using a new computer‐based algorithm for accurate component calculating.
Methods
Children with epilepsy receiving kPN were included. A computer‐based algorithm was established on the basis of guidelines of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN): fat intake not exceeding 4 g/kg/day, age‐adequate supply of protein, electrolytes, vitamins, and trace elements, but reduced carbohydrates. Primary outcome was successfully reaching relevant ketosis, defined as beta‐hydroxybutyrate plasma level of ≥ 2 mmol/L. Efficacy was defined as seizure reduction ≥50% in de novo kPN and maintenance of response in children already on a ketogenic diet (KD). Safety was assessed by adverse effects, laboratory findings, and the appropriateness of nutritional intake.
Results
Seventeen children (median 1.84 years) were studied, of which 76% (13/17) were already on an oral ketogenic diet. Indications for kPN were surgery, status epilepticus, vomiting, food refusal, and introduction of enteral feeding in neonates. The parenteral fat/nonfat ratio was mean 0.9 (±0.3; range 0.6–1.5). Relevant ketosis was reached in 10 children (median 2.9 mmol/L), but not in 7 (median = 1.4 mmol/L). In de novo kPN, significant response was observed in 50% (2/4); in patients previously responding to the KD (77%, 10/13), response was maintained. A significant correlation between the degree of ketosis and seizure reduction (correlation coefficient = 0.691; p = .002) was observed. Only mild and transient adverse events occurred during kPN.
Significance
KPN with fat intake of 3.5–4.0 g/kg/day was safe and effective. KPN was tailored according to guidelines and individual nutritional needs. In nearly half of the patients, ketosis was lower than during oral KD. Despite this, seizures remained controlled.
Keywords: Children, Computer‐based algorithm, Epileptic encephalopathies, Ketogenic diet, Parenteral nutrition
Key Points.
Fat intake of 3.5–4 g/kg/day used in parenteral ketogenic nutrition was effective and safe
A computer‐based algorithm based on the guidelines of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) helped tailor the ketogenic parenteral nutrition according to age and nutritional needs
Relevant clinical ketosis (≥2 mmol/L) was achieved/maintained in the majority of children
There was a significant correlation between the degree of ketosis and seizure reduction (correlation coefficient = 0.691; p = .002)
Only mild and transient adverse events were observed during the use of ketogenic parenteral nutrition up to 41 days
Introduction
The ketogenic diet (KD) is an established nonpharmacological treatment option for medically intractable childhood epilepsies. The KD is high in fat, adequate in protein, and low in carbohydrate intake.1, 2 Usually, the KD is initiated by enteral feeding with an established fat to nonfat ratio that shifts metabolism to use ketone bodies as an energy source. Ketogenic parenteral nutrition (kPN) is indicated when enteral intake of a KD is impaired and ketosis for seizure control must be maintained. Ketogenic PN is also needed when the KD must be initiated and an enteral application is temporarily impossible. Various medical conditions, such as severe acute illness with gastrointestinal affection, status epilepticus (SE), surgery, and food refusal, may impair the intake of enteral foods.3
The composition of PN and nutritional intake vary according to the age of the child. According to the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN),4 the standard for the highest parenteral intake of fat was determined as 3–4 g/kg (in the United States the cut‐off was set at 4 g/kg).5 In addition, PN must be tailored to both age and individual nutritional needs. During KD treatment, maintenance of adequate ketosis is the primary goal. To date, information on efficacy and safety of kPN during treatment with the KD is limited: so far, only 5 case reports and 1 retrospective study provided data on a variety of approaches in infants for initiation and maintenance of a kPN.5, 6, 7, 8, 9, 10
In the present study, we describe our experience using kPN in children with epilepsy.
Methods
Study design
This prospective observational study investigated the efficacy and safety of a kPN in pediatric epilepsy patients (age <18 years) using a computer‐based algorithm. Data were retrieved from a longitudinal observational electronic database that contains the complete medical records of all children and adolescents with epilepsy aged 18 years or younger who have been treated with the KD at the study center since 2008. The data consist of a retrospective data set on patients’ demographics and medical history and prospectively collected data on seizure and developmental outcomes as well as tolerability of the KD.
For the purpose of the present study, data from all patients who received kPN were extracted from this database. The study was approved by the ethics committee of the Medical University of Vienna (MUW) (No. 542/2007).
Patients and indications for kPN
KPN was used in children who were already treated with an enteral KD but were temporarily unable to absorb enteral feedings (severe acute illness with gastrointestinal affection, status epilepticus, food refusal, children who required complete bowel rest for surgery), or children with a de novo indication for the KD who were unable to tolerate enteral feeding (severe neonatal encephalopathy with epilepsy, status epilepticus).
Standardized feeding regimen and a computer‐based algorithm for calculating the ketogenic PN
The composition of the kPN was calculated individually for each child using a computer‐based algorithm based on ESPGHAN guidelines,4 with fat intake not exceeding 4 g/kg/day, age‐adequate intake of protein, electrolytes, vitamins, and trace elements, and low supply of carbohydrates.
The first step was to measure weight and length of the child and to determine the protein and liquid requirements appropriate for age (Fig. 1).
Figure 1.
