

Summary
Advance in the exome‐wide sequencing analysis contributes to identifying hundreds of genes that are associated with early‐onset epileptic encephalopathy and neurodevelopmental disorders. On the basis of massive sequencing data, functional interactions among different genes are suggested to explain the common molecular pathway underlying the pathogenic process of these disorders. However, the relevance of such interactions with the phenotypic severity or variety in an affected individual remains elusive. In this report, we present a 45‐year‐old woman with neurofibromatosis type 1 (NF1), infantile‐onset epileptic encephalopathy, and severe developmental delay. Whole‐exome sequencing identified de novo pathogenic mutations in NF1 and the Schaaf‐Yang syndrome‐associated gene, MAGEL2. Literature‐curated interaction data predicted that NF1 and MAGEL2 proteins were closely connected in this network via their common interacting proteins. Direct conversion of fibroblasts into neurons in vitro showed that neuronal cells from 9 patients with NF1 expressed significantly lower levels of MAGEL2 (54%, p = 0.0047) than those from healthy individuals. These data provide the first evidence that pathogenic mutations of NF1 deregulate the expression of other neurodevelopmental disease‐associated genes. De novo mutations in multiple genes may lead to severe developmental phenotypes through their cumulative effects or synergistic interactions.
Keywords: Early‐onset epileptic encephalopathy, Whole‐exome sequencing, De novo mutation, Neurofibromatosis type 1, MAGEL2, Direct conversion, Functional interaction
Neurofibromatosis type 1 (NF1) is a neurocutaneous syndrome with autosomal dominant patterns of inheritance.1 Affected individuals show characteristic skin lesions of café‐au‐lait spots, multiple tumors, and variable degree of mental disability with or without autistic features. The NF1 gene is located at chromosome 17q11.2, encoding 2,839 amino acid protein neurofibromin.2 NF1 mutations cause the hyperactive extracellular signal‐regulated kinase (ERK) pathway, thereby leading to accelerated cell growth and tumor formation.3 Hyperactive signals in synaptic ERK pathways are associated with both epilepsy and mental development in childhood.4 Previous studies demonstrated that the prevalence (0.76%) of infantile spasm (IS) or early‐onset epileptic encephalopathy (EOEE) in NF1 has been reported to be higher than those (0.02–0.05%) in general populations.5 Neuroimaging studies also revealed that cortical and subcortical lesions were occasionally found in NF1 patients.6 However, specific brain lesions or genetic backgrounds have not been disclosed for NF1 in the majority of cases with IS/EOEE. We experienced a case of a 45‐year‐old female with NF1, EOEE, and severe developmental delay. Atypical phenotypes in the present case were studied on the basis of genetic as well as biological backgrounds.
Materials and Methods
This study was approved by the Institutional Review Board at Kyushu University (#461‐00). Written informed consent was obtained from the parents. Experiments were conducted under a stringent compliance to the institutional guideline and our experimental protocol (23‐53). Case report and experimental procedures for whole‐exome sequencing (WES), in vitro conversion of neurons, quantitative polymerase chain reactions (PCR), and bioinformatics are presented in Appendix S1 and Figs. S1, S2).
Results
De novo mutations in NF1 and MAGEL2
De novo mutations in NF1 (NM001042492.2: c.4835 + 1G>T) and MAGEL2 (NM019066.2: c.219C>G, c.224delC) were validated with Sanger sequencing (Figs. 1A,B). The former mutation occurred at the splicing junction of exon 36 in NF1, disrupting the functional expression of neurofibromin. The latter was mapped to the coding region of MAGEL2 and was considered to produce a truncated form of MAGEL2. We further investigated whether the mutated allele was located on the paternal allele.7 Methylation‐sensitive digestion with SmaI followed by PCR amplification and direct sequencing of the flanking region revealed that the mutated allele remained intact after the SmaI digestion (Fig. 1B). We thus confirmed that the de novo MAGEL2 mutation occurred at the maternal allele, which was fully methylated in lymphocytes. Microarray‐based comparative genome hybridization (CMA ver. 8.1 at Baylor MGL; data not shown) excluded that this case carried pathogenic copy number variants (CNVs).
Figure 1.
