

Abstract
Breast adenoid cystic carcinoma (AdCC), a rare type of triple-negative breast cancer (TNBC), has been shown to be driven by MYB pathway activation, most often underpinned by the MYB-NFIB fusion gene. Alternative genetic mechanisms, such as MYBL1 rearrangements, have been reported in MYB-NFIB-negative salivary gland AdCCs. Here we report on the molecular characterization by massively parallel sequencing of four breast AdCCs lacking the MYB-NFIB fusion gene. In two cases, we identified MYBL1 rearrangements (MYBL1-ACTN1 and MYBL1-NFIB), which were associated with MYBL1 overexpression. A third AdCC harbored a high-level MYB gene amplification, which resulted in MYB overexpression at the mRNA and protein levels. RNA-sequencing and whole-genome sequencing revealed no definite alternative driver in the fourth AdCC studied, despite high levels of MYB expression and the activation of pathways similar to those activated in MYB-NFIB-positive AdCCs. In this case, a deletion encompassing the last intron and part of exon 15 of MYB, including the binding site of ERG-1, a transcription factor that may down-regulate MYB, and the exon 15 splice site, was detected. In conclusion, we demonstrate that MYBL1 rearrangements and MYB amplification likely constitute alternative genetic drivers of breast AdCCs, functioning through MYBL1 or MYB overexpression. These observations emphasize that breast AdCCs likely constitute a convergent phenotype, whereby activation of MYB/MYBL1 and their downstream targets can be driven by the MYB-NFIB fusion gene, MYBL1 rearrangements, MYB amplification or other yet to be identified mechanisms.
Keywords: adenoid cystic carcinoma, breast, MYB, MYBL1, MYB-NFIB fusion gene
INTRODUCTION
Breast adenoid cystic carcinoma (AdCC) is a rare type of triple-negative breast cancer (TNBC, i.e. estrogen receptor (ER), progesterone receptor (PR) and HER2-negative) [1,2]. Whilst differing from conventional TNBCs [1,3], breast AdCCs share with salivary gland AdCCs similar molecular profiles and an identical genetic driver: the MYB-NFIB fusion gene [1,3-7], which functions through MYB overexpression due to the loss of miRNA binding sites or super-enhancer translocations [5,8].
Salivary gland AdCCs lacking the MYB-NFIB fusion gene may harbor MYBL1 rearrangements [9-11]. MYBL1 encodes for the A-MYB protein, which shares with c-MYB (encoded by MYB) extensive homology and downstream target genes, converging in the activation of similar downstream pathways. Consistent with this notion, salivary gland AdCCs harboring either rearrangements display similar transcriptomic profiles [11].
Here we performed a comprehensive genomic analysis of four MYB-NFIB-negative breast AdCCs, indicating that MYBL1 rearrangements and MYB gene amplification likely constitute alternative oncogenic drivers of this rare form of TNBC.
MATERIAL AND METHODS
Case selection
Four MYB-NFIB-negative breast AdCCs (Figure 1, supplementary material, Table S1) were retrieved from the authors’ institutions and centrally reviewed (Supplementary material and methods). Whole-exome sequencing (WES) data from AdCC11 and AdCC12 and three MYB-NFIB-positive AdCC controls were reported previously [3]. Patient consent was obtained where appropriate, according to the protocols approved by the local Institutional Review Boards.
Figure 1. Histologic and immunohistochemical features and MYB/MYBL1 fluorescence in situ hybridization results of four breast adenoid cystic carcinomas (AdCCs).