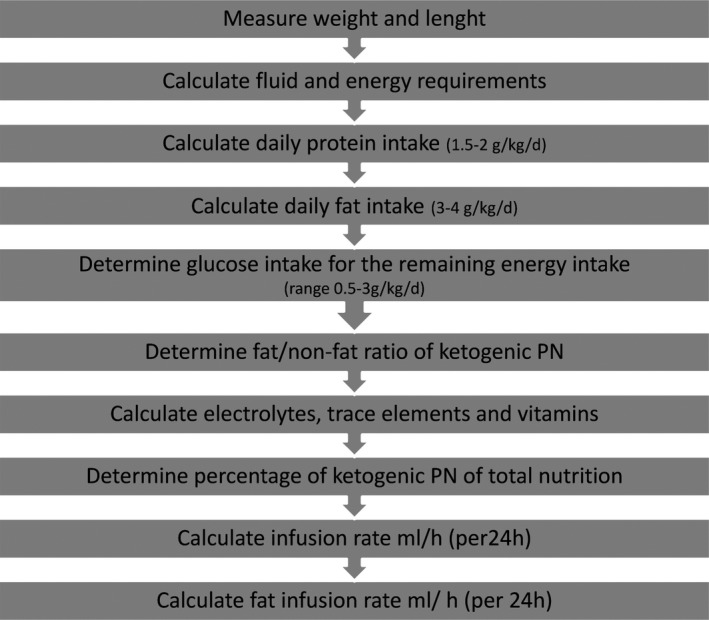
Computer‐based algorithm for calculating a ketogenic parenteral nutrition. The individual components were calculated according to the ESPGHAN guidelines.4
Next, fat intake was established between 3 and 4 g/kg/day. The remaining components (electrolytes, trace elements, and vitamins) were calculated according to age. The remaining calories were allocated to carbohydrates. Finally, the fat/nonfat ratio was determined. Glycerol was not calculated (see Figs. 1 and 2).
Figure 2.
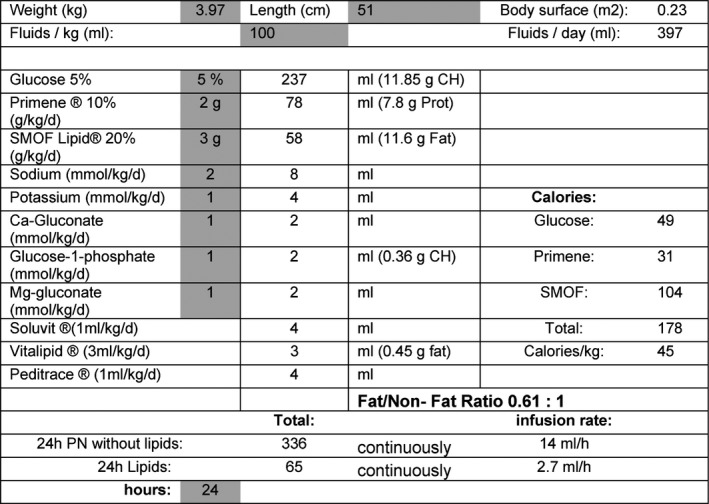
Example of the treatment algorithm of a ketogenic parenteral nutrition day in an 8‐week‐old infant with percutaneous endoscopic gastrostomy (PEG) implantation. Prior enteral KD consisted of a 3:1 fat/nonfat ratio. During ketogenic PN, only a maximum fat/nonfat ratio of 0.61:1 was achievable for this child and was the lowest fat/nonfat ratio of all children in this study. The child suffered from Ohtahara syndrome based on vanishing white matter disease (Case 17).
The kPN was mixed in a laminar air flow under sterile conditions and administered to the children via central or peripheral line.
The following products were used: Glucose solutions 50 and 100 mg/mL as appropriate (B. Braun 50 mg/mL and B. Braun 10%; B. Braun Melsungen, Germany), amino acids (Primene 10% pur; Baxter, Vienna, Austria), lipids (Clinoleic; Baxter, Vienna, Austria; Intralipid 20% and SMOF‐lipid 200 mg/mL; Fresenius Kabi, Graz, Austria), electrolytes (Sodium Chloride and Glucose‐1‐Phosphate; Fresenius Kabi, Graz, Austria; Potassium Chloride and Calcium‐Gluconicum; B. Braun Melsungen, Germany; Magnesium Gluconicum; G. L. Pharma, Austria), vitamins and trace elements (Peditrace, Soluvit, Vitalipid; Fresenius Kabi, Graz, Austria).
At initiation of the kPN, calories were calculated according to the ESPGHAN guidelines for fat and protein intake. 5%–10% intravenous glucose (median = 1.7 g/kg/day; interquartile range [IQR] 1.5–2.4 g/kg/day, range 0.5–3.8 g/kg/day) was administered for the remaining calories instead of calculating a higher fat intake per kilogram per day, as described by other authors.3, 5 With these fixed amounts for a fat intake between 3 and 4 g/kg/day and high protein intake, lower fat to nonfat ratios for kPN compared with an oral KD were be expected.
The target level for clinically relevant ketosis was defined as a beta‐hydroxybutyrate plasma level of ≥2 mmol/L.
For transition from kPN to enteral KD, ketogenic enteral feeds were gradually introduced with a fat/nonfat ratio of 1:1 and increased on an individual basis up to 3:1 or until beta‐hydroxybutyrate levels were >5 mmol/L.
Efficacy
When the kPN was used in children who had been already treated with an enteral KD, treatment response was defined as the maintenance of the previously achieved ≥50% reduction compared to baseline, defined as the 3 months before the initiation of enteral KD. In cases where kPN was the initial treatment, response was defined as the time to stop seizures in patients with SE and the absolute reduction in seizure frequency ≥50% after 3 months of KD treatment, including treatment with kPN and subsequent enteral KD as compared to baseline (defined as seizure frequency 1 month before the initiation of the treatment) in all other patients.
