

The most common autosomal recessive spastic paraplegia is caused by mutations in SPG11. Denora et al. report the first postmortem neuropathological analysis of two unrelated patients with SPG11, and demonstrate clinical and pathological overlap between this disease and ALS. Abnormal neuronal lipid accumulation is identified as a hallmark of SPG11.
Keywords: spastic paraplegia 11, spatacsin, amyotrophic lateral sclerosis, lipofuscin, lysosome
The most common autosomal recessive spastic paraplegia is caused by mutations in SPG11. Denora et al. report the first postmortem neuropathological analysis of two unrelated patients with SPG11, and demonstrate clinical and pathological overlap between this disease and ALS. Abnormal neuronal lipid accumulation is identified as a hallmark of SPG11.
Abstract
The most common form of autosomal recessive hereditary spastic paraplegia is caused by mutations in the SPG11/KIAA1840 gene on chromosome 15q. The nature of the vast majority of SPG11 mutations found to date suggests a loss-of-function mechanism of the encoded protein, spatacsin. The SPG11 phenotype is, in most cases, characterized by a progressive spasticity with neuropathy, cognitive impairment and a thin corpus callosum on brain MRI. Full neuropathological characterization has not been reported to date despite the description of >100 SPG11 mutations. We describe here the clinical and pathological features observed in two unrelated females, members of genetically ascertained SPG11 families originating from Belgium and Italy, respectively. We confirm the presence of lesions of motor tracts in medulla oblongata and spinal cord associated with other lesions of the central nervous system. Interestingly, we report for the first time pathological hallmarks of SPG11 in neurons that include intracytoplasmic granular lysosome-like structures mainly in supratentorial areas, and others in subtentorial areas that are partially reminiscent of those observed in amyotrophic lateral sclerosis, such as ubiquitin and p62 aggregates, except that they are never labelled with anti-TDP-43 or anti-cystatin C. The neuropathological overlap with amyotrophic lateral sclerosis, associated with some shared clinical manifestations, opens up new fields of investigation in the physiopathological continuum of motor neuron degeneration.
Introduction
Hereditary spastic paraplegias (HSPs) are neurodegenerative diseases, which include a large spectrum of inherited disorders presenting with lower limb spasticity as the common clinical feature (Harding, 1983). They are usually classified according to their clinical presentation, pattern of inheritance, age at onset and, more recently, their putative physiopathological mechanism (Finsterer et al., 2012; Fink, 2013; Lo Giudice et al., 2014; Tesson et al., 2015). Traditionally, and according to clinical criteria, which remains the most operative for nosographic classification, two forms of the disease are considered: the pure or uncomplicated forms, which represent the most frequent HSPs in European countries, and the complex or complicated forms, which are more frequent in recessively inherited HSP and therefore in countries with a higher rate of consanguinity (Boukhris et al., 2009; Ruano et al., 2014). Besides the spasticity, the latter form is often characterized by one or more additional neurological symptoms, such as peripheral neuropathy, dementia or intellectual disability, cerebellar ataxia, optic atrophy, deafness, retinopathy, etc. (Harding, 1983). The variety of inheritance modes (autosomal dominant, autosomal recessive, mitochondrial and X-linked) and of mutated genes (>70) contributes to further heterogeneity of these neurodegenerative diseases (Tesson et al., 2015).
One of the most common (14 to 26%) autosomal recessive spastic paraplegias (AR-HSP) is caused by mutations in the SPG11/KIAA1840 gene on chromosome 15q (SPG11; MIM 610844) (Stevanin et al., 2007, 2008; Boukhris et al., 2009; Crimella et al., 2009; Denora et al., 2009; Schüle et al., 2009; Coutinho et al., 2013; Ishiura et al., 2014; Pensato et al., 2014). This form is, in most cases, characterized by a combination of progressive spasticity, cognitive impairment, peripheral neuropathy and a thin corpus callosum on brain MRI (Hehr et al., 2007; Stevanin et al., 2008; Denora et al., 2009). Atypical cases of autosomal recessive juvenile amyotrophic lateral sclerosis (ALS) with long disease duration (Orlacchio et al., 2010; Daoud et al., 2012) or with Charcot-Marie-Tooth disease (Montecchiani et al., 2016) have also been found with mutations in SPG11. The nature of the vast majority of SPG11 mutations found to date suggests a mechanism of loss-of-function of the encoded protein, spatacsin. The function of spatacsin is indeed largely unknown but its interaction with AP5Z1 (SPG48) (Słabicki et al., 2010), a member of the adaptor-protein complex 5, suggests that it plays a role in intracellular trafficking (Hirst et al., 2013) and endolysosomal homeostasis in particular (Hirst et al., 2015). An abnormal anterograde trafficking has also been demonstrated in neurons derived from induced pluripotent stem cells of SPG11 patients (Pérez-Branguli et al., 2014). Recently, its requirement for lysosome turnover and autophagy has also been evidenced (Chang et al., 2014; Renvoisé et al., 2014; Varga et al., 2015). Finally, the consequences of its knock-down in zebrafish (Martin et al., 2012; Southgate et al., 2010), together with the early age at onset in most patients, suggest a role for this gene during development of the CNS.
Neuropathological reports of cases with spastic paraplegia are sparse and often without genetic–aetiological considerations. We report here the clinical and pathological description of two unrelated female autopsy cases, members of genetically ascertained SPG11 families originating from Belgium and Italy, respectively. We show that motor neuron degeneration, particularly in spinal cord and medulla oblongata, presents with histological lesions and immunolabelling partially reminiscent of those observed in ALS. This illustrates the overlap between HSPs and ALS and should help to improve our understanding of the physiopathological continuum of motor neuron degeneration. We also show that lysosomal-like granules accumulate in agreement with the potential role of spatacsin.
Materials and methods
Neuropathological studies
Full neuropathological examination was conducted according to classical methods, including frozen and paraffin sections for the Belgian case (Patient BG-2). Only paraffin sections from frontal cortex, basal ganglia, cerebellum, medulla oblongata and cervical spinal cord were available for the Italian case (Patient IT). Brain sampling followed the ethical rules of each country.