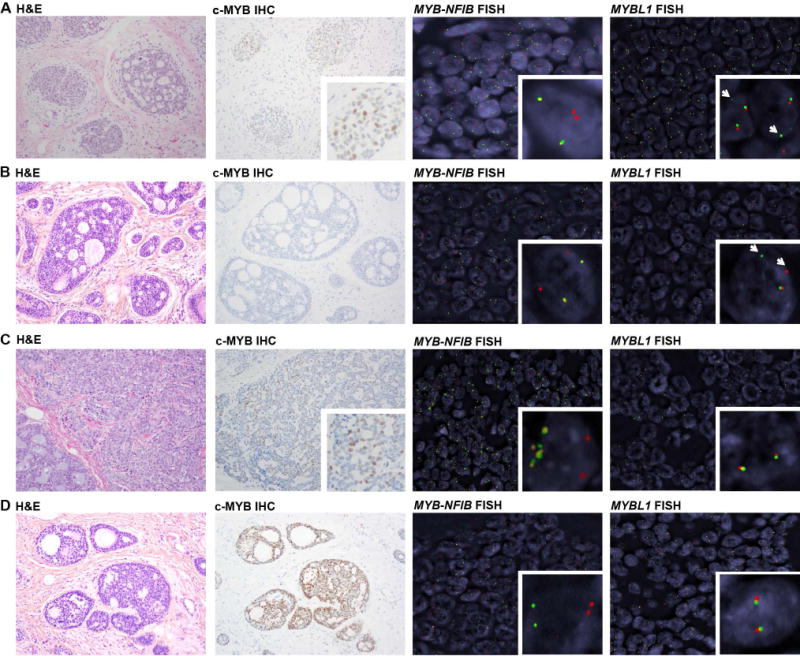
Representative micrographs depicting histologic features (magnification 200X), c-MYB immunohistochemistry (magnification 200×), MYB-NFIB three-color FISH (green: 5′ MYB, orange: 3′ MYB, red: 3′ NFIB) and MYBL1 dual-color break-apart FISH (green: 5′ MYBL1, red: 3′ MYBL1) of four MYB-NFIB-negative breast AdCCs. (A) AdCC35, displaying c-MYB expression, and rearrangement of MYBL1 as demonstrated by a single green 5′ signal (arrowhead) and loss of the red 3′ signal, indicative of an unbalanced translocation. (B) AdCC11, lacking c-MYB protein expression and displaying rearrangement of MYBL1 as demonstrated by a break-apart signal pattern of MYBL1 by FISH (arrowheads). (C) AdCC34, displaying c-MYB protein expression and MYB DNA amplification indicated by multiple green/orange signals in the form of small clusters, with an average of 5 MYB signals per cell), whereas ~2 copies were observed for NFIB (red). (D) AdCC12, displaying c-MYB protein expression and no altered signal patterns in MYB-NFIB and MYBL1 FISH. FISH, fluorescence in situ hybridization; H&E, hematoxylin & eosin; IHC, immunohistochemistry. Note that the same H&E micrograph of AdCC12 is also depicted in supplementary material, Figure S1.
Immunohistochemistry
Immunohistochemistry for ER, PR, HER2 and c-MYB(clone EP769Y) was performed as previously described [2,3,7] (Supplementary materials and methods, and Table S2).
Microdissection and nucleic acid extraction
DNA and/or RNA samples of AdCC11 and AdCC12, and of MYB-NFIB-positive controls were extracted from fresh-frozen tissue as previously described [3,12]; those of AdCC34 and AdCC35 were retrieved from formalin-fixed paraffin-embedded tissue samples, following microdissection (Supplementary materials and methods).
Whole-genome and targeted capture massively parallel sequencing
Tumor-normal DNA samples of AdCC11 and AdCC12 were subjected to whole-genome sequencing (WGS), and those of AdCC34 to Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay [12,13], which targets the entire coding regions of 410 cancer genes [12,13](Supplementary materials and methods, and Table S3). Sequencing data analyses were performed as previously described (Supplementary materials and methods). To validate the somatic mutations identified by WGS, a re-analysis of the previously reported WES results [3] revealed a validation rate >90% (Supplementary materials and methods). WGS and MSK-IMPACT data have been deposited in the NCBI Sequence Read Archive under Accession Nos. SRP108137 and SRP108155, respectively.