Safety
Lipid patterns as well as blood count and liver function parameters were assessed. Adverse effects were evaluated using an adverse effect questionnaire filled in by caregivers that (included questions on the presence (yes/no answers)) of constipation, nausea, vomiting, flatulence, abdominal pain, diarrhea, dehydration, high ketone bodies, compliance to food, supplementation of vitamins and calcium, feeding schedules, growth, and laboratory data.11
Follow‐up
Follow‐up assessments included the documentation of concomitant medications, seizure count according to seizure diaries, and the administration of adverse effects questionnaires as well as laboratory investigations and video‐sleep electroencephalograms (EEGs) when indicated.
During kPN, monitoring was on a daily basis. Follow‐up data were collected for the whole period of parenteral and enteral KD treatment, at least at 1 month after initiation of the diet and every 3 months thereafter. In‐between visits were scheduled according to patients’ needs.
Data analysis
Primary outcome was the level of ketosis as defined by beta‐hydroxybutyrate during kPN. We defined beta‐hydroxybutyrate levels of ≥2 mmol/L as clinically relevant ketosis.11
Secondary outcome parameters were the efficacy on seizure frequency (i.e., maintenance of ≥50% seizure reduction in patients who had been pretreated successfully with an enteral KD) and time to stop seizures in those with SE and de novo kPN and safety measured by both adverse effects and laboratory findings (GOT, GPT, GGT, LDH, triglyceride, total cholesterol, HDL, LDL, and VLDL).
Data analysis was performed using the IBM Statistical Package for Social Science (IBM Corp., Version 22.0. Armonk, NY, USA). Given the non‐normal distribution of data, nonparametric statistical data description was used (median, interquartile range, minimum and maximum values). For comparisons between groups, mean and confidence intervals were used where appropriate. We assessed the correlation between the degree of ketosis achieved (beta‐hydroxybutyrate plasma levels) during kPN and the percentage of seizure reduction using Pearson's correlation coefficient. Patients who achieved clinically relevant ketosis were compared with children who failed to reach this target with respect to seizure reduction using Fisher's exact test.
Results
Patient characteristics
From March 2013 until April 2016, 130 children were treated with the KD at the study center, and 13% (17/130) needed initial or intermittent kPN (Table 1).
Table 1.
Patient characteristics and outcome
| ID/Sex | Age (y) | On KD before | Duration ketogenic PN (d) | AED at trial start | Etiology | Epilepsy syndrome | Indication for PN | Ketosis >2 mmol/L | Long‐term outcome response at 3 mo |
|---|---|---|---|---|---|---|---|---|---|
| 01/M | 0.03 | No | 4 | Ca‐folic acid, vitamin B6 | Unknown | Ohtahara syndrome | Gradual enteral feeding | No | No response |
| 02/F | 0.15 | No | 3 | Ca‐folic acid, vitamin B6 | Unknown | Ohtahara syndrome | Gradual enteral feeding | Yes | 50% seizure reduction |
| 03/M | 1.79 | 1 y | 2 | LEV, TPM | Perinatal asphyxia | West syndrome | Surgery (PEG implantation) | Yes | 82% seizure reduction |
| 04/F | 3.27 | 16 wk | 10 | CLB, TPM | Alpers disease | Convulsive status epilepticus | Ventilation | No | No response |
| 05/M | 6.49 | 2 y | 4 | TPM | Prematurity, IVH | Lennox‐Gastaut syndrome | Illness, vomiting | No | 56% seizure reduction |
| 06/M | 0.72 | 25 wk | 3 | TPM, LEV | Sturge‐Weber syndrome | Focal epilepsy | Epilepsy surgery | Yes | Seizure free |
| 07/M | 3.35 | No | 1 | TPM | Unknown | Lennox‐Gastaut syndrome | Food refusal at start | Yes | 86% seizure reduction |
| 08/M | 10.80 | No | 41 | LEV, MDZ | Mitochondriopathy (POLG mutation) | Convulsive status epilepticus | Ventilation | No | 10% seizure reduction |
| 09/M | 0.46 | 2 wk | 19 | LEV, TPM | Unknown | Partial migrating seizures | Prolonged generalized seizures | Yes | No response |
| 10/M | 4.16 | 2.5 y | 2 | TPM, LEV, CLB | Sturge‐Weber syndrome | Focal epilepsy | Epilepsy surgery | No | 50% seizure reduction |
| 11/M | 0.78 | 16 wk | 2 | VGB | Gaba‐transaminase deficiency | West syndrome | Surgery (PEG implantation) | No | 90% seizure reduction |
| 12/F | 1.98 | 1.5 y | 4 | PGB, VitB6, TPM | PDHC‐deficiency | West syndrome | Illness, vomiting | Yes | 75% seizure reduction |
| 13/M | 3.95 | 3.5 y | 2 | TPM, LEV, VBG | Unknown | West syndrome | Surgery (PEG implantation) | Yes | 90% seizure reduction |
| 14/M | 1.84 | 1 y | 3 | TPM, LEV, NZP | Unknown | West syndrome | Surgery (PEG implantation) | Yes | Seizure free |
| 15/M | 1.58 | 7 mo | 5 | LEV, VGB | Prematurity, HIE | West syndrome | Epilepsy surgery | Yes | Seizure free |
| 16/F | 1.97 | 8 mo | 2 | LEV | FCD | West syndrome | Epilepsy surgery | No | No response |
| 17/F | 0.15 | 1 mo | 1 | FBM, LEV | Vanishing white matter disease | Ohtahara syndrome | Surgery (PEG implantation) | Yes | 80% seizure reduction |
AED, antiepileptic drugs; CBZ, carbamazepine; F, female; FCD, focal cortical dysplasia; HIE, hypoxic ischemic encephalopathy; ID, identification; IVH, intraventricular hemorrhage; KD, ketogenic diet; LEV, levetiracetam; M, male; mo, months; NZP, nitrazepam; PB, phenobarbital; PDHC, pyruvate dehydrogenase complex; PEG, percutaneous endoscopic gastrostomy implantation; PN, parenteral nutrition; TPM, topiramate; VGB, vigabatrin; vit B6, vitamin B6; VPA, valproic acid; wk, weeks; y, years; ZS, zonisamide.