Frozen sections were stained for myelin (Spielmeyer), cytology (Nissl), fibrillary glia (Holzer) and neutral fats (Sudan III). Paraffin sections were stained for cytology (Nissl, haematoxylin and eosin, Masson’s trichrome), axons (Bodian silver impregnation), myelin (Klüver-Barrera) and carbohydrate macromolecules (periodic acid-Schiff stain).
Stored formalin-fixed samples of the lumbar spinal cord as well as deparaffinized spinal ganglia were further processed by an additional 24 h fixation in 4% glutaraldehyde for conventional electron microscopy. After post-fixation in 2% osmium tetroxide, blocks were embedded in Araldite®. Ultrathin sections were stained with uranyl acetate and lead citrate and examined under an FEI CM10 Philips transmission electron microscope.
Immunostaining was performed using the citrate buffer epitope retrieval method. Endogenous peroxidase was quenched by incubation for 20 min at room temperature in a phosphate-buffered saline (PBS)/0.1% Triton™ X-100 (Sigma) solution containing 10% methanol and 0.003% H2O2. Microscope slides carrying brain sections were washed three times and incubated in the blocking solution (0.1% PBS, 4% Triton™ X-100, 4% normal goat serum, 2% bovine serum albumin) for 1 h at room temperature. Sections were then incubated for 48 h at 4 °C with specific antibodies (Supplementary Table 1) diluted in blocking solution against the following proteins: cystatin C, ubiquitin, calbindin 28 kD, neurofilaments SMI31 and SMI32, hyperphosphorylated protein tau AT8, amyloid-β17–24 4G8, TDP-43, GFAP, spatacsin, FUS, p62 and cathepsin D. Sections were washed three times, incubated for 2 h at room temperature with the corresponding biotinylated appropriate (anti-mouse or anti-rabbit) secondary antibody (1:250; Vector Laboratories) diluted in blocking solution and washed another three times. Bound antibodies were visualized using the ABC amplification system (Vectastain ABC kit, Vector Laboratories) with 3,3'-diaminobenzidine tetrahydrochloride (DAB Metal Concentrate; Biogenex) as the substrate. The sections, nuclearly counterstained with haematoxylin or not, were dehydrated twice in ethanol and xylene solutions and mounted with Eukitt®.
Neuronal loss, gliosis and atrophy were semiquantitatively assessed and estimated as severe, moderate or absent by two different observers.
Genetic analyses
Mutation screening of Patient IT (Italy) used an amplicon-based Sanger sequencing method focused on the SPG11 gene, as previously reported (Stevanin et al., 2007).
DNA of the autopsy Patient BG-2 (Belgium) was not available but DNA from her affected sister Patient BG-3 (Supplementary Fig. 1) was subjected to whole exome sequencing using the SureSelect 50Mb capture kit (Agilent) followed by massive parallel sequencing in the Illumina HighSeq2000 sequencer. Sequence alignment to the human genome and variant annotation were performed as described elsewhere (Gonzalez et al., 2014).
Results
Clinical outline of Italian and Belgian patients
The Italian proband Patient IT (Supplementary Fig. 1) was female, born by normal full-term delivery, after an uneventful pregnancy. Her parents, of Italian ancestry, were healthy and unrelated. Her initial growth and development were normal. Onset of the disease manifested at the age of 10 years as a learning disability (Table 1). At 12 years of age, she developed a mild motor disability, which progressed to a severe spastic-ataxic gait at age 18 years; walking was possible with one stick. At age 31 years she was wheelchair-bound. Sagittal and axial T1-weighted and coronal T2-weighted brain MRI (Supplementary Fig. 2A and B), performed at age 14, showed corpus callosum thinning, diffuse white matter abnormalities in frontal, occipital, temporal and periventricular regions and atrophy of frontal and occipital brain regions. MRI examination performed at age 24 years (Supplementary Fig. 2C) confirmed the severe evolution of the white matter and cortical abnormalities. Neurological examination at 27 years, when she was wheelchair-bound, showed lower and upper limb spasticity with brisk reflexes as well as extrapyramidal rigidity, cerebellar ataxia in the four limbs, dementia and dysarthria, and distal amyotrophy suggesting axonal peripheral neuropathy. No fasciculations were seen at that time. Electroneuromyographic (ENMG) and nerve conduction velocity recordings performed in the following years revealed slightly reduced amplitudes of compound motor action potentials, while amplitudes of sensory nerve action potentials and motor and sensory nerve conduction velocities (38 m/s in the tibial nerve, 48 m/s in the median nerve) were normal. ENMG of the tibialis anterior muscle showed chronic denervation with long durations and large amplitudes of motor unit action potentials. The patient died severely demented at 32 years of age of cardiopulmonary arrest; a brain autopsy was performed 48 h after death. Focused SPG11 gene analysis was performed by direct sequencing in this patient.
Table 1.
Clinical characteristics of the SPG11 patients
| Patient /gender | Age at motor onset (y) | Age at death (y) | Disease duration (y) | Current age (y) | Intellectual disability (y) | Dementia (y) | Spastic ataxic gait (y) | Epileptic seizures (y) | Bulbar dysphagia (y) | MRI features (Y) | Other clinical features |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ITa,b/F | 10 | 32 | 22 | Deceased | 10 | 27 | 12 | Not recorded | Not recorded | Thin corpus callosum, periventricular white matter abnormalities, fronto- temporal and cerebellar atrophy (14 and 24) | Distal amyotrophy, axonal peripheral neuropathy, cerebellar dysarthria |
| BG-2a/F | 10 | 46 | 36 | Deceased | 10 | <45 | 10 | 20 | 45, PEG tube needed | Diffuse periventricular white matter abnormalities, cerebral atrophy (36) | Cerebellar dysarthria, pes cavus, claw hands, quadriparesis |
| BG-1/M | 18 | Alive | 35 | 53 | 18 | Not recorded | 18 | Not recorded | Not recorded | Not available | Distal amyotrophy, claw hands |
| BG-3b/F | 10 | Alive | 41 | 51 | 10 | Not recorded | 10 | 42 | Not recorded | Thin corpus callosum (25) | Cerebellar dysarthria, saccadic eye movements, pes cavus, distal amyotrophy, claw hands, sporadic generalized epileptic seizures |
Patients BG-1, BG-2 and BG-3 belong to the same sibship. PEG = percutaneous endoscopic gastrostomy.
aAutopsy confirmed patients.
bDNA extraction.