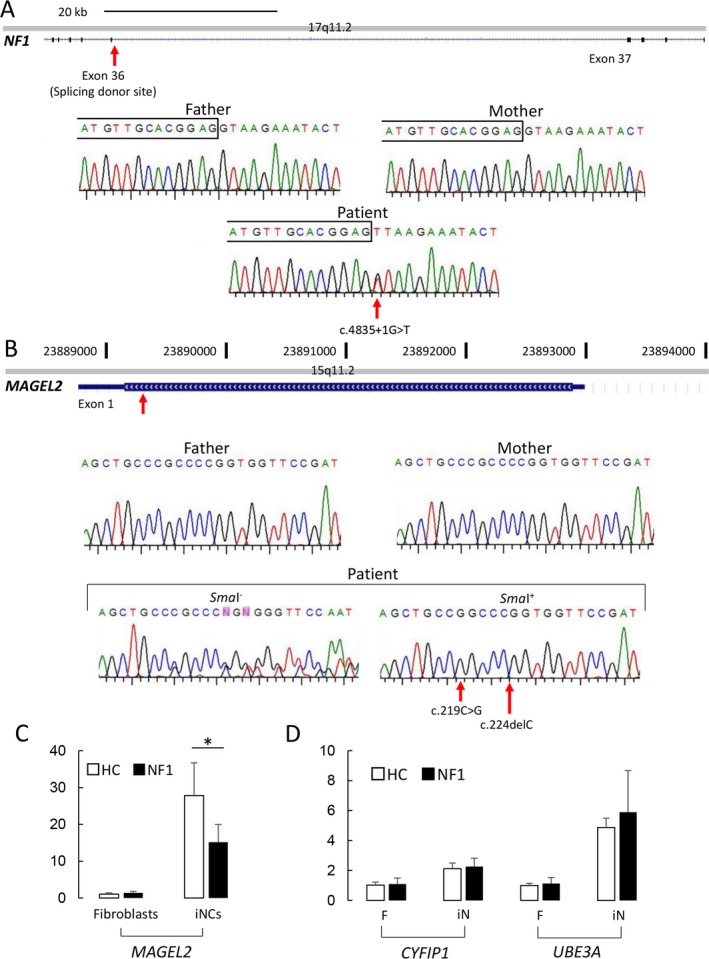
De novo mutations identified in the present case and functional interaction between NF1 and MAGEL2. (A) De novo mutation at the splicing junction of exon 36 in NF1. The sequence chromatograms of father, mother, and the patient are shown. Red arrow indicates that this mutation occurred at chr17:29592358 (NM_001042492.2:c.4835 + 1G>T). Boxed letters above the sequencing data denote exonic sequences. (B) MAGEL2 mutation in the present case. Aligned data illustrate that the mutation occurred de novo in this patient at chr15:23892666, 23892671 (NM_019066.4:c.219C>G, c.224delC). Sequencing results before (SmaI−) and after the SmaI digestion (SmaI+) indicate that this mutation occurred at the methylated (or maternally inherited) allele. (C) MAGEL2 mRNA expression before and after neuronal induction. White and black bar plots represent the relative expression levels in indicated cells from healthy controls (n = 9) and NF1 patients (n = 9), respectively. (D) CYFIP1 and UBE3A expressions in fibroblasts and induced neurons. The gene expression profiles were quantitated in vitro using the cells from healthy controls and NF1 patients (n = 9 for each group). Values in C and D are shown as mean ± SD of each group. Asterisk indicates the p value of less than 0.05 (Student's t test).
Functional interaction between NF1 and MAGEL2
The de novo MAGEL2 mutation in this case was unlikely a primary cause of the patient's developmental phenotypes. We considered the double mutations of NF1 and MAGEL2 as an extremely rare genetic event8 (Table 1). We alternatively interpreted this event to be possibly relevant with the unusual NF1 phenotype of this case. To explore whether NF1 and MAGEL2 might work as genetic modifiers for each other, we used STRING, a protein‐protein interaction database (http://string-db.org/). This open database predicted that NF1 and MAGEL2 were directly or indirectly connected via common binding proteins in a functional network consisting of 35 proteins (nodes) and 84 interactions (edges) (Fig. S2A). Given the number of edges expected to be 48, these 35 proteins were considered to have 1.8‐fold enriched protein‐protein interactions (p = 2.25 × 10−6). Thus, this network was suggested to have functional enrichment in certain molecular pathways. Indeed, the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses showed that signaling molecules associated with “chromatin binding (GO:0003682),” “RNA polymerase (GO:0003899 and KEGG:3020),” and “RAS signaling pathways (KEGG:4014)” were enriched in this network (Figs. S2B,C).