Seventy‐one percent (12/17) of these patients were boys. The median age was 1.8 years (IQR 0.7–3.4, minimum 1 month, maximum 10.8 years). Etiology was known in 64.7% (11/17) and unknown in 35.3% (6/17). The median number of antiepileptic drugs (AEDs) used prior to the initiation of the KD was one (min. 0, max. 5) (Table 1). At kPN start, 88.2% (15/17) of patients received concomitant AEDs (median 2, IQR 1–2, min. 0, max. 3), 2 children received vitamin B6, calcium, and folic acid supplementation.
The indications for kPN in our cohort are shown in Table 1: surgery (n = 4 epilepsy surgery, n = 5 percutaneous endoscopic gastrostomy [PEG] implantations), status epilepticus (n = 3), illness with vomiting (n = 2), food refusal (n = 1), and gradual introduction of enteral feeding in neonates with Ohtahara syndrome (n = 2).
Seventy‐six percent (13/17) of our patients had already received the KD for a median period of 3 months (IQR 2 weeks–1 year, range 0.00–2.00 years) before the switch to kPN became necessary.
The kPN was de novo in 24% (4/17) of the children: in two neonates with Ohtahara syndrome, conventional parenteral feeding was switched to parenteral KD while they were still on a mixed enteral and parenteral feeding regimen. In one infant with drug‐resistant SE, de novo kPN was initiated because the child was unable to tolerate enteral feeds, and in one child with Lennox‐Gastaut syndrome, kPN was started because of food refusal at KD initiation.
The duration of kPN was median 3 days (IQR 2–4 days, range 1–41 days).
PN was administered via central line in 29% (5/17) and via peripheral line in 71% (12/17).
Intralipid was used in 29% (5/17), Clinoleic in 41% (7/17), and SMOF‐Lipid in 29% (5/17). Fifty‐nine percent (10/17) of the children received only kPN; 41% (7/17) of the children received a combination of enteral KD and kPN. An example of an exclusively parenteral ketogenic prescription is given in Figure 2.
Ketogenic ratio of ketogenic PN versus enteral KD
The fat/nonfat ratio of exclusively parenteral KD was mean 0.9:1 (SD = ±0.28; range 0.6–1.5), the lipid intake was median 3 g/kg/day (IQR = 3–3; range 3–4), the protein intake was median 2 g/kg/day (IQR = 1.5–1.5; range 1.5–2), and the carbohydrate intake was median 1.66 g/kg/day (IQR = 1.5–2.4; range 0.5–3.8) (Figure 3).
Figure 3.
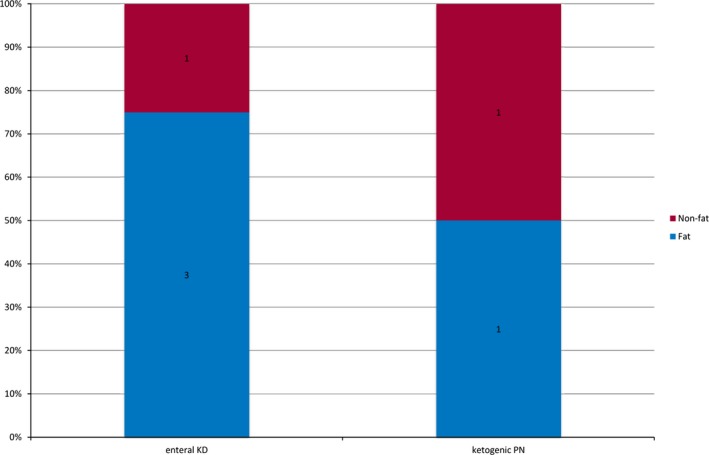
Fat/nonfat ratio during enteral KD and during ketogenic PN (n = 17). KD, ketogenic diet; PN, parenteral nutrition. The fat to nonfat ratio is defined by calculating grams of fat versus grams of proteins and carbohydrates.
When combined, the proportion of parenteral intake varied between 38% and 86%. The fat/nonfat ratio of combined parenteral and enteral KD was 1.22:1 (= mean; SD = ±0.45; range 0.6–2.1). Lipid intake of a combined kPN and enteral KD was median 3.1 g/kg/day (IQR = 3–4.6; range 1.6–7.2), protein intake was median 1.6 g/kg/day (IQR = 1.5–2.0; range 1.3–6.5), and carbohydrate intake was median 1.6 g/kg/day (IQR = 1.5–2.4; range 1.5–4.4).