F = female; M = male; y = years.
Proband BG-2 was also female (Supplementary Fig. 1). Her parents of Belgian origin were healthy and related. Onset of the disease, at age 10 years, was characterized by incoordination of all four limbs and intentional tremor; subsequently, a stiff gait was noted. She had mild intellectual disability. She developed a spastic cerebellar ataxic gait that progressed rapidly (Table 1). At age 20 years she was wheelchair-bound. She had sporadic generalized epileptic seizures. The EEG was normal at that time. MRI of the brain was performed at age 36 years. T2-weighted images showed diffuse white matter lesions in periventricular areas and diffuse atrophy of the brain. Neurological examination at age 36 years showed pyramidal signs with brisk reflexes in all four limbs and bilateral Babinski sign, cerebellar ataxia, distal atrophy in all four limbs and bilateral pes cavus. By age 45 years she had complete quadriparesis, claw hands and bulbar palsy with dysphagia and dysarthria. Severe feeding problems arose due to the dysphagia and a percutaneous endoscopic gastrostomy tube was needed. No ENMG examination was performed at this stage of the disease. She developed bradypsychia and dementia. She died at the age of 46 years, severely demented. Brain and spinal cord autopsy was performed 24 h after death.
Patient BG-1, brother of proband Patient BG-2, is still alive at age 53 years and also has mild intellectual disability (Table 1). The disease started at 18 years of age. He developed an ataxic gait and later on, a stiffness in the lower limbs. Distal amyotrophy was seen in all four limbs, with claw hands. By age 29 years, he was wheelchair-bound due to a severe spastic ataxic gait. No dementia and no bulbar palsy were seen in this patient.
Patient BG-3, sister of proband Patient BG-2 is still alive at age 51 years. She also has mild intellectual disability. Onset of the disease was at age 10 years (Table 1). First, she had a wide-based cerebellar ataxia. Later on a spastic gait developed with distal amyotrophy in all limbs. She had pes cavus. She became wheelchair-bound at the age of 20 years. She had dysarthric speech and saccadic eye movements by age 24 years. The distal muscles became severely atrophied and she developed complete bilateral drop-foot and claw hands. Sporadic generalized epileptic seizures were seen. Her EEG was normal. An ENMG examination performed in the upper limbs at age 42 years showed normal motor and sensory nerve conduction velocity responses (49 m/s and 53 m/s in the right ulnar and median nerves) with normal amplitudes of the action potentials. Signs of chronic neurogenic degeneration with large motor units (polyphasic or giant motor unit potentials) were seen in the thenar and hypothenar muscles and in the first interosseous muscles. No clinical signs of bulbar involvement were observed in this patient. Cognitive function remained normal. MRI of the brain performed at age 25 years showed a thin corpus callosum. Whole exome sequence analysis was performed in this patient. Both Belgian parents are deceased.
Genetic analyses
Whole exome sequencing in Patient BG-3 revealed the homozygous truncating mutation c.6739_6742delGAGT (p.Glu2247Leufs*14) in exon 36 of SPG11. This mutation was reported previously in other cases (Stevanin et al., 2008) and was confirmed by Sanger sequencing (Supplementary Fig. 1).
Direct Sanger sequencing of all exons of the SPG11 gene in Patient IT revealed two unreported heterozygous truncating mutations; c.2358_2359delinsTT (p.Glu786_Gly787delinsAspfs*) in exon 13 and c.4868delT (p.Leu1623Tyrfs*17) in exon 28. The two mutations were heterozygous in healthy parents, the c.4868delT being found in the father and the c.2358-2359delinsTT in the mother (Supplementary Fig. 1).
Macroscopic and myelinic modifications of brain autopsies
The autopsy of Patient IT was limited to brain and upper cervical spinal cord. The brain was not weighed, but the examination confirmed the data obtained by MRI, with atrophy of the cerebellum, particularly of the vermis. The corpus callosum was also thin.
The autopsy of Patient BG-2 was also limited to the brain, which weighed 950 g, and to the thoraco-lumbo-sacral spinal cord. Macroscopic examination revealed mild atrophy of the frontal lobe, severe thinning of the corpus callosum, dilatation of the ventricular system and a greyish aspect of the white matter, especially in subcortical areas. There was some depigmentation of the substantia nigra, a slight atrophy of the pons and atrophy of the pyramids in the medulla oblongata as well as a thinning of the spinal cord. There was no cerebellar atrophy but an artificial discoloration of the inner granular cell layer; the so-called état glacé.
Myelin stains on frozen and paraffin sections in the Belgian Patient BG-2 showed a myelin pallor of the white matter in the cerebral hemispheres and a demyelination of the corpus callosum (Supplementary Fig. 2E). There was also demyelination of the pyramidal pathways in the medulla oblongata and the spinal cord (crossed and uncrossed tracts), of the dorsal and ventral spinocerebellar tracts and also of the fasciculus gracilis. Myelin staining of spinal cord of Patient IT (Klüver-Barrera stain alone or associated with Bodian silver impregnation) showed degeneration of crossed and uncrossed pyramidal tracts. The atrophy also involved the dorsal spinocerebellar tracts and the fasciculus gracilis in the posterior columns (Supplementary Fig. 2D).