RNA-sequencing
RNA-sequencing of AdCC11, AdCC12 and AdCC35, and of the MYB-NFIB-positive AdCC5 was performed to identify fusion transcripts [14](Supplementary materials and methods). RNA-sequencing data have been deposited in the NCBI Sequence Read Archive under Accession No. SRP108156.
Fluorescence in situ hybridization (FISH)
The MYB-NFIB fusion gene and MYBL1 rearrangements were evaluated by FISH in all cases as previously described [2](Supplementary materials and methods).
RT-qPCR
RT-qPCR was performed to compare the expression levels of the 5′ and 3′ portions/exons of MYB and MYBL1 using TaqMan Assay-on-Demand, as described previously [3], in all cases, MYB-NFIB-positive AdCC6 and AdCC8 [3], and three breast cancer cell lines with known MYB mRNA expression levels (Supplementary materials and methods)
Gene set enrichment analysis (GSEA)
Expression levels using normalized RNA-sequencing RPKM values were used for single sample GSEA (ssGSEA; https://genepattern.broadinstitute.org/gp/, Supplementary materials and methods).
RESULTS AND DISCUSSION
Histopathological characteristics of MYB-NFIB-negative breast AdCCs
The four FISH-proven MYB-NFIB-negative AdCCs were of triple-negative phenotype and did not differ histologically from MYB-NFIB-positive AdCCs (Figure 1, supplementary material, Figure S1). c-MYB protein expression, which has been documented in AdCCs lacking the MYB-NFIB fusion gene [15], was detected at varying levels (AdCC35, H-score 43; AdCC12, H-score 75; AdCC34, H-score 120) in all cases but AdCC11 (Figure 1, supplementary material, Figure S2).
AdCC35 harbors a MYBL1-NFIB fusion gene and MYBL1 overexpression
RNA-sequencing of AdCC35 revealed an in-frame MYBL1-NFIB fusion gene, resulting in a chimeric transcript composed of exons 1-14 of MYBL1 and exon 9 of NFIB (Figure 2A). Both DNA binding and c-terminal regulatory domains of MYBL1 were retained, akin to previously reported MYB or MYBL1 rearrangements [10,11,16]. FISH analysis with MYBL1 break-apart probes confirmed the MYBL1 rearrangement at the genomic level (Figure 1A), and RT-qPCR and RNA-sequencing revealed differential mRNA levels of the 5′ and 3′ portions of MYBL1, consistent with gene breakage (Figure 3A–B). MYBL1 was overexpressed as compared to MYB-NFIB-positive AdCCs [3] and the breast cancer cell lines tested, and MYB expression was low (Figure 3A and 3B). As in a previously described MYBL1-rearranged salivary gland AdCC [9], c-MYB protein expression was observed (Figure 1A, supplementary material, Figure S2A), potentially caused by cross-reactivity between the c-MYB antibody and the rearranged A-MYB protein, due to the extensive homology between MYB and MYBL1 genes and their respective proteins.
Figure 2. MYBL1 rearrangements identified by RNA-sequencing in two breast adenoid cystic carcinomas (AdCCs).
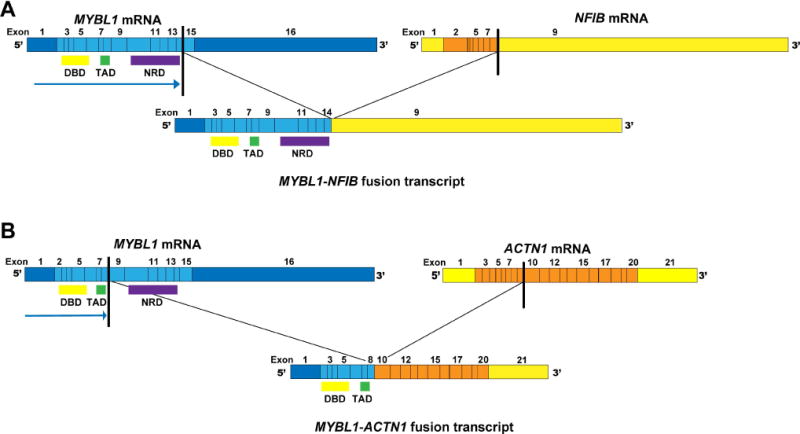
MYBL1 fusion transcripts are illustrated, including the exons and domains involved. The breakpoints of each 5′ (MYBL1) and 3′ partner genes are represented as black lines. (A) AdCC35, displaying a MYBL1-NFIB fusion transcript, in which the c-terminal negative regulatory domain is retained, creating a ‘long’ fusion transcript as described in salivary gland AdCCs [10]. (B) AdCC11, displaying a MYBL1-ACTN1 fusion transcript, in which the c-terminal negative regulatory domain is lost, creating a ‘short’ fusion transcript. DBD, DNA binding domain; NRD; negative regulatory domain; TAD, transactivation domain.