Seizure outcome
Seventy‐six percent (13/17) of the patients already had an ongoing enteral KD before kPN became necessary, and 77% (10/13) had previously shown significant seizure reductions (≥50%) that were maintained throughout kPN with no seizure exacerbation in all of them.
Twenty‐three percent (3/13) had not responded to enteral KD so far, but still within the evaluation period, they remained nonresponders throughout kPN.
Of the 4 children with de novo kPN, 50% (2/4) were responders (50% and 86% reduction in seizure frequency at 3 months after start). One of the 2 neonates with Ohtahara syndrome responded within the first 2 weeks on combined kPN and enteral KD; the other neonate did not respond. One child with drug‐resistant Lennox‐Gastaut syndrome was started on a kPN because of food refusal at KD initiation, was switched to enteral KD after 30 h, started to tolerate enteral feedings after 4 days, and in the long term showed a seizure reduction of 86% when again on enteral KD.
In one infant with mitochondriopathy and drug‐resistant SE, de novo kPN was initiated during intensive care, ventilation, and high gastric residual volumes. A moderate level of ketosis was achieved within the first day (0.7 mmol/L), and SE was interrupted within the first 3 days. However, series of prolonged generalized tonic‐clonic seizures (GTCS) continued. As oral feeding gradually became possible again, ketone bodies increased. The ketogenic PN was successfully transitioned to enteral KD after 41 days. However, reduction in seizure frequency was only 10% at 3 months after initiation. Consequently, the enteral KD was weaned after 12 weeks (for the relation of ketone bodies to the percentage of ketogenic PN, see Fig. 4).
Figure 4.
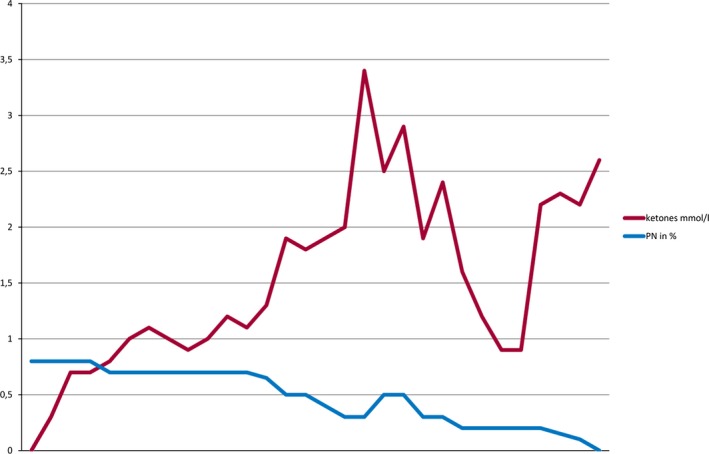
Relation of beta‐hydroxybutyrate levels to the percentage of ketogenic PN. KD, ketogenic diet; PN, parenteral nutrition.
Impact of ketosis
During kPN, beta‐hydroxybutyrate levels were median 2.1 mmol/L (IQR 1, 4–3, 3 mmol/L; range 0.5–5 mmol/L). Clinically relevant ketosis (≥2 mmol/L beta‐hydroxybutyrate) was established/maintained in only 59% of the patients (10/17), with median plasma levels of 2.9 mmol/L (IQR 2.3–3.6 mmol/L; range 2–5 mmol/L). In the remaining 41% (7/17), clinically relevant ketosis was not achieved: the median level of ketosis in these children was 1.4 mmol/L (IQR 1.2–1.6 mmol/L; range 0.5–1.7 mmol/L). When kPN was withdrawn and children were on full enteral feedings again, beta‐hydroxybutyrate levels increased to median 3.7 mmol/L (IQR 3–4.3 mmol/L; range 1.4–4.8 mmol/L) and were >2 mmol/L in 16/17 infants.
In children with de novo kPN, ketosis was achieved within 24 h (in one only with 0.7 mmol/L).
There was a significant correlation between the degree of ketosis achieved (beta‐hydroxybutyrate plasma levels) during the kPN and the percentage of seizure reduction (correlation coefficient = 0.691; p = .002) (Figs. 4 and 5) maintained during kPN or achieved after ketogenic PN and transition to enteral KD.
Figure 5.
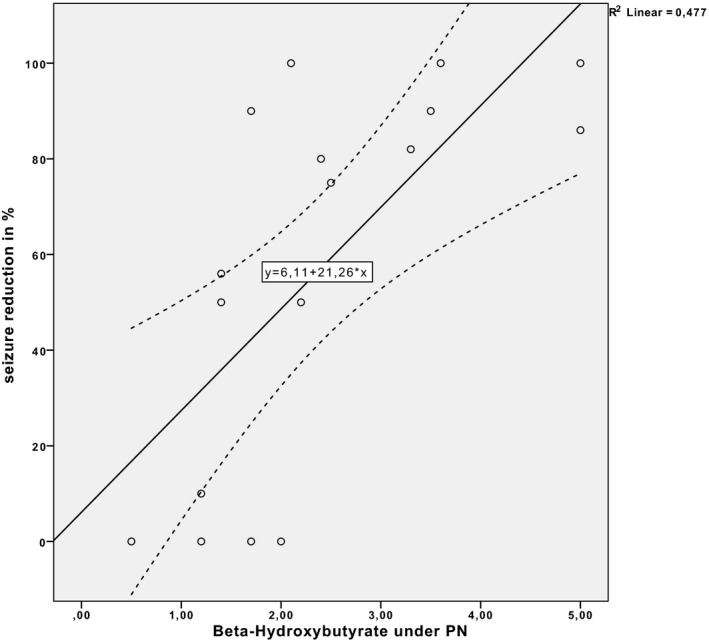
Correlation between degrees of ketosis achieved (beta‐hydroxybutyrate plasma levels) during ketogenic PN and the percentage of seizure reduction. Beta‐hydroxybutyrate plasma levels are given in millimoles per liter; seizure reduction as a percentage is given with respect to seizure reduction to baseline (p = .002).