Table 1.
Clinical features of the present case in comparison with those of neurofibromatosis type 1 and Schaaf‐Yang syndrome
| Present case | Neurofibromatosis type 1 | Schaaf‐Yang syndrome | |
|---|---|---|---|
| Mutated gene | NF1, MAGEL2 | NF1 | MAGEL2 |
| (Band Locus) | (17q11.2) | (15q11.2) | |
| Phacomatosis and associated lesion | |||
| Café‐au‐lait spots | + | + | − |
| Neurofibroma | + | + | − |
| Lisch nodules of iris | − | + | − |
| Dysmorphism | |||
| Bitemporal narrowing of facial appearance | − | − | + |
| Almond‐shaped palpebral fissures | − | − | + |
| Small hands | + | − | + |
| Joint contractures | − | − | + |
| Tumorigenesis and skeletal problem | |||
| Brain tumor | |||
| Abnormal MRI signal | + | + | − |
| Bone fracture | − | + | − |
| Neurological sign | |||
| Feeding problem | − | − | + |
| Hypotonia | + | + | + |
| Developmental delay | + | + | + |
| Autism spectrum disorder | + | + | + |
| Seizure | + | + | − |
Altered expression of MAGEL2 in NF1 patient‐derived neurons
To validate the possible functional interplays between NF1 and MAGEL2, we examined whether pathogenic mutations in NF1 might influence neuronal expression of MAGEL2. We determined the transcriptional activation of NF1 and MAGEL2 using the direct conversion system of fibroblast into neuron in vitro. Quantitative analyses on mRNA expression showed that MAGEL2 expressions in fibroblasts did not differ between the patients and controls. The neuronal conversion induced robust increase in the MAGEL2 expression (Fig. 1C). Notably, the neuronal expression of MAGEL2 was decreased to 54% of that in healthy controls (n = 9 for each group, p = 0.0047, Student's t test; Fig. 1C). The neuronal expression of MAGEL2 in the present case was 41% of that in healthy controls (p = 0.0189, Student's t test; data not shown). Neuronal conversion also induced higher expressions of imprinted genes (UBE3A and CYFIP1) at the chromosome 15p11.2 region than those in fibroblasts (Fig. 1D). However, induced neurons from NF1 patients and healthy controls expressed these genes at comparable levels. These data confirmed the epistatic regulation of MAGEL2 expression by NF1 and illustrated the specific effects of the NF1 mutations on transcriptional activation of MAGEL2 during the neuronal conversion.
To test whether NF1 mutations might affect other genes associated with EOEE, we randomly selected 6 EOEE‐associated genes (CDKL5, CHD2, ARX, KCNT1, SCN1A, and TRIM8)9, 10, 11, 12, 13 and assessed their expression profiles in both fibroblasts and induced neurons (Fig. S3). As expected, in vitro conversion of fibroblasts into neurons resulted in 2‐ to 5‐fold higher expression of these six genes than those in fibroblasts. When we compared their expression levels in neurons, only KCNT1 was expressed at a significantly lower level (69%) in NF1 neurons than that in healthy controls (p = 0.011, Student's t test; Fig. S3). All the other genes were expressed in neurons from NF1 patients at similar levels to those in controls. These data suggested that NF1 mutation attenuated the expression of a subset of EOEE‐associated genes.
Discussion
We presented a Japanese woman who had dysmorphic appearance, severe intellectual disability, and intractable epilepsy with a history of infantile‐onset seizures. This is the first case of NF1 carrying another de novo mutation in a gene that is known to cause different neurodevelopmental disorders. Previous studies demonstrated that NF1 patients are susceptible to the onset of epileptic encephalopathy, such as IS and EOEE.14
The WES and subsequent analyses led us to the following two discussions: First, the severe phenotypes of this patient resulted solely from the NF1 mutation regardless of the MAGEL2 mutation. Second, the double mutations in NF1 and MAGEL2 exerted cumulative or synergistic effects to produce more severe phenotypes than those expected for individuals with a single gene mutation in either NF1 or MAGEL2.