Figure 3. MYB and MYBL1 gene expression levels in breast adenoid cystic carcinomas (AdCCs) defined by RT-qPCR and RNA-sequencing.
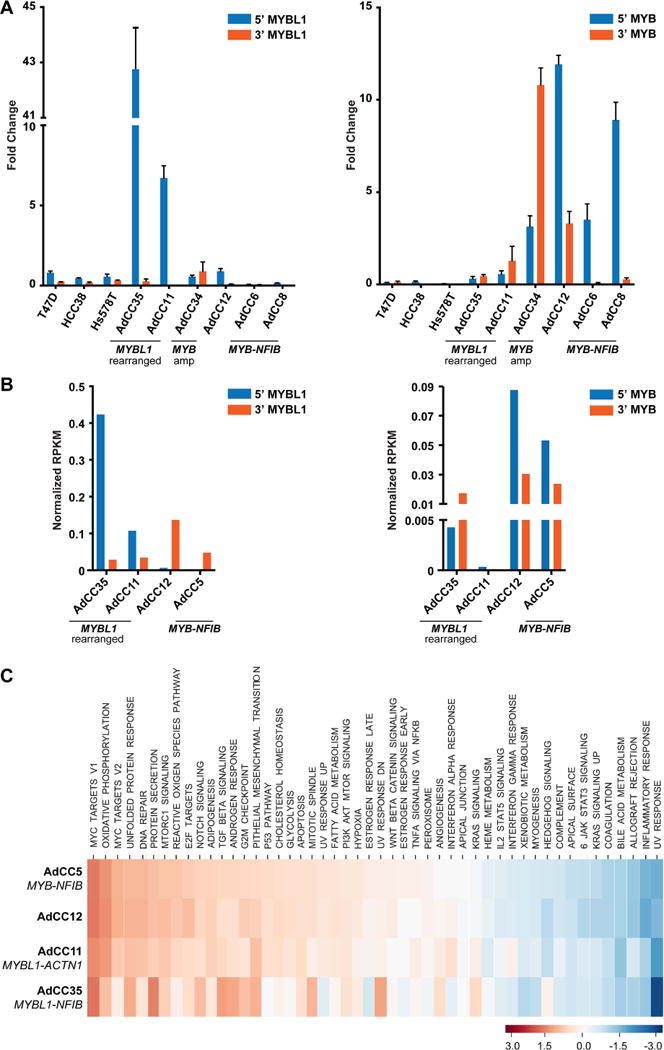
(A, B) The gene expression levels of the 5′ and 3′ portions of the MYBL1 (left) and MYB (right) transcripts in four MYB-NFIB-negative breast AdCCs are compared to those of AdCCs previously documented to harbor the MYB-NFIB fusion gene (AdCC5, AdCC6 and/or AdCC8) [1,3] and/or three breast cancer cell lines (T47D, HCC38 and Hs578T), as defined by RT-qPCR (A), error bars, SD of mean (n = 3 experimental replicates); (B), RNA-sequencing using normalized RPKM values). (C) Single sample gene set enrichment analysis (ssGSEA) was performed to assess pathways activated in the four breast AdCCs analyzed by RNA-sequencing. The calculated enrichment score (ES) for each pathway in each case is illustrated, where each column represents a case and each line represents a pathway. For comparison, the MYB-NFIB-positive AdCC5 [3] was included. Amp, amplification, RPKM, reads per kilobase per million mapped reads. ssGSEA, single-sample gene set enrichment analysis.
AdCC11 harbors a MYBL1-ACTN1 fusion gene and MYBL1 overexpression
WGS of AdCC11 revealed a low mutational burden (0.52/Mb) and lack of TP53 mutations, which are frequently found in TNBCs [17](Figure 4A, supplementary material, Table S4). RNA-sequencing of this case resulted in the identification of an in-frame MYBL1-ACTN1 fusion gene, which comprised exons 1-8 of MYBL1 and exons 10-21 of ACTN1, resulting in loss of the C-terminal regulatory domain of A-MYB (Figure 2B). MYBL1 rearrangements in salivary gland AdCCs have been described to occur with multiple partners [10,11], but ACTN1 has not been previously reported in this context. Consistent with these findings, FISH validated the MYBL1 rearrangement (Figure 1B), and RT-qPCR and RNA-sequencing analyses showed high expression levels of the 5′ of MYBL1 and low levels of MYB.
Figure 4. Genomic and transcriptomic features of the MYB-NFIB-negative breast adenoid cystic carcinomas AdCC11, AdCC12 and AdCC34.
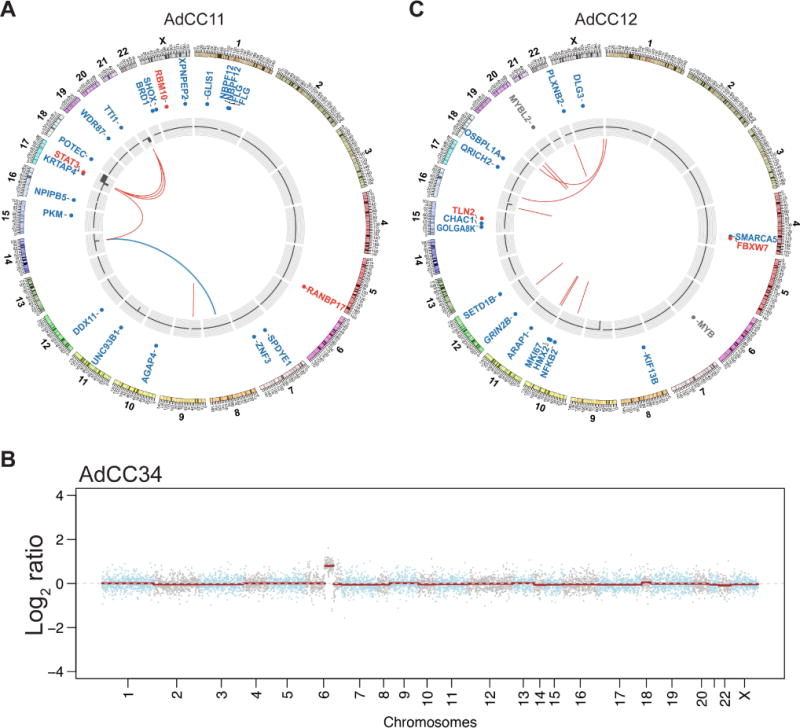
(A) Circos plot of AdCC11 depicting the chromosomes on the outer ring, the non-synonymous somatic mutations, copy number alterations and the fusion genes in the center. The MYBL1-ACTN1 fusion gene is illustrated in blue, and likely-pathogenic mutations in red. (B) Copy number plot of AdCC34, depicting amplification of 6q23.3, encompassing the MYB gene. The Log2 ratios are plotted along the y-axis and the chromosome positions are plotted along the x-axis. (C) Circos plot of AdCC12 depicting the chromosomes on the outer ring, the non-synonymous somatic mutations, copy number alterations and the fusion genes in the center. Likely-pathogenic mutations are shown in red. The MYB large deletion and the MYBL2 mutation identified are also shown (gray).