Among the patients who reached clinically relevant ketosis (beta‐hydroxybutyrate levels ≥2 mmol/L) during kPN, 90% (9/10) showed seizure reductions of ≥50%, whereas only 43% (3/7) of those with beta‐hydroxybutyrate levels <2 mmol/L showed seizure reductions ≥50% (Fisher exact test, p = .06) (Fig. 5).
In de novo kPN, the 2 responders reached the target beta‐hydroxybutyrate level of ≥2 mmol/L within 24 h (2, 2 mol/L and 5 mmol/L, respectively), whereas the nonresponders did not reach target levels (0, 5 and 1, 2 mmol/L, respectively).
Safety
No adverse effects were observed according to adverse effects questionnaires administered during parenteral KD. However, in 6 patients side effects associated with the enteral KD were reported several months later (constipation in 4, diarrhea in 2, noncompliance to ketogenic food in 1). In the majority of individuals, laboratory values on lipid patterns remained within the age‐dependent ranges; transient increases, which were resolved after stopping kPN, were observed for triglycerides in 3, cholesterol in 5, high‐density lipoproteins (HDL) in 6, and low‐density lipoproteins (LDL) in 4 children (Fig. 6). Liver function parameters were not altered in the majority of individuals and only mildly increased (GGT in 1 child, GOT in 3 children, GPT in 2, LDH in 4) and normalized after stopping kPN (GGT median 16 U/L; IQR 11–27 U/L, range 9–125 U/L; median GOT 28 U/L, IQR 20–43 U/L, range 12–max. 102 U/L; median GPT 25 U/L, IQR 19–44 U/L, range 5–156 U/L; median LDH 248 U/L, IQR 213–319 U/L, range 156–499 U/L).
Figure 6.
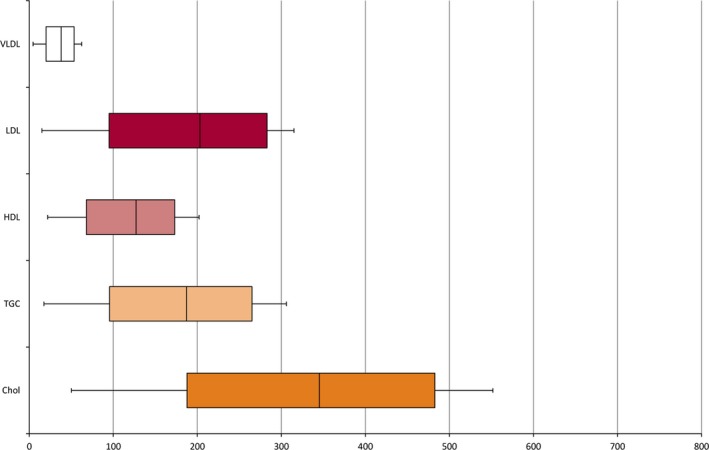
Lipid patterns under ketogenic parenteral nutrition. Chol, cholesterol; HDL, high‐density lipoproteins; LDL, low‐density lipoproteins; TGC, triglycerides; VLDL, very low‐density lipoproteins. Data are described in median and quartiles.
The child in whom the kPN was applied for 41 days showed no gastrointestinal side effects and only mild increase of laboratory markers, which resolved after kPN (cholesterol 164, triglycerides 113, HDL 33, LDL 108, VLDL 23, GGT 17, GOT 28, GPT 25, and LDH 224).
Discussion
Our data confirm that ketosis can be maintained successfully by kPN as demonstrated by plasma beta‐hydroxybutyrate levels ≥2 mmol/L in the majority of our patients. Nutritional intake was in line with the ESPGHAN guidelines for parenteral nutrition in infants and children.
In children who were responders to enteral KD before initiation of kPN, response could be maintained and no seizure exacerbation was observed.
Children started on kPN showed a 50% responder rate. However, response in SE was moderate because SE was interrupted within the first 3 days, but series of prolonged GTCS continued, and seizure reduction after 3 months was only 10%.
We did not observe any relevant adverse effects of the kPN during short‐ or long‐term application (up to 41 days) but only mildly and transiently altered lipid patterns and liver function tests.
A new treatment algorithm
The aim of PN during KD treatment in children with refractory epilepsy is to maintain ketosis and seizure control when enteral feeding is temporarily impossible. So far, few data on kPN have been published.5, 6, 7, 8, 9, 10 These data showed the safety of kPN in children, but did not provide a basis for a standardized approach to prescribe kPN. Therefore, we developed a computer‐based algorithm for calculating kPN according to the ESPGHAN guidelines.4 This algorithm guarantees the limitation of fat intake to a maximum of 4 g/kg/day, an age‐related intake for proteins, electrolytes, trace elements, and vitamins, but allows only small amounts of carbohydrates. Thereby, the kPN results in a lower fat/nonfat ratio than an enteral KD with a fat/nonfat ratio of 4:1 alone.