CNS neuronal loss and astrogliosis
Cytological stains (haematoxylin and eosin, Masson’s trichrome, cresyl violet) revealed neuronal loss in various regions of the CNS in both patients (Table 2). In both cases, the frontal cortex was severely altered, particularly the gyrus cinguli, the frontal gyri, mainly in the third layer and, to a lesser extent, the temporal gyri. Only a few, although shrunken, Betz neurons remained in the precentral gyrus. In the thalamus, focal neuronal loss was severe in the mediodorsal nucleus and the lateral dorsal nucleus in both patients. The caudate and putamen nuclei were only moderately affected in Patient IT and were spared in Patient BG-2. Lesions in the brainstem were heterogeneous. The reticular formation was spared, but moderate (nucleus X and principal olivary nucleus) to severe neuronal loss (hypoglossal, gracilis and cuneate nuclei) was observed in other brainstem regions in the two cases. In Patient BG-2, neuronal loss was evidenced in the ventro-lateral part of the zona compacta of the substantia nigra but not in the pontine locus coeruleus (these structures were not available for Patient IT). The cerebellar cortex was only atrophied in Patient IT with loss of Purkinje and granule cells (Supplementary Fig. 2F). Anti-calbindin 28 kD immunolabelling in Patient IT confirmed the rarefaction of Purkinje cells (data not shown). Deep cerebellar nuclei were not examined in the Italian patient but there was no abnormality in the Belgian patient (data not shown). Finally, the spinal cord was severely affected in the two cases.
Table 2.
Neuropathological characteristics of SPG11 patients
| Brain region |
Neuronal loss
|
Astrocytic reactions
|
||
|---|---|---|---|---|
| Patient IT | Patient BG2 | Patient IT | Patient BG2 | |
| Frontal cortex | Severe | Severe | Severe | Moderate |
| Basal ganglia | ||||
| Caudate | Moderate | Unaffected | Moderate | Unaffected |
| Putamen | Moderate | Unaffected | Moderate | Unaffected |
| Pallidum | Unaffected | Unaffected | Unaffected | Unaffected |
| Thalamus | Severe | Severe (mediodorsal nucleus) | Severe | Severe |
| Brainstem | ||||
| Substantia nigra | NE | Moderate | NE | Moderate |
| Locus coeruleus | NE | Unaffected | NE | Unaffected |
| Pontine nuclei | NE | Unaffected | NE | Unaffected |
| Reticular formation | Unaffected | Unaffected | Unaffected | Unaffected |
| Nucleus cuneatus | Severe | Severe | Moderate | Moderate |
| Nucleus gracilis | Severe | Severe | Moderate | Moderate |
| Nucleus ambiguous | Moderate | Moderate | Moderate | Moderate |
| Nucl XII | Severe | Severe | Moderate | Moderate |
| Inferior olive | Moderate | Moderate | Moderate | Moderate |
| Cerebellum | ||||
| Molecular layer | Severe | Unaffected | Severe | Unaffected |
| Purkinje cells | Severe | Unaffected | Severe | Unaffected |
| Granule cells | Severe | Unaffected | Severe | Unaffected |
| Dentate nucleus | NE | Unaffected | Moderate | Moderate |
| Spinal cord | Severe | Severe | Severe | Severe |
NE = not examined.
Astrocytic reaction was evidenced by Holzer stain in Patient BG-2 and by anti-GFAP immunohistochemistry in two patients (Table 2). A mild fibrillary gliosis in the frontal and temporal white matter and in the middle third of the corpus callosum on a vertico-frontal section was observed in Patient BG-2 while astrogliosis was more severe in Patient IT, particularly in frontal cortex, thalamus, cerebellum and spinal cord, and was moderate in basal ganglia and brainstem.
Neutral fat staining, performed in Patient BG-2 only, in the absence of frozen tissues from Patient IT, was unremarkable.
Cytological anomalies
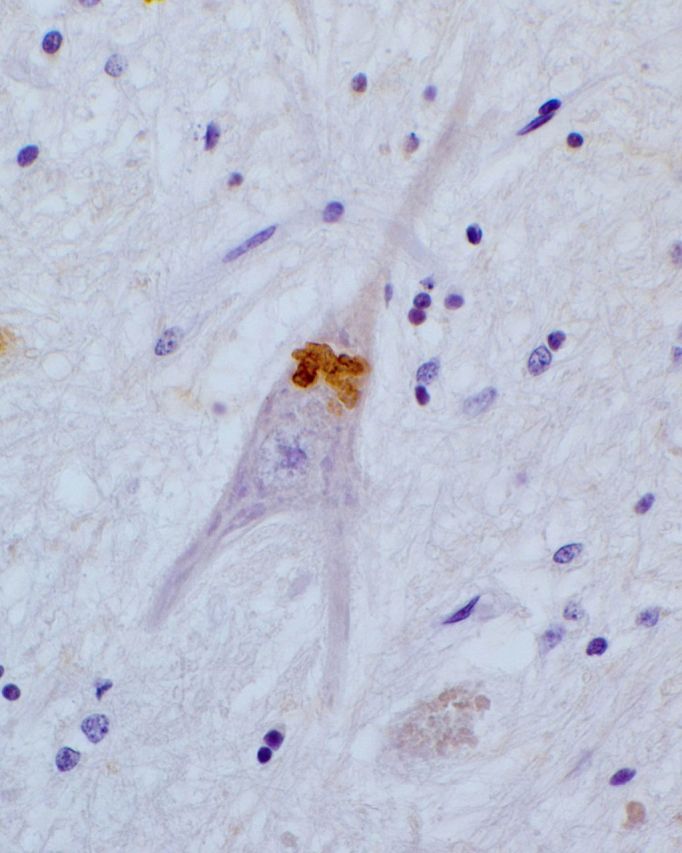
Masson’s trichrome, Sudan black, Klüver-Barrera (Fig. 1B) or Nissl staining revealed abundant lipofuscin granules in several parts of the brain and particularly in cerebral, pontine and medullar neurons of both patients. In addition, granular eosinophilic (Fig. 1A) and PAS+ (periodic acid Schiff) structures different from classical lipofuscin granules by size and polycyclic shape were found in the perikaryon of remaining motor neurons of hypoglossal nucleus and the anterior horns of the spinal cord. Anti-cathepsin D evidenced in both patients abundant granules, reminiscent of lysosomes, with variable shape and size in neurons from several brain regions, more precisely in subtentorial areas (Supplementary Fig. 3A). Ubiquitin (Fig. 1C and Supplementary Fig. 3C) and p62 (Fig. 1D and Supplementary Fig. 3B) immunoreactive coarse granules or beaded inclusions were also found in remaining perikarya of motor neurons in medulla oblongata and spinal cord and in spinal ganglionic cells. The anti-ubiquitin and anti-p62 antibodies did not reveal such structures in cortex, basal ganglia or cerebellum, however none of these inclusions were immunostained by anti-cystatin C (Fig. 1F) or anti-TDP43 (Fig. 1E and Supplementary Fig. 3D).
Figure 1.

Neuronal rarefactions and presence of inclusions in the hypoglossal nucleus of Patient BG-2. Similar features were observed in remaining neurons of the anterior horns of the spinal cord (data not shown). Large eosinophilic inclusions are observed in the neuronal perikarya (arrows) after haematoxylin and eosin (A) or Klüver-Barrera (B, blue) staining. Numerous inclusions (arrows) are immunoreactive for ubiquitin (C) or p62 (D). No labelling of the inclusions was evidenced with an anti-TDP-43 (E) or an anti-cystatin C antibody (F). Scale bars = 50 µm.
Figure 2.

Spinal ganglion of Patient BG-2. Haematoxylin and eosin staining highlighted large, mostly eosinophilic inclusions in the perikarya of the ganglion cell neurons (A, arrows). Similar irregular inclusions were observed in the neuronal perikarya by Klüver-Barrera staining (B, arrows) or by an anti-ubiquitin (C) or and anti-p62 (D) antibody immunolabelling. The inclusions were not labelled by an anti-TDP-43 antibody (E, arrows). Giant bodies or ‘retention bodies’ (arrows) were observed at electron microscopy of a ganglion cell (F, low magnification; G, high magnification of a giant body). Classical lipofuscin granules were also present in these giant bodies (H). Scale bars: immunohistochemistry = 50 µm; electron microscopy = 1 µm.
The labelling against SMI32, in Patient IT, revealed abnormal accumulation of neurofilaments in motor neurons of pontine nuclei and of spinal cord (data not shown). No abnormal immunoreactivity against SMI31 neurofilaments was detected in the two cases, except for a few sparse spheroids and axon swellings in Patient IT.
The mutations of the SPG11 gene described in both patients are expected to lead to the loss-of-function of the spatacsin protein by nonsense-mediated mRNA decay. In agreement with this hypothesis, no immunolabelling with antibodies directed against spatacsin developed and characterized by us (Murmu et al., 2011), was obtained in brain paraffin sections from the present cases (data not shown).
No Lewy bodies, neurofibrillary tangles, senile plaques or intranuclear inclusions were observed either by histological (haematoxylin and eosin, Masson’s trichrome or Bodian silver impregnation) or immunohistochemical staining (phosphorylated alpha-synuclein, hyperphosphorylated tau protein AT8 and amyloid-β 4G8 labelling). In addition, no anti-FUS immunoreactivity was observed, neither in spinal motor neurons nor in the remaining neurons of the hypoglossal nucleus, in contrast to positive anti-FUS inclusions observed in a control case with frontotemporal dementia carrying a FUS mutation (data not shown).
Dorsal root ganglia exploration in Patient BG-2
Ganglion neurons were also affected (such material was not available for Patient IT). We observed, by haematoxylin and eosin staining, eosinophilic inclusions associated with refringent non-eosinophilic droplets (Fig. 2A). These inclusions were also labelled by lipophilic Klüver-Barrera staining (Fig. 2B). Their immunohistochemical characterization showed that they were strongly ubiquitin-positive (Fig. 2C), and some were also p62-labelled (Fig. 2D). As in the case of motor neurons of cortex or spinal cord, they were not stained by anti-TDP-43 (Fig. 2E).
Ultrastructural analysis of neuronal inclusions
Electron microscopy of deparaffinized spinal ganglia neurons of Patient BG-2 (Fig. 2F–H) showed many abnormal electron dense storage materials: few giant bodies or ‘retention bodies’ (Fig. 2F arrows and Fig. 2G) (3–5 µm × 4–8 µm) with closely clustered lipofuscin granules associated with multiple lipid droplets (Fig. 2G) and a variety of heterogeneous aggregates (Fig. 2H).
In formalin-fixed anterior horns, motor neuronal perikarya showed large amounts of lipofuscin granules with granular and osmiophilic amorphous aggregates but very rare small-sized lipid droplets (Fig. 3A). Unusual electron-lucent areas with no limiting membrane were closely intermingled with lipofuscin in some neurons and they contained variable amounts of cross- and longitudinal sectioned fibrillary filaments (Fig. 3B arrows). The latter had a densified and coarse tubular appearance with occasional distortions at higher magnifications (data not shown). A few deposits appeared partly wrapped in lipofuscin granules. These abnormal deposits were not seen in neurons in which lipid droplets appeared numerous and larger, such as usually observed. Finally, similar features were not seen in control spinal cord neurons containing numerous lipofuscin granules and in a proven case of ALS due to a C9orf72 abnormal repeat expansion (data not shown).
Figure 3.

Electron micrographs of a motor neuron of the anterior horn of the lumbosacral spinal cord. (A) Presence of electron-lucent zones (arrows) intermingled with lipofuscin. The area indicated by the horizontal arrow in (A) is shown at a higher magnification in (B, arrows).
Additionally, no intranuclear inclusions were seen in neurons and glial cells of spinal anterior horn and ganglia specimens.
Discussion
We report here the first neuropathological description of two patients with HSP from genetically proven SPG11 families. The two cases share similar lesions, including cortical atrophy, white matter involvement with myelin pallor and severe astrogliosis. They also share a demyelination of the pyramidal pathways and loss of motor neurons in medulla oblongata and anterior horns of spinal cord. The thalamus of the two cases was also affected, especially the mediodorsal nucleus of Patient BG-2.
In contrast, there were some differences in severity of the lesions between the two cases in the basal ganglia and cerebellum (Table 2). The cerebellar cortex of Patient IT was severely affected after 22 years of disease duration (onset at age 10 years), while it was spared in Patient BG-2 after 36 years of disease duration (onset also at age 10 years) in spite of a cerebellar syndrome in both patients. Patient BG-2 showed moderate gliosis of dentate nucleus that could however account for the observed cerebellar ataxia. Why the involvement of specific neuronal populations and the severity of the lesions varied between the two cases remain an unresolved question. Other genetic or environmental factors might have played a role.
Other clinicopathological studies of HSP cases have been reported, but without genetic investigations. More particularly, two reported cases share similar clinical and neuropathological characteristics with ours (Wakabayashi et al., 2001; Kuru et al., 2005). After 37 years of disease duration, the first patient had a slowly progressive spastic tetraplegia with mental deterioration and severe amyotrophy. Marked cerebral atrophy with thin corpus callosum was shown by brain MRI. The second patient had spastic paraplegia, mental retardation and amyotrophy with sensory disturbances after a disease duration of 26 years. Interestingly, while the cerebellum was atrophied in the first case, it was normal in the second case, which mimics the heterogeneity also found in our two SPG11 patients. A severe thinning of the corpus callosum was present in both autopsies. In these two published cases, the remaining neurons also presented abundant eosinophilic and lipofuscin granules, which were partially identified as anti-ubiquitin positive in one case (Wakabayashi et al., 2001), but were not fully characterized. As between our two cases, some differences between the two Japanese cases themselves could result from the circumstances of death or different time in post-mortem examination. In addition, we cannot exclude different genetic conditions in the two Japanese cases as the SPG11 gene was not tested.
Overlapping findings between our SPG11 cases and ALS
Our SPG11 patients partially shared various clinical and neuropathological features of ALS.
The neuropathological features, especially observed in subtentorial areas, namely medulla oblongata and spinal cord, including the dorsal columns, are reminiscent of findings already observed in ALS, thus emphasizing a putative physiological link between SPG11 mutations and some forms of ALS. In agreement with this observation, SPG11 mutations were recently reported in autosomal recessive juvenile ALS with unusual long disease durations (Orlacchio et al., 2010; Daoud et al., 2012). The brainstem and spinal cord autopsy of one of these juvenile ALS patients mutated in SPG11 showed minor lesions after 33 years of disease duration (Orlacchio et al., 2010). Neuronal loss and astrogliosis were limited to glossopharyngeal and hypoglossal nuclei and to the anterior horn of the spinal cord, and a myelin pallor was reported in the anterolateral columns. The remaining motor neurons of the spinal cord showed central chromatolysis, pigmentary degeneration and hyaline inclusions that were not characterized using immunohistochemistry markers. Neither Bunina bodies nor skein-like inclusions were observed.
In our SPG11 cases, Bunina bodies were also not evidenced by the anti-cystatin-C antibody. Peculiar observations were, however, found in our patients; the supratentorial structures—namely frontotemporal cortex, basal ganglia and the thalamic mediodorsalis nucleus—showed neuronal degeneration without the immunohistochemical landmark of ALS pathology as we did not observe ubiquitin, p62, or TDP-43 immunoreactivity either in basal ganglia or in the neocortex. In medulla oblongata and spinal cord, however, ubiquitin and p62-positive granules were found. Indeed, the accumulation and/or aggregation of neurofilaments in motor neuron perikarya and axons (axonal spheroids) together with the abnormal topographic location of neurofilaments in the cell body are classical features associated with familial and sporadic ALS (Delisle and Carpenter, 1984; Xiao et al., 2006).
Moreover, a clinical overlap between complicated HSP and ALS was overt in Patient BG-2. In the end stage of her disease, there was a severe bulbar palsy in addition to severe cerebellar dysarthria. There were severe feeding problems and a percutaneous endoscopic gastrostomy tube was needed. There was an evolution to a proximal and distal amyotrophy and a spastic quadriparesis. All this suggests a clinical evolution toward a complicated form of HSP with involvement of the lower motor neuron and a more severe ALS-like phenotype. However, fasciculations were seen at that time and no ENMG was performed at that stage, which could have supported the diagnosis of ALS.
SPG11 pathology includes abnormal aggregation and lipid storage
Lipofuscin-positive granules were abnormally abundant in supratentorial neurons of our SPG11 patients and in the anterior horn associated with granular osmiophilic amorphous aggregates. Interestingly, granular intracytoplasmic eosinophilic/PAS+ structures were exclusively found in motor neurons in spinal cord and hypoglossal nucleus. All these deposits were restricted to neurons and were labelled with anti-cathepsin-D as lysosomal-like structures in supratentorial areas, and by anti-ubiquitin and anti-p62 in spinal cord neurons and ganglion cells. Although we showed, by electron microscopy, unusual fibrillary filaments intermingled with lipofuscin deposits, no relationship could be established with the immunostained inclusions in the absence of immuno-electron microscopy.
The abundance of pigment granules in neurons seems to represent a cytopathological hallmark of SPG11. Recent studies showed that SPG11 and SPG15 proteins interact with the late endosomal/lysosomal adaptor protein complex AP-5 (Hirst et al., 2013) and their involvement in this pathway is consistent with the abnormal lysosomal storage evidenced in fibroblasts from SPG11, SPG15 and SPG48 patients (Renvoisé et al., 20014; Hirst et al., 2015). Very recently, an Spg11 knockout mouse model was reported to recapitulate the major clinical features of the human disease (Varga et al., 2015). Briefly, the Spg11 knockout mice developed symptoms reminiscent to spastic paraplegia with progressive loss of cortical motor neurons and Purkinje cells, as we observed in the human pathology (present study). Degenerating neurons accumulated autofluorescent material, which was dark and osmiophilic at the electron microscopy level, consistent with the accumulations observed in our human brain samples. The neuronal inclusions in the knockout mouse model were partially immunostained with anti-p62 and with the anti-lysosomal membrane protein LAMP1, supporting the observation of lysosome/autophagy defects. In addition, morphological analysis of neurons from Spg15 knockout mice also showed accumulation of large intraneuronal deposits of membrane-surrounded material, which co-stained with the lysosomal marker LAMP1 (Khundadze et al., 2013). Although differences exist between human and mouse brains, the aformentioned observations indicate that the invalidation of Spg11 in the mouse mimics the pathology we observed in SPG11 patients and this is relevant for future studies aimed at identifying potential therapeutic avenues.
Taken together, these results argue in favour of a lysosomal impairment in SPG11 and SPG15. The abnormal deposits observed in our SPG11 cases are also reminiscent of other pathological hallmarks of motor neuron diseases associated with mutations in other genes. Motor neuron inclusions (e.g. granular lesions, ubiquitin, TDP-43, and/or p62 positive structures) may represent a pivotal feature of these diseases. TDP-43 accumulation is observed in patients with mutations in GRN (progranulin), C9orf72, DCTN1 (dynactin), OPTN (optineurin), VCP (valosin-containing protein), ANG (angiogenin) (Da Cruz and Cleveland, 2011), but not in our SPG11 cases. On the other hand, TDP-43 proteinopathy with widespread spinal cord and cerebral skein-like and round neuronal cytoplasmic inclusions has recently been observed in patients with NIPA1 (SPG6) mutation associated with ALS clinical features (Martinez-Lage et al., 2012). Regarding p62 deposits, the evidence of their involvement in neurodegenerative diseases is growing (Zatloukal et al., 2002). The p62 protein, after mono- or poly-ubiquitination, is associated with autophagy (Rubinsztein et al., 2012). The cytological association between LC3 and p62 markers in neuronal inclusions may indicate autophagic degradation of misfolded proteins. Inhibition of autophagy and increasing levels of p62 are generally correlated and p62 itself is degraded by autophagy (Zatloukal et al., 2002; Sasaki, 2011; Martinez-Lage et al., 2012). In Lewy body disease, it was shown that p62 deposition precedes ubiquitination (Kuusisto et al., 2003) but p62, ubiquitin and TDP-43 deposition kinetics remains unknown; further biochemical studies and cellular and animal model analyses are needed to elucidate these kinetics. In our SPG11 patients, the deposition of p62 and ubiquitination were only observed in subtentorial neurons and dorsal root ganglia. The differences in composition of the aggregates according to neuronal populations remain unanswered and we can hypothesize that the immunohistochemical discrepancy between p62 and TDP-43 deposits observed in the present SPG11 cases and as reported by others (King et al., 2011) could be explained by two different hypotheses. Different kinetics of deposition of these proteins may vary according to the subpopulations of neurons. Alternatively, SPG11 could represent a subtype of motor neuron pathology in which TDP-43 does not aggregate, as it has been observed in the human pathology and in a mouse model of ALS/frontotemporal lobar degeneration due to multivesicular body protein (CHMP2B) gene mutation (Ghazi-Noori et al., 2012).
In conclusion, this study, performed in two distinct sibships, is the first report of brain and spinal cord autopsy in complex forms of hereditary spastic paraplegia with SPG11 mutations. We demonstrate the neuropathological link between HSP and ALS in terms of neurodegeneration topology and show for the first time the existence of abnormal accumulations in neurons of SPG11, a disorder that can then be added to the growing list of diseases associated with aggregates. Further studies, using animal and cellular models may determine if the aggregation of proteins and lysosomal abnormalities with abnormal lipidic deposits may precede axonal dying back, which was proposed as a pathological mechanism associated with HSP (Behan and Maia 1974; Deluca et al., 2004).
Supplementary Material
Acknowledgements
The contribution of the members of the different laboratories is gratefully acknowledged with a special mention for Mrs Inge Bats for the photographic work-up.
Glossary
Abbreviations
- ALS
amyotrophic lateral sclerosis
- HSP
hereditary spastic paraplegias
Funding
This work was supported by the Agence Nationale de la Recherche (to G.S.), the Verum Foundation (to A.B. and G.S.), the Roger de Spoelberch Foundation (to A.B.), the ERA-Net for Research Programmes on Rare Diseases (E-rare “Neurolipid”, to G.S.), the European Union with the European Research Council (ERC, Starting grant No. 311149 to F.D.) and the Seventh Framework Programme (FP7, Omics call, to A.B.). This study benefited from the programme ‘Investissements d’avenir’ ANR-10-IAIHU-06 and ANR-11-INBS-0011-NeurATRIS (Translational Research Infrastructure for Biotherapies in Neurosciences).
Supplementary material
Supplementary material is available at Brain online.
References
- Behan WM, Maia M. Strümpell’s familial spastic paraplegia: genetics and neuropathology. J Neurol Neurosurg Psychiatry 1974; 37: 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhris A, Stevanin G, Feki I, Denora P, Elleuch N, Miladi MI, et al. Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 2009; 75: 527–36. [DOI] [PubMed] [Google Scholar]
- Chang J, Lee S, Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest 2014; 124: 5249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P, Ruano L, Loureiro JL, Cruz VT, Barros J, Tuna A, et al. Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 2013; 70: 746–55. [DOI] [PubMed] [Google Scholar]
- Crimella C, Arnoldi A, Crippa F, Mostacciuolo ML, Boaretto F, Sironi M, et al. Point mutations and a large intragenic deletion in SPG11 in complicated spastic paraplegia without thin corpus callosum. J Med Genet 2009; 46: 345–51. [DOI] [PubMed] [Google Scholar]
- Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol 2011; 21: 904–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A, et al. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging 2012; 33: 5–9. [DOI] [PubMed] [Google Scholar]
- Delisle MB, Carpenter S. Neurofibrillary axonal swellings and amyotrophic lateral sclerosis. J Neurol Sci 1984; 63: 241–50. [DOI] [PubMed] [Google Scholar]
- Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol 2004; 30: 576–84. [DOI] [PubMed] [Google Scholar]
- Denora PS, Schlesinger D, Casali C, Kok F, Tessa A, Boukhris A, et al. Screening of ARHSP-TCC patients expands the spectrum of SPG11 mutations and includes a large scale gene deletion. Hum Mutat 2009; 30: 500–19. [DOI] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 2013; 126: 307–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J, Löscher W, Quasthoff S, Wanschiitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegia with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurosci Neurol 2012; 318: 1–18. [DOI] [PubMed] [Google Scholar]
- Ghazi-Noori S, Froud KE, Mizielinska S, Powell C, Smidak M, Fernandez de Marco M, et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 2012; 137: 819–32. [DOI] [PubMed] [Google Scholar]
- Gonzalez G, Koyanagi KO, Aoki K, Kitaichi N, Ohn S, Kaneko H, et al. Intertypic modular exchanges of genomic segment by homologous recombination at universally conserved segments in human adenovirus species D. Gene 2014; 547: 10–17. [DOI] [PubMed] [Google Scholar]
- Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet 1983; 1: 1151–5. [DOI] [PubMed] [Google Scholar]
- Hehr U, Bauer P, Winner B, Schule R, Olmez A, Koehler W, et al. Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol 2007; 62: 656–65. [DOI] [PubMed] [Google Scholar]
- Hirst J, Borner GHH, Edgar J, Hein MY, Mann M, Buchholz F, et al. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell 2013; 24: 2558–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Edgar JR, Esteves T, Darios F, Madeo M, Chang J, et al. Loss of AP-5 results in accumulation of aberrant endolysosomes: defining a new type of lysosomal storage disease. Hum Mol Genet 2015; 24: 4984–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura H, Takahashi Y, Hayashi T, Saito K, Furuya H, Watanabe M, et al. Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses. J Hum Genet 2014; 59: 163–72. [DOI] [PubMed] [Google Scholar]
- Khundadze M, Kollmann K, Koch N, Biskup C, Nietzsche S, Zimmer G, et al. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet 2013; 10e1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A, Maekawa S, Bodi I, Troakes C, Al-Sarraj S. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathol 2011; 31: 239–49. [DOI] [PubMed] [Google Scholar]
- Kuru S, Sakai M, Konagaya M, Yoshida M, Hashizume Y. Autopsy case of hereditary spastic paraplegia with thin corpus callosum showing severe gliosis in the cerebral white matter. Neuropathol 2005; 25: 346–52. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol 2003; 62: 1241–53. [DOI] [PubMed] [Google Scholar]
- Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014; 261: 518–39. [DOI] [PubMed] [Google Scholar]
- Martin E, Yanicostas C, Rastetter A, Naini SM, Maouedj A, Kabashi E, et al. Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol Dis 2012; 48: 299–308. [DOI] [PubMed] [Google Scholar]
- Martinez-Lage M, Molina-Porcel L, Falcone D, McCluskey L, Lee VM-Y, Van Deerlin VM, et al. TDP-43 pathology in a case of hereditary spastic paraplegia with a NIPA1/SPG6 mutation. Acta Neuropathol 2012; 124: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecchiani C, Pedace L, Lo Giudice T, Casella A, Mearini M, Gaudiello F, et al. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain 2016; 139: 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murmu RP, Martin E, Rastetter A, Esteves T, Muriel M-P, El Hachimi KH, et al. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol Cell Neurosci 2011; 47: 191–202. [DOI] [PubMed] [Google Scholar]
- Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S, et al. Spatacsin mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010; 133: 591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 2014; 137: 1907–20. [DOI] [PubMed] [Google Scholar]
- Pérez-Brangulí F, Mishra HK, Prots I, Havlicek S, Kohl Z, Saul D, et al. Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum Mol Genet 2014; 18: 4859–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoisé B, Chang J, Singh R, Yonekawa S, Fitz Gibbon EJ, Mankodi A, et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol 2014; 1: 379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano L, Melo MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014; 3: 174–84. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol CB 2012; 22: R29–34. [DOI] [PubMed] [Google Scholar]
- Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2011; 70: 349–59. [DOI] [PubMed] [Google Scholar]
- Schüle R, Schlipf N, Synofzik M, Klebe S, Klimpe S, Hehr U, et al. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry 2009; 80: 1402–4. ) [DOI] [PubMed] [Google Scholar]
- Słabicki M, Theis M, Krastev DB, Samsonov S, Mundwiller E, Junqueira M, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol 2010; 10: e1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southgate L, Dafou D, Hoyle J, Li N, Kinning E, Critchley P, et al. Novel SPG11 mutations in Asian kindreds and disruption of spatacsin function in the zebrafish. Neurogenetics 2010; 11: 379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008; 131: 772–84. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 2007; 39: 366–72. [DOI] [PubMed] [Google Scholar]
- Tesson C, Koht J, Stevanin G. Delving into the complexity of hereditary spastic paraplegias; how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum Genet 2015; 134: 511–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, et al. In Vivo evidence for lysosome depletion and impared autophagic clearance in hereditary spastic paraplegia. PLoS Genet 2015; 10: e1005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Kobayashi H, Kawasaki S, Kondo H, Takahashi H. Autosomal recessive spastic paraplegia with hypoplastic corpus callosum, multisystem degeneration and ubiquitinated eosinophilic granules. Acta Neuropathol 2001; 101: 69–73. [DOI] [PubMed] [Google Scholar]
- Xiao S, McLean J, Robertson J. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys 2006; 1762: 1001–12. [DOI] [PubMed] [Google Scholar]
- Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, et al. p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002; 160: 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.