The first perspective might be valid taking the allele‐specific expression of MAGEL2 into account. Indeed, truncating MAGEL2 mutations proved to be critical for dysmorphic appearance and hypothalamic dysfunctions exclusively when the mutations occurred in the paternal alleles.7 Despite these facts, experimental studies have shown that MAGEL2 was widely expressed in embryonic as well as in the adult brains.15 Moreover, allele‐specific expression of imprinted genes varies over time and by region in neuronal subpopulations.15 Recent studies have shown that MAGEL2 and NF1 were expressed from the maternal allele in embryonic tissues under certain conditions, suggesting that Prader‐Willi syndrome–associated genes in the chromosomal region at 15q11–q13 have epigenetic flexibility.16, 17 With our findings of lower expression of MAGEL2 in neurons from NF1 patients, we considered that the MAGEL2 mutation on the maternal allele caused only negligible effects on neuronal phenotype, whereas it could reach the pathogenic level when the additional mutation in NF1 coincided. Therefore, it cannot be safely concluded that the MAGEL2 mutation in the maternal allele was irrelevant to the neurodevelopmental phenotypes in this case.
MAGEL2 protein belongs to a family of melanoma‐associated antigen (MAGE) domain‐containing molecules, which has been characterized as highly expressed genes/proteins in various types of tumors.18 Among MAGE family proteins, NRAGE was shown to interact with p75 neurotrophin receptor and to promote nerve growth factor (NGF)‐dependent apoptosis, suggesting that NRAGE could be a component of intracellular signaling pathways.19 Similarly, NF1 is involved in the NGF‐dependent survival of embryonic sensory and sympathetic neurons.20 These data supported our hypothesis that MAGE family proteins and NF1 may cooperatively regulate the maturation of neurons under certain molecular pathways, as the network in this study illustrated.
The interaction database and subsequent bioinformatics analysis revealed that the NF1‐ and MAGEL2‐containing network was significantly enriched in the proteins associated with particular cellular functions, such as RAS signaling. Although physiological functions of MAGEL2 in neurons remain unknown, this result raised a new possibility that MAGEL2 may regulate the RAS signals cooperatively with NF1 in the developing brain. Thus, the hypomorphic mutations in the two genes may cause more profound effects on neuronal dysfunctions than those resulting from the mutation in either gene.
According to the protein interaction database, NF1 and MAGEL2 were unlikely to constitute a protein complex. Rather, our experimental data supported evidence that NF1 acts as an epistatic regulator of MAGEL2 expression. In line with these data, we conceptualized that the de novo mutation of MAGEL2 exaggerated neuronal dysfunctions owing to the NF1 mutation and hyperactive RAS‐ERK signaling. Thus, the severe neurological phenotype of our case can be regarded as an extended model of digenic inheritance or the transheterozygote at two loci.21, 22 To determine their additive or synergistic effects on RAS signaling and neurological deficits, more studies using genetically engineered mice will be necessary.
Missing pieces are left for our future study to investigate whether and how recessive mutations inherited from the patient's parents could modify the phenotypes of other cases with atypical phenotypes of NF1. Accumulation of genome‐wide analysis data for NF1 patients with IS and EOEE will clarify these issues.
Disclosure
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1. Dysmorphic appearance of the present case.
Figure S2. Bioinformatics analyses on interaction between NF1 and MAGEL2.
Figure S3. Expression of epileptic encephalopathy‐associated genes in fibroblasts and induced neurons.
Appendix S1. Materials and methods.
Acknowledgments
We thank Huda Y. Zoghbi, Christian P. Schaaf (Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital), and Ryutaro Kira (Fukuoka Children's Hospital) for helpful discussion, and Kenjiro Gondo (Gondo Pediatric Clinic) for continual support of the patient and her family. This study was supported by JSPS KAKENHI grant numbers 15K0962 (Y.S.), 16K09991 (M.S.), evidence‐based early diagnosis and treatment strategies for neuroimmunological diseases from the Ministry of Health, Labour and Welfare of Japan, the Life Science Foundation of Japan, the Takeda Science Foundation, Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics, and the Japan Epilepsy Research Foundation (Y.S.).
Biography
Satoshi Akamine is a graduate student in the Department of Pediatrics, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan.

References
- 1. Garg S, Green J, Leadbitter K, et al. Neurofibromatosis type 1 and autism spectrum disorder. Pediatrics 2013;132:1642–1648. [DOI] [PubMed] [Google Scholar]
- 2. Cawthon RM, Weiss R, Xu GF, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990;62:193–201. [DOI] [PubMed] [Google Scholar]
- 3. Bollag G, Clapp DW, Shih S, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 1996;12:144–148. [DOI] [PubMed] [Google Scholar]
- 4. Pavlowsky A, Chelly J, Billuart P. Emerging major synaptic signaling pathways involved in intellectual disability. Mol Psychiatry 2012;17:682–693. [DOI] [PubMed] [Google Scholar]
- 5. Ruggieri M, Iannetti P, Clementi M, et al. Neurofibromatosis type 1 and infantile spasms. Childs Nerv Syst 2009;25:211–216. [DOI] [PubMed] [Google Scholar]
- 6. Kulkantrakorn K, Geller TJ. Seizures in neurofibromatosis 1. Pediatr Neurol 1998;19:347–350. [DOI] [PubMed] [Google Scholar]
- 7. Schaaf CP, Gonzalez‐Garay ML, Xia F, et al. Truncating mutations of MAGEL2 cause Prader‐Willi phenotypes and autism. Nat Genet 2013;45:1405–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fountain MD, Aten E, Cho MT, et al. The phenotypic spectrum of Schaaf‐Yang syndrome: 18 new affected individuals from 14 families. Genet Med 2017;19:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mirzaa GM, Paciorkowski AR, Marsh ED, et al. CDKL5 and ARX mutations in males with early‐onset epilepsy. Pediatr Neurol 2013;48:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012;380:1674–1682. [DOI] [PubMed] [Google Scholar]
- 11. Barcia G, Fleming MR, Deligniere A, et al. De novo gain‐of‐function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012;44:1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Claes L, Ceulemans B, Audenaert D, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat 2003;21:615–621. [DOI] [PubMed] [Google Scholar]
- 13. Epi4K Consortium , Epilepsy Phenome/Genome Project , Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Darke K, Edwards SW, Hancock E, et al. Developmental and epilepsy outcomes at age 4 years in the UKISS trial comparing hormonal treatments to vigabatrin for infantile spasms: a multi‐centre randomised trial. Arch Dis Child 2010;95:382–386. [DOI] [PubMed] [Google Scholar]
- 15. Boccaccio I, Glatt‐Deeley H, Watrin F, et al. The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader‐Willi region. Hum Mol Genet 1999;8:2497–2505. [DOI] [PubMed] [Google Scholar]
- 16. Matarazzo V, Muscattelli F. Natural breaking of the maternal silence at the mouse and human imprinted Prader‐Willi locus: a whisper with functional consequences. Rare Dis 2013;1:e27228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rieusset A, Schaller F, Unmehopa U, et al. Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with Prader‐Willi syndrome, has functional consequences. PLoS Genet 2013;9:e1003752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991;254:1643–1647. [DOI] [PubMed] [Google Scholar]
- 19. Salehi AH, Roux PP, Kubu CJ, et al. NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor‐dependent apoptosis. Neuron 2000;27:279–288. [DOI] [PubMed] [Google Scholar]
- 20. Vogel KS, Brannan CI, Jenkins NA, et al. Loss of neurofibromin results in neurotrophin‐independent survival of embryonic sensory and sympathetic neurons. Cell 1995;82:733–742. [DOI] [PubMed] [Google Scholar]
- 21. Lupski JR. Digenic inheritance and Mendelian disease. Nat Genet 2012;44:1291–1292. [DOI] [PubMed] [Google Scholar]
- 22. Schaffer AA. Digenic inheritance in medical genetics. J Med Genet 2013;50:641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dysmorphic appearance of the present case.
Figure S2. Bioinformatics analyses on interaction between NF1 and MAGEL2.
Figure S3. Expression of epileptic encephalopathy‐associated genes in fibroblasts and induced neurons.
Appendix S1. Materials and methods.