In contrast to our regimen, other centers allowed higher lipid intakes up to 4.5 g/kg/day5: Jung and coworkers reported a fat/nonfat ratio of 4:1 corresponding to a lipid intake of 4.1–4.5 g/kg/day (50 g fat per 1,000 kcal, with an average of 83–90 kcal/kg/day).10
Enteral ketogenic diet (KD) is usually initiated with a fixed fat/nonfat ratio (2.5 up to 4.0) that is well tolerated.1, 2 In contrast, the administration of similarly high amounts of lipids intravenously is not recommended. Owing to this limitation, a target fat/nonfat ratio of 2.5–4:1 cannot be achieved with a kPN. However, the new algorithm enables clinicians to calculate the highest fat/nonfat ratio possible.
In the present study, we reached the target level of ketosis in 59% of our patients, whereas levels were not reached in 41%, likely because of the relatively limited fat intake via kPN. When kPN was switched back to full enteral feedings, beta‐hydroxybutyrate levels increased to a median of 3.7 mmol/L, reaching target levels in all infants but one.
Using de novo kPN at start of ketogenic treatment resulted in target ketosis levels within 24 h and a seizure reduction of ≥50% at 3 months in 50% of the children. SE was stopped within 3 days, and the child achieved ketosis <2 mmol/L within the first 24 h; however, series of GTCS persisted. Consequently, in SE the role of kPN has to be investigated further. To date, only two cases of SE treated with kPN have been described: in one, ketosis was reached within 24 h and seizures stopped within 70 h.8 In the other, ketosis was reached within the first 3 days (2.4 mmol/L) without seizure control.9
Our responder rate of 50% obtained with de novo kPN is in accordance with the current literature for enteral KD.12, 13 Seizure reduction was maintained during kPN in all of the children having previously responded to an enteral ketogenic regimen (10/13); 23% (3/13) were still within the evaluation period. No seizure exacerbation was observed under the kPN, even when target ketosis levels were not achieved. However, we found a significant correlation between the degree of ketosis during kPN and the percentage of seizure reduction. Patients achieving clinically relevant ketosis responded significantly better. This is of interest, because there is discussion whether ketone bodies are the key factor for efficacy, because most studies did not find an association between levels of ketosis and clinical response.14, 15 Moreover, fine‐tuning of the KD by increasing the fat intake or the fat/nonfat ratio has not been reported to additionally reduce seizure frequency.16 On the other hand, strict initiation of a classical KD with high fat/nonfat ratios is well established to lead to better seizure control.17, 18 In addition, trials with enteral KD often aim at similar target levels (≥2–5 mmol/L) for safety reasons.11, 14 In our study, target levels of ketosis were more easily obtained during transition to enteral KD. Therefore, early enteral feeding has to be encouraged. On the basis of data from enteral KDs, we expected that adding medium‐chain triglyceride (MCT) oils would booster ketosis,19 but we were not able to observe this in our study.
In our cohort, we could not identify other factors associated with efficacy, such as younger age,14, 20 likely because of the sample size.
We did not observe relevant adverse effects, and laboratory parameters remained stable throughout kPN applied for up to 41 days, with only mild and transient changes in blood lipids and liver function tests. Several short‐ or long‐term complications have been reported in the context of PN, such as elevated liver enzymes and parenteral nutrition‐associated cholestasis (PNAC). Especially, neonates seem to be highly susceptible to developing PNAC, as outlined by Shin et al.21 Cases with altered lipid profiles and liver function parameters have been reported.5, 7, 10, 22 The absence of relevant adverse effects is likely the result of the limitation of fat intake and optimization of nutritional intake owing to our algorithm.
Strengths and limitations
We demonstrate both efficacy and safety of kPN administered to a cohort of 17 seriously ill patients with severe and pharmacoresistant epilepsy. We describe a newly developed algorithm to optimize kPN and minimize the risk for errors compared to manual calculations.
This is the largest cohort ever studied in kPN, but the group is heterogeneous and the sample size limits drawing definite conclusions. More data on the long‐term use of kPN in children with epilepsy are still required. Future research should focus on determining which patients need urgent KD during critical care and SE and, hence, should receive kPN as soon as possible. Another question is how kPN can be bolstered in order to achieve higher levels of ketosis rapidly.
Conclusions
In critically ill children, kPN adhering to nutritional guidelines can maintain high ketosis in half of the patients, and clinical response is similar to that of enteral KD. On the basis of our results, we suggest setting the upper limit of parenteral fat at 4 g/kg/day and keeping protein intake according to age‐appropriate guidelines.4, 23 However, ketosis is lower using kPN than during enteral application of the KD. Rapid (re)introduction of enteral feeding is therefore recommended to increase the ketogenic ratio and the proportion of fat.
Acknowledgments and Conflict of Interest
Anastasia Dressler received travel reimbursement and speaker honoraria from SHS, Nutricia, and Vitaflo; Martha Feucht received travel reimbursement and speaker honoraria from SHS and Nutricia; Petra Trimmel‐Schwahofer has received travel reimbursement from SHS and Nutricia. None were related to this article. The other authors have no conflicts of interest to disclose. This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
We thank Prof. C. Male, Medical University Vienna, and E. van der Louw, Erasmus Medical Center Sophia Children's Hospital, for their valuable comments.
Biography
Dr. Anastasia Dressler, Assistant Professor, Medical University Vienna, Department of Pediatrics and Adolescent Health.

References
- 1. Kossoff EH, McGrogan JR. Worldwide use of the ketogenic diet. Epilepsia 2005;46:280–289. [DOI] [PubMed] [Google Scholar]
- 2. Freeman JM, Kossoff EH, Hartman AL. The ketogenic diet: one decade later. Pediatrics 2007;119:535–543. [DOI] [PubMed] [Google Scholar]
- 3. Zupec‐Kania B, Neal E, Schultz R, et al. An update on diets in clinical practice. J Child Neurol 2013;28:1015–1026. [DOI] [PubMed] [Google Scholar]
- 4. Koletzko B, Goulet O, Hunt J, et al. Guidelines on Paediatric Parenteral Nutrition of the European Society of Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) and the European Society for Clinical Nutrition and Metabolism (ESPEN), Supported by the European Society of Paediatric Research (ESPR). J Pediatr Gastroenterol Nutr 2005;41(suppl 2):S1–S87. [DOI] [PubMed] [Google Scholar]
- 5. Zupec‐Kania BA, Aldaz V, Montgomery ME, et al. Enteral and parenteral applications of ketogenic diet therapy. Infant, Child, Adolesc Nutr 2011;3:274–281. [Google Scholar]
- 6. Rosenthal E, Weissman B, Kyllonen K. Use of parenteral medium‐chain triglyceride emulsion for maintaining seizure control in a 5‐year‐old girl with intractable diarrhea. J Parenteral Enteral Nutr 1990;14:543–545. [DOI] [PubMed] [Google Scholar]
- 7. Roan M. Management of long‐term ketogenic parenteral nutrition. Infant, Child, Adolesc Nutr 2011;3:282–287. [Google Scholar]
- 8. Lin JJ, Lin KL, Chan OW, et al. Intravenous ketogenic diet therapy for treatment of the acute stage of super‐refractory status epilepticus in a pediatric patient. Pediatr Neurol 2015;52:442–445. [DOI] [PubMed] [Google Scholar]
- 9. Chiusolo F, Diamanti A, Bianchi R, et al. From intravenous to enteral ketogenic diet in PICU: a potential treatment strategy for refractory status epilepticus. Eur J Paediatr Neurol 2016;20:843–847. [DOI] [PubMed] [Google Scholar]
- 10. Jung DE, Kang H‐C, Lee JS, et al. Safety and role of ketogenic parenteral nutrition for intractable childhood epilepsy. Brain Dev 2012;34:620–624. [DOI] [PubMed] [Google Scholar]
- 11. Kossoff EH, Zupec‐Kania BA, Amark PE, et al. Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group. Epilepsia 2009;50:304–317. [DOI] [PubMed] [Google Scholar]
- 12. Seo JH, Kim HD. Cultural challenges in using the ketogenic diet in Asian countries. Epilepsia 2008;49(suppl 8):50–52. [DOI] [PubMed] [Google Scholar]
- 13. Kossoff EH, Rho JM. Ketogenic diets: evidence for short‐ and long‐term efficacy. Neurotherapeutics 2009;6:406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dressler A, Trimmel‐Schwahofer P, Reithofer E, et al. The ketogenic diet in infants—advantages of early use. Epilepsy Res 2015;116:53–58. [DOI] [PubMed] [Google Scholar]
- 15. Dressler A, Stocklin B, Reithofer E, et al. Long‐term outcome and tolerability of the ketogenic diet in drug‐resistant childhood epilepsy—the Austrian experience. Seizure 2010;19:404–408. [DOI] [PubMed] [Google Scholar]
- 16. Selter JH, Turner Z, Doerrer SC, Kossoff EH. Dietary and medication adjustments to improve seizure control in patients treated with the ketogenic diet. J Child Neurol 2015;30:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seo JH, Lee YM, Lee JS, et al. Efficacy and tolerability of the ketogenic diet according to lipid:nonlipid ratios—comparison of 3:1 with 4:1 diet. Epilepsia 2007;48:801–805. [DOI] [PubMed] [Google Scholar]
- 18. Kossoff EH, Bosarge JL, Miranda MJ, et al. Will seizure control improve by switching from the modified Atkins diet to the traditional ketogenic diet? Epilepsia 2010;51:2496–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neal EG, Chaffe H, Schwartz RH, et al. A randomized trial of classical and medium‐chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 2009;50:1109–1117. [DOI] [PubMed] [Google Scholar]
- 20. Kossoff EH, Hedderick EF, Turner Z, et al. A case‐control evaluation of the ketogenic diet versus ACTH for new‐onset infantile spasms. Epilepsia 2008;49:1504–1509. [DOI] [PubMed] [Google Scholar]
- 21. Shin JI, Namgung R, Park MS, et al. Could lipid infusion be a risk for parenteral nutrition‐associated cholestasis in low birth weight neonates? Eur J Pediatr 2008;167:197–202. [DOI] [PubMed] [Google Scholar]
- 22. Hong AM, Turner Z, Hamdy RF, et al. Infantile spasms treated with the ketogenic diet: prospective single‐center experience in 104 consecutive infants. Epilepsia 2010;51:1403–1407. [DOI] [PubMed] [Google Scholar]
- 23. Deutsche Gesellschaft für Ernährung (DGE), Österreichische Gesellschaft für Ernährung (ÖGE), Schweizerische Gesellschaft für Ernährung (SGE) . Referenzwerte für die Nährstoffzufuhr. 2. Aufl. ed. Bonn: Neuer Umschau Buchverlag; 2015.