AdCC34 harbors MYB gene amplification and MYB overexpression
AdCC34, which expressed c-MYB protein and lacked MYB-NFIB fusion gene and MYBL1 rearrangements by FISH (Figure 1C, supplementary material, Figure S2B), was subjected to MSK-IMPACT, revealing only one synonymous mutation affecting TSC2 (E392E; supplementary material, Table S4) and a simple copy number profile. Importantly, however, AdCC34 harbored a focal amplification on 6q23.3 encompassing MYB (Figure 4B). This was confirmed by FISH (mean of 5(range 3-10) copies of MYB per tumor cell; Figure 1C), and found to be associated with high MYB mRNA (Figures 3A and 3B) and MYB protein expression levels (supplementary material, Figure S2B). MYB amplification has not been previously described in AdCCs of any anatomical site. Based on the lack of potentially pathogenic somatic mutations and of MYB and MYBL1 rearrangements, it is plausible that MYB overexpression was the result of MYB gene amplification, a novel potential mechanism of MYB activation in breast AdCCs.
AdCC12 expresses MYB and displays a gene expression profile consistent with MYB/MYBL1 activation, despite the lack of MYB and MYBL1 rearrangements
RNA-sequencing, WGS and FISH of AdCC12 revealed no fusion genes or copy number alterations affecting MYB, MYBL1 or other potential candidates (Figure 1D, Figure 4C). MYB overexpression was, however, identified by immunohistochemistry (Figure 1, supplementary material, Figure S2C), RT-qPCR (Figure 3A) and RNA-seq (Figure 3B). ssGSEA revealed that AdCC12 shared with MYBL1-rearranged AdCC35 and AdCC11 and the MYB-NFIB-positive AdCC5 similar transcriptomic profiles enriched for the MYC and NOTCH signaling pathways (Figure 3C). These findings suggest that MYB activation may be driven by yet additional genetic mechanisms.
WGS analysis revealed a low mutational burden (0.73/Mb). We identified, however, a large deletion mapping to the last intron and part of exon 15 of MYB (c.2170-1153_2218), encompassing the binding sites of the transcription factors ERG-1 and VDR, a DNase I hypersensitive site and the initial parts of exon 15, including the splice site (supplementary material, Figure S3 and Table S4). Up-regulation of ERG-1 has been shown to result in down-regulation of MYB [18], hence loss of its binding site could have potentially resulted in MYB overexpression. In addition, this large deletion could account for the higher mRNA levels of the 5′ part than the 3′ part of MYB (Figures 3A and 3B). We have also identified an intronic single-nucleotide deletion within intron 1 of MYBL2 (Figure 4C; supplementary material, Table S4), affecting a locus between the promoter and promoter flanking regions (c.20+1403delG). MYBL2, which was highly expressed in AdCC12 (supplementary material, Figure S4) and has been implicated in the biology of different human malignancies by regulating cell cycle and proliferation [19], is the third member of the MYB-family genes, encoding the B-MYB protein. B-MYB, however, is less closely related to either c-MYB or A-MYB than the latter two are related between themselves. Given that the functional impact of non-coding variants has been increasingly recognized in cancer [20], further studies are warranted to investigate whether these MYB and/or MYBL2 alterations would be pathogenic and activate the MYB pathway akin to MYB or MYBL1 rearrangements.
Taken together, we have identified MYBL1 rearrangements and MYB amplification as alternative genetic drivers of MYB-NFIB-negative breast AdCCs. Our analysis corroborates the previous findings of MYB and MYBL1 rearrangements causing overexpression of MYB and MYBL1, respectively, whereas a subset of AdCCs lack MYB/MYBL1 rearrangements but still display relatively high mRNA levels of either gene and activation of similar pathways. Moreover, we demonstrate that not only rearrangements, but also gene amplification may be the genetic alteration resulting in MYB overexpression in breast AdCCs.
Our study has important limitations. The number of cases analyzed is small because breast MYB-NFIB-negative AdCCs are vanishingly rare. Owing to limited amount of tissue, we could only perform a targeted massively parallel sequencing assay in AdCC34, which does not rule out the possibility of co-existing fusion genes. Despite these limitations, our data support the contention that MYB/MYBL1 activation likely constitutes the common mechanism driving breast AdCCs, to which various underlying genomic alterations may converge. Further investigation of larger cohorts of AdCCs lacking the MYB-NFIB fusion gene, MYBL1 rearrangements and MYB gene amplification are warranted to elucidate the drivers of AdCCs lacking these genetic alterations.
Supplementary Material
Figure S1. Histologic features of the MYB-NFIB-negative breast adenoid cystic carcinomas included in this study
Figure S2. c-MYB protein expression in MYB-NFIB-negative adenoid cystic carcinomas as defined by immunohistochemistry
Figure S3. Copy number plot of the MYB gene locus in AdCC12
Figure S4. MYBL2 gene expression in adenoid cystic carcinomas as defined by RNA-sequencing
Table S1. Clinico-pathologic characteristics of four breast adenoid cystic carcinomas included in this study
Table S2. List of antibodies, clones, dilutions, antigen retrieval methods and scoring systems
Table S3. Sequencing statistics of whole-genome sequencing (WGS) and targeted massively parallel sequencing (MSK-IMPACT) analyses performed
Table S4. List of somatic mutations identified by whole-genome sequencing (AdCC11, AdCC12; mutations affecting exonic and selected intronic regions are shown) or by targeted massively parallel sequencing (MSK-IMPACT; AdCC34)
Acknowledgments
Funding: Research reported in this publication was funded in part by the Adenoid Cystic Carcinoma Research Foundation, the Breast Cancer Research Foundation and a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (No P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest: The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
BW, AV-S and JSR-F conceived the study; OM, SB, AV-S and JSR-F provided samples; FCG, FP, NF, ME, SB, AV-S and JSR-F performed histopathologic review; FCG and NF performed tissue microdissection; JK, FCG, LGM, AL, FP, NF and RG-M carried out experiments; CKYN, RSL, PS and RK performed bioinformatics analysis; JK, FCG, LGM, CKYN, PS, ANF, EK, LN, BW and JSR-F discussed and interpreted the results. JK, FCG, JSR-F and BW wrote the first draft. All authors read, edited and approved the final manuscript.
References
* Cited only in supplementary material.
- 1.Marchiò C, Weigelt B, Reis-Filho JS. Adenoid cystic carcinomas of the breast and salivary glands (or ‘The strange case of Dr Jekyll and Mr Hyde’ of exocrine gland carcinomas) J Clin Pathol. 2010;63:220–228. doi: 10.1136/jcp.2009.073908. [DOI] [PubMed] [Google Scholar]
- 2.Fusco N, Geyer FC, De Filippo MR, et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod Pathol. 2016;29:1292–1305. doi: 10.1038/modpathol.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martelotto LG, De Filippo MR, Ng CK, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol. 2015;237:179–189. doi: 10.1002/path.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Persson M, Andren Y, Mark J, et al. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A. 2009;106:18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephens PJ, Davies HR, Mitani Y, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965–2968. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wetterskog D, Lopez-Garcia MA, Lambros MB, et al. Adenoid cystic carcinomas constitute a genomically distinct subgroup of triple-negative and basal-like breast cancers. J Pathol. 2012;226:84–96. doi: 10.1002/path.2974. [DOI] [PubMed] [Google Scholar]
- 8.Drier Y, Cotton MJ, Williamson KE, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48:265–272. doi: 10.1038/ng.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujii K, Murase T, Beppu S, et al. MYB, MYBL1, MYBL2, and NFIB gene alterations and MYC overexpression in salivary gland adenoid cystic carcinoma. Histopathology. 2017 doi: 10.1111/his.13281. [DOI] [PubMed] [Google Scholar]
- 10.Mitani Y, Liu B, Rao PH, et al. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res. 2016;22:725–733. doi: 10.1158/1078-0432.CCR-15-2867-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brayer KJ, Frerich CA, Kang H, et al. Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov. 2016;6:176–187. doi: 10.1158/2159-8290.CD-15-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piscuoglio S, Ng CK, Murray M, et al. Massively parallel sequencing of phyllodes tumours of the breast reveals actionable mutations, and TERT promoter hotspot mutations and TERT gene amplification as likely drivers of progression. J Pathol. 2016;238:508–518. doi: 10.1002/path.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piscuoglio S, Burke KA, Ng CK, et al. Uterine adenosarcomas are mesenchymal neoplasms. J Pathol. 2016;238:381–388. doi: 10.1002/path.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West RB, Kong C, Clarke N, et al. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol. 2011;35:92–99. doi: 10.1097/PAS.0b013e3182002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wysocki PT, Izumchenko E, Meir J, et al. Adenoid cystic carcinoma: emerging role of translocations and gene fusions. Oncotarget. 2016;7:66239–66254. doi: 10.18632/oncotarget.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnaraju K, Hoffman B, Liebermann DA. The zinc finger transcription factor Egr-1 activates macrophage differentiation in M1 myeloblastic leukemia cells. Blood. 1998;92:1957–1966. [PubMed] [Google Scholar]
- 19.Liang HB, Cao Y, Ma Q, et al. MYBL2 is a potential prognostic marker that promotes cell proliferation in gallbladder cancer. Cell Physiol Biochem. 2017;41:2117–2131. doi: 10.1159/000475454. [DOI] [PubMed] [Google Scholar]
- 20*.Rheinbay E, Parasuraman P, Grimsby J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017;547:55–60. doi: 10.1038/nature22992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Sapino A, Sneige N, Eusebi V. Adenoid cystic carcinoma. In: Lakhani SR, Ellis IO, Schnitt SJ, et al., editors. WHO classification of tumours of the breast. 4th. IARC press; Lyon: 2012. pp. 56–57. [Google Scholar]
- 22*.Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19:403–410. doi: 10.1111/j.1365-2559.1991.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 23*.Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–2795. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24*.Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 25*.Piscuoglio S, Ng CK, Murray MP, et al. The genomic landscape of male breast cancers. Clin Cancer Res. 2016;22:4045–4056. doi: 10.1158/1078-0432.CCR-15-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 30*.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Layer RM, Chiang C, Quinlan AR, et al. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Zhang J, White NM, Schmidt HK, et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016;26:108–118. doi: 10.1101/gr.186114.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35*.McPherson A, Hormozdiari F, Zayed A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36*.Shugay M, Ortiz de Mendibil I, Vizmanos JL, et al. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics. 2013;29:2539–2546. doi: 10.1093/bioinformatics/btt445. [DOI] [PubMed] [Google Scholar]
- 37*.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 38*.Tilli TM, Castro Cda S, Tuszynski JA, et al. A strategy to identify housekeeping genes suitable for analysis in breast cancer diseases. BMC Genomics. 2016;17:639. doi: 10.1186/s12864-016-2946-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Histologic features of the MYB-NFIB-negative breast adenoid cystic carcinomas included in this study
Figure S2. c-MYB protein expression in MYB-NFIB-negative adenoid cystic carcinomas as defined by immunohistochemistry
Figure S3. Copy number plot of the MYB gene locus in AdCC12
Figure S4. MYBL2 gene expression in adenoid cystic carcinomas as defined by RNA-sequencing
Table S1. Clinico-pathologic characteristics of four breast adenoid cystic carcinomas included in this study
Table S2. List of antibodies, clones, dilutions, antigen retrieval methods and scoring systems
Table S3. Sequencing statistics of whole-genome sequencing (WGS) and targeted massively parallel sequencing (MSK-IMPACT) analyses performed
Table S4. List of somatic mutations identified by whole-genome sequencing (AdCC11, AdCC12; mutations affecting exonic and selected intronic regions are shown) or by targeted massively parallel sequencing (MSK-IMPACT; AdCC34)