

Abstract
Epigenetic mechanisms provide an adaptive layer of control in the regulation of gene expression that enables an organism to adjust to a changing environment. Epigenetic regulation increases the functional complexity of deoxyribonucleic acid (DNA) by altering chromatin structure, nuclear organization, and transcript stability. These changes may additively or synergistically influence gene expression and result in long-term molecular and functional consequences independent of the DNA sequence that may ultimately define an individual’s phenotype. This paper (1) describes histone modification, DNA methylation, and expression of small noncoding ribonucleic acid (RNA) species; (2) reviews the most common methods used to measure these epigenetic changes; and (3) presents factors that need to be considered when choosing a specific tissue to evaluate for epigenetic changes.
Keywords: epigenetics, DNA methylation, histone modification, small untranslated RNA
The consensus sequence of the human genome was published 10 years ago, and information on variation within disease susceptibility genes has increased. Genome search methods (e.g., candidate gene association studies, family-based studies, genome-wide association studies) have uncovered many genetic variants associated with a trait or disease phenotype. However, for any given study, any combination of these variants accounts for only a relatively small proportion of the total phenotypic variance observed in a sample. Therefore, alternative sources of phenotypic variation in heritable traits must exist.
Epigenetics (epi meaning above; that is, above the level of the DNA) is defined as changes in phenotype or gene expression caused by a mechanism other than alterations in the underlying DNA sequence (Berger, Kouzarides, Shiekhattar, & Shilatifard, 2009). These changes may remain for the duration of the cell’s life, persist through cell division, and be transmitted in a specific cell lineage for multiple generations. Epigenetics provides an adaptive layer of control in the regulation of gene expression to enable the organism to adjust to a changing environment. This additional layer of control is influenced by developmental stage, tissue type, environmental conditions, and disease status of the individual (De Bustos et al., 2009; Keshet et al., 2006; Mikkelsen et al., 2007). Epigenetic mechanisms can alter chromatin structure, nuclear organization, and transcript stability. These changes, alone or in combination, influence gene expression and result in long-term molecular and functional consequences.
For example, in a seminal article by Weaver et al. (2004), maternal care of newborn rats was associated with epigenetic changes that resulted in altered stress reactivity in adult offspring. Inbred newborn rats reared by mothers who provided higher rates of licking and grooming and arched-back nursing behaviors displayed fewer fear behaviors than pups reared by mothers with lower rates of these maternal care behaviors. This association persisted after pups were cross-fostered to adoptive mothers with different maternal care styles and suggested that the variation in maternal care style was responsible for individual differences in the long-term stress responses of these pups. Subsequent investigation found that maternal care differentially altered the expression of the glucocorticoid receptor gene involved in the stress response pathway. Epigenetic mechanisms, specifically the methylation of the promoter region of this gene and acetylation of histone proteins, altered the ability of a transcription factor to bind to the promoter that resulted in decreases in gene expression.
In addition to critical developmental periods, epigenetic alterations may accumulate throughout the individual’s lifetime. Recent studies have investigated the relationship between aberrant epigenetic events on health and predisposition to chronic diseases such as cancer. Cancer development is characterized by the progressive loss of normal physiologic regulation of cellular proliferation which results in uncontrolled cell growth. One of the most recognized epigenetic states that promotes oncogenesis is the inactivation of tumor suppressor genes by hypermethylation of CpG islands located in the promoter of the gene (Rodríguez-Paredes & Esteller, 2011). For example, the Breast Cancer 1 (BRCA1) gene encodes the breast cancer type 1 susceptibility protein that participates in DNA repair (Friedenson, 2007). An increase of DNA methylation of the CpG island located in the promoter region of BRCA1 prevents expression of BRCA1 (Esteller et al., 2000). As a result, damaged DNA is not properly repaired which increases the risk of cancer. While cancer has been the focus of the majority of epigenetic studies, epigenetic alterations have also been described in other diseases including asthma (Ober & Vercelli, 2011), autism (Nguyen, Rauch, Pfeifer, & Hu, 2010), and diabetic neuropathy (Bell et al., 2010).
As the field of epigenetics matures, linkages will emerge between environmentally induced epigenetic changes and health and disease. These associations may prove useful clinically as biomarkers to establish disease risk, monitor disease progression and guide interventions (Rodriguez-Paredes & Esteller, 2011). For example, cancer-specific hypermethylation of CpG dinucleotides in saliva has been associated with oral cancer and may be useful as a tool to predict the incidence of oral cancer (Viet & Schmidt, 2008).
Future studies may identify associations that contribute to interindividual variability in the severity of patients’ symptoms. Epigenetic processes may program cells for alterations in biological pathways in advance of the development of a clinically significant phenotype. Therefore, understanding how to measure these epigenetic processes will assist in the elucidation of mechanisms that produce specific phenotypic changes and identify therapeutic targets.
Any evaluation of epigenetic changes should be interpreted within the context of other genomic analyses because it is the interaction between genetic variations and epigenetic regulatory events that may ultimately define an individual’s phenotype. The purpose of this paper is to describe three of the best characterized forms of epigenetic regulation: histone modification, DNA methylation, and noncoding ribonucleic acid (RNA) expression. In addition, we describe the most common methods used to measure these epigenetic changes. Finally, we discuss the factors to consider when choosing a specific tissue to evaluate for epigenetic changes.
Histone Modifications
Histone modification is defined as a posttranslational modification to one of the amino acid side chains in a histone protein. Epigenetic regulation of chromatin structure through histone modifications enables cells to alter gene expression by modifying a transcription factor’s ability to access DNA (Felsenfeld & Groudine, 2003). Almost every human cell contains approximately 6.4 billion nucleotides divided among 46 chromosomes (i.e., one pair of 22 autosomes, two sex chromosomes). As a linear structure, DNA is too long to fit within the nucleus of a cell. As illustrated in Figure 1, a precise combination of proteins produces hierarchical levels of protein–DNA structures that are responsible for organizing, stabilizing, and compacting DNA within the nucleus (Luger & Hansen, 2005).
Figure 1.
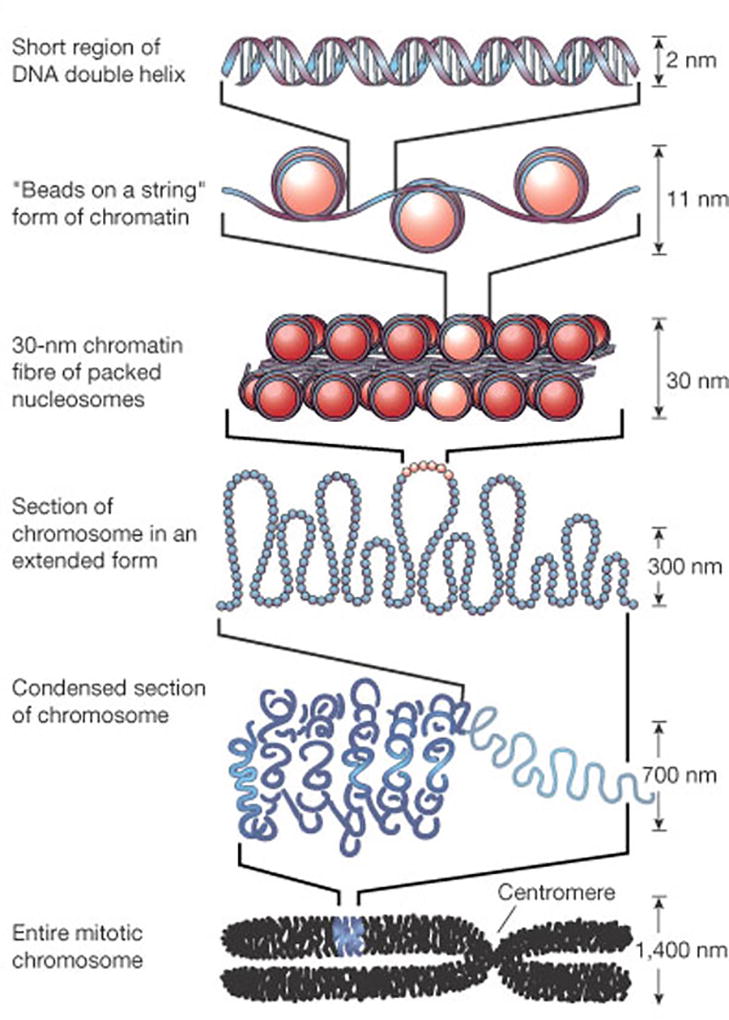
Chromatin organization. Chromatin is arranged into nucleosomes as its first level of organization. A nucleosome is made up of histone proteins and double-stranded deoxyribonucleic acid (DNA) that wraps around a histone octomer 1.7 times in a left-handed coil. Hydrogen bonds hold the phosphodiester backbone of DNA to the amino acid side chains of the histones. Each nucleosome is separated from the next by a short segment of DNA. The conformation resembles a “beads-on-a-string” structure. Further condensation of chromatin structure is facilitated by histone “tails” that extend from the nucleosome and help to stack nucleosomes into a more compact chromatin structure by linking them to neighboring nucleosomes. These circular stacks of nucleosomes compact further to form a solenoid that results in a chromatin fiber that measures 30 nanometers in diameter. As the final chromatin structure, chromosomes are thought to be the result of a mesh formed through fibers connected by cross-linked proteins or as a result of hierarchical packaging of chromatin around a central axis mediated by structural maintenance of chromosome proteins. Reprinted by permission from Macmillan Publishers Ltd: Felsenfeld & Groudine, 2003.
In a low-ordered structure, chromatin is organized into discrete units called nucleosomes. A nucleosome is made up of eight histone proteins and a 150-nucleotide sequence of DNA. The N-terminal regions of the histones extend from the nucleosome and are referred to as histone tails. These histone tails, composed of amino acids, are subject to a variety of posttranslational modifications (Figure 2; Mersfelder & Parthun, 2006). The functional consequences of these modifications include changes in gene transcription, DNA repair, DNA replication, and chromatin condensation through alterations in the chemical properties and physical conformation of histones (Kouzarides, 2007).
Figure 2.
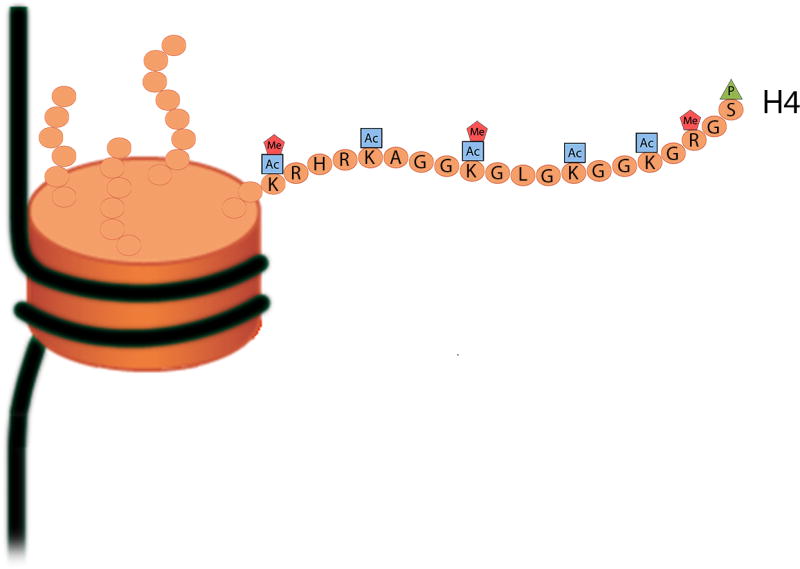
Posttranslational modifications of the N-terminal region of histone H4. The majority of the known histone modifications affect the amino acids that make up the unstructured N-terminal region of the histone core protein. Covalent modifications of the human H4 histone protein include phosphorylation (P), acetylation (Ac), and methylation (Me). Single letter abbreviations for amino acid residues: A = alanine; G = glycine; H = histidine; K = lysine; L = leucine; R = arginine; S = serine.
The functional consequence of any single modification depends on the type of modification (e.g., acetylation, phosphorylation) and the site at which it occurs (e.g., amino acid, location in the histone tail). Modifications may act individually, sequentially, and/or in combination to define a “histone code.” This histone code can modify access to specific DNA sequences and produce distinct regulatory events (Jenuwein & Allis, 2001). An individual modification may exclude the possibility of other modifications occurring in the same location. Histone modifications can influence the binding of proteins to sites on adjacent amino acid side chains (Fischle et al., 2005) or alter an enzyme’s ability to recognize a neighboring site (Clements et al., 2003).
Histone modifications are thought to function through two different mechanisms. First, covalent modification of amino acid side chains may result in either nucleosome–nucleosome interactions or alterations in the contact between the histone and DNA (Grewal & Moazed, 2003). For example, one of the best-studied histone modifications is acetylation of lysine (Shogren-Knaak et al., 2006). Histone tails contain several lysine residues. Lysine has an aliphatic backbone with a terminal amino group. At physiologic pH, lysine has a positive charge that is strongly attracted to negatively charged DNA. These ionic qualities favor the winding of DNA around histones, which results in a condensed chromatin structure. When histones are acetylated by acetyltransferases, the positive charge is removed from lysine and DNA loosens from the histone. Histone deacetylases remove the acetyl group from lysine, which restores the positive charge and promotes a closed chromatin structure.
Second, histone modifications may actively recruit or repel nonhistone gene regulatory proteins (Bhaumik, Smith, & Shilatifard, 2007). Specialized structural domains within these recruited proteins recognize histone modifications (Higgs, Vernimmen, Hughes, & Gibbons, 2007) and enable these proteins to preferentially associate with modified histone tails. Some modifications may inhibit binding of these proteins, promoting a closed chromatin structure, which represses transcription (Jacobs & Khorasanizadeh, 2002; Margueron, Trojer, & Reinberg, 2005).
Various regions of the eukaryotic genome assume plastic and dynamic chromatin structures. The extent of compaction dictates the expression pattern of the genes contained within a specific segment of DNA. Regions of highly condensed chromatin contain genes that are refractory to expression. As chromatin structures become less condensed, the binding sites for transcriptional proteins are more accessible and the genes within these regions can be transcribed (Hu, Kireev, Plutz, Ashourian, & Belmont, 2009). Histone modifications are dynamic events that occur rapidly in response to cellular signaling and alter compaction of a specific region of DNA. The status of modifications both in the 5’ regulatory region and throughout the length of the gene alters access of DNA-binding proteins to DNA. As a result, gene expression varies from silent to active depending on the amount and types of histone modifications that are present (Barski et al., 2007).
Measurement of Histone Modifications
Chromatin immunoprecipitation (ChIP)-based methods are used to measure the presence of histone modifications in specific gene regions (Park, 2009). First, cells from a tissue sample are treated with an agent, such as formaldehyde, to cross-link DNA to nearby histone proteins so that the nucleotide sequence associated with the protein can be determined. Then, cells are lysed and chromatin is broken into small fragments through sonication. These fragments consist of short lengths of DNA and covalently bound proteins. Antibodies for a specific protein of interest (e.g., acetylated histone core protein X) are added and bind to their respective epitopes. The Fc region of the antibody is used to precipitate the protein–DNA fragment complex out of solution. Once the antibody–protein–DNA complexes are isolated, the cross-linking is reversed and the proteins are separated from the DNA. The DNA is assayed with sequencing (ChIP-Seq) or microarray (ChIP-Chip) technologies to determine the nucleotide sequence where the protein was bound.
Any modification to histone proteins can be examined with ChIP as long as an antibody is available. When ChIP is combined with microarray technology, the entire genome can be interrogated for a specific histone modification in a DNA–protein interaction. However, the quality of the data depends on several factors including the specificity and affinity of the antibody used and the effectiveness of the precipitation reaction. Many histone modifications do not have an antibody available. In addition, chromatin proteins may block an antibody’s access to histone modifications, which would produce false negative findings. Though beyond the scope of this review, several variations of ChIP were developed to overcome many of these limitations (Mito, Henikoff, & Henikoff, 2005; O'Neill & Turner, 2003; van Steensel, Delrow, & Henikoff, 2001).
DNA Methylation
DNA methylation refers to the covalent addition of a methyl group to one of the DNA nucleotides. DNA methylation influences the ability of transcription factors and other DNA-binding proteins to recognize a nucleotide sequence that regulates gene expression. This process is associated with repression of gene transcription (Jones & Baylin, 2007). While DNA methylation is believed to occur in all organisms, the specific nucleotide and the position of the nucleotide that becomes methylated differs among species (Hattman, 2005). In mammals, methylation occurs predominantly on cytosine at the carbon 5 position of the pyrimidine ring to form 5-methylcytosine (Ehrlich & Wang, 1981).
DNA methylation is an essential process for normal development and cellular differentiation (Robertson, 2005). All nucleated cells within an organism contain DNA that is identical in sequence. DNA methylation enables cells to differentiate and maintain lineage by either downregulating or turning off promoters for genes that are not to be expressed by particular tissues. As these cells replicate, DNA methylases enable cells to retain their differentiated identity by transmitting their DNA methylation pattern during mitosis.
DNA methylation plays a number of roles in promoting genome stability and maintenance including genomic imprinting, X-chromosome inactivation, and suppression of repetitive elements (Avner & Heard, 2001; Wilkins, 2005). Changes in the methylation patterns of DNA during a cell’s lifetime provide an adaptive ability for the organism to adjust to changes in the environment. In addition, deregulation of DNA methylation processes is associated with several diseases including the development of cancer (Jones & Baylin, 2007). For example, compared to normal somatic cells, cancer cells are relatively hypomethylated (Robertson, 2005). Loss of normal DNA methylation results in genomic instability and decreased growth controls which permit adaptation and metastasis of tumor cells.
In humans, methylation occurs on the cytosine nucleotide when it lies immediately 5’ to a guanine nucleotide. Since phosphate groups link nucleotides within the DNA molecule, the cytosine–guanine sequence is commonly referred to as the CpG dinucleotide. The CpG sequence is paired to the GpC sequence on the complementary strand and methylation is symmetrical. Therefore, cytosine paired with a guanine on the opposite strand is also methylated.
CpG dinucleotides are aggregated in the promoter region of many genes and are referred to as CpG islands. These CpG islands are defined as sequences comprising greater than 50% guanine and cytosine with a ratio of CpG to GpC on the same strand of at least 0.6 (Gardiner-Garden & Frommer, 1987). A common form of DNA damage is the hydrolysis of the cytosine nucleotide. Hydrolysis of cytosine results in the mutation of cytosine to uracil and the release of ammonia (i.e., deamination of cytosine). Both methylated and unmethylated cytosines are subject to spontaneous deamination under physiologic conditions. Deamination of unmethylated cytosine to uracil is repaired accurately by DNA repair mechanisms, leaving the CpG dinucleotide intact. However, deamination of methylated cytosine results in the creation of thymine, which DNA repair mechanisms do not recognize. Over time, this DNA damage-and-repair cycle appears to have resulted in a nonrandom decreased prevalence of the CpG dinucleotide in the human genome. The CpG dinucleotides that have resisted mutation are positioned in regions of DNA where a change in sequence would be deleterious to the organism. For example, within the promoter region of a gene, mutation of a cytosine may affect recognition by sequence-specific transcription factors. Altered expression patterns of that gene may change the organism’s phenotype and fitness for survival.
If DNA methylation occurs in the promoter region, gene expression is decreased (Egger, Liang, Aparicio, & Jones, 2004). Conversely, active gene transcription occurs in the absence of DNA methylation (Egger et al., 2004; Tazi & Bird, 1990). DNA methylation represses gene expression through two mechanisms (Singal & Ginder, 1999). First, methylation of regulatory regions of a gene may impede transcription factors from physically binding to the DNA (Hark et al., 2000). Transcription factors bind to specific sequences of DNA in response to extracellular signals and internal programs. Following binding, transcription machinery is recruited to the site and the gene is expressed at a higher rate. When DNA methylation occurs, gene expression is repressed because transcription cannot be initiated.
Second, when cytosines within the promoter region are methylated, methyl-CpG-binding proteins have a greater affinity for the promoter sequence than transcription factors. Binding of the methyl-CpG-binding proteins to CpGs within the promoter physically impedes the binding of transcription factors so the gene cannot be expressed. In addition, the methyl-CpG-binding proteins recruit additional proteins, which bind to each other and form a complex (Bird, 2002; Jin, Jiang, Rauch, Li, & Pfeifer, 2005). These complexes have histone deacetylase activity and alter the structure of chromatin into a closed conformation (Boyes & Bird, 1991). Therefore, transcription factors are not able to gain access to the promoter region, and transcription of that gene is repressed.
Measurement of DNA Methylation Status
The common approaches used to evaluate DNA methylation are high-performance liquid chromatography (HPLC), bisulfite sequencing, and CpG island microarrays. HPLC measures the total amount of methylation present in the DNA. Bisulfite sequencing and CpG island microarrays allow for locus-specific assessment of methylation (Laird, 2010).
High-Performance Liquid Chromatography (HPLC)
HPLC is one of the most common methods used to separate proteins and small molecules and is a form of column chromatography (Snyder & Dolan, 2006). In addition to collecting fractions enriched for specific molecules, HPLC generates a chromatogram that measures the number of molecules in a given fraction. This quantitative property enables HPLC to determine the total amount of methylated cytosine in a DNA sample (Kuo, McCune, Gehrke, Midgett, & Ehrlich, 1980). In order to use HPLC to quantify the level of methylated cytosine, DNA must be denatured and digested into single nucleotides (Ramsahoye, 2002). Then, HPLC is used to separate nucleotides based on size. The height of each peak on the chromatogram corresponds to the amount of that nucleotide present in the genomic DNA. In this way, HPLC provides a high-throughput measure of total DNA methylation in a sample and can be used to determine differences in methylation between cell types or tissues. However, since the DNA is fully digested into nucleotides, HPLC cannot reveal the location of methylation in the genome. Therefore, other methods are required to determine the methylation status of specific genes or alleles.
Locus-specific Methods for Detection of DNA Methylation
Hybridization- and sequencing-based applications are used to reveal the location of methylated and unmethylated cytosines at a specific locus following methylation-dependent changes to the DNA. The methyl group of 5-methylcytosine is located within the major groove of the DNA backbone. Therefore, it does not interfere with base pairing. As a result, traditional hybridization-based molecular biology techniques are not adequate to evaluate the methylation status of cytosines. Current DNA methylation detection techniques rely on the introduction of methylation-dependent changes to the DNA sequence. These techniques include immunoprecipitation, digestion with restriction endonucleases, and sodium bisulfite treatment. The gold standard for assaying DNA methylation within a specific locus is bisulfite sequencing (Clark, Harrison, Paul, & Frommer, 1994; Frommer et al., 1992).
Bisulfite sequencing
Bisulfite sequencing provides accurate data regarding 5-methylcytosine content at the single nucleotide level of resolution. Through this process, sodium bisulfite is used to convert unmethylated cytosine to uracil (Hayatsu, Wataya, Kai, & Iida, 1970). Methylated cytosine is protected from conversion and remains cytosine. Then the region of interest can be amplified by polymerase chain reaction (PCR) and sequenced through conventional means. The locations of all methylated cytosines can then be determined because they are the only loci where cytosines remain. Following bisulfite treatment, the two DNA strands are no longer complementary. Therefore, one pair of primers specific for bisulfite-converted DNA must be designed for each strand that will be sequenced.
PCR primers for bisulfite-converted DNA templates must be designed carefully to achieve accurate results (Li & Dahiya, 2002). Following PCR amplification, uracil is amplified as thymine. Methylated cytosines are amplified as unmethylated cytosines. Primers should not include CpG dinucleotides, and thymine must be substituted in place of all cytosines that are not located within a CpG dinucleotide. If CpGs are included within the primer sequence, methylation of the cytosine may interfere with hybridization to the target sequence, introducing measurement bias.
Bisulfite sequencing has several limitations (Laird, 2010). The success of this method is dependent on complete conversion of unmethylated cytosine into uracil. Incomplete conversion may produce false positive results if the unconverted nucleotides are located within the region of interest. Because most cytosine will be converted to thymine during PCR, the complexity of the DNA sequence is reduced (i.e., DNA is comprised of three nucleotides instead of four). Therefore, optimizing PCR may be more difficult than in nonbisulfite-converted DNA samples. Additionally, prolonged incubation with sodium bisulfite may degrade DNA, and single nucleotide polymorphisms within the region of interest may complicate interpretation of the results.
Furthermore, there are several limitations related to the current state of bioinformatics (Laird, 2010). The current generation of sequencing technology limits the length of the region that can be examined. Sequencing generates large data files for short reads. Therefore, extensive computing resources and bioinformatics tools are required to align and analyze data on the genomic scale. Data processing and analysis are complicated due to the need for a bisulfite-converted reference genome. As new sequencing technologies are developed, locus-specific amplification may not be required and whole-genome coverage will become feasible.
CpG island microarray
In order to interrogate larger areas of DNA including genome-wide coverage and overcome many of the limitations of bisulfite sequencing, investigators routinely use CpG island microarrays. Microarrays provide high-throughput assessment of methylation status of up to hundreds of thousands of CpG sites in parallel using hybridization (Figure 3; 2002). For a detailed review on CpG island microarray see the paper by Gitan, Shi, Chen, Yan, & Huang (2002).
Figure 3.

Following amplification of the region of interest by polymerase chain reaction (PCR), array-based methods rely on hybridization of a sample of deoxyribonucleic acid (DNA) to oligonucleotides contained within the array. Sodium bisulfite treatment of DNA produces a change in the nucleotide sequence of all unmethylated cytosines. Amplification of a specific locus of interest is performed, and a “tag” is incorporated into the PCR products using modified PCR primers that feature one of two unique sequence tails (i.e., one primer detects the methylated state and has one specific tag, while the other primer detects the unmethylated state and has a different tag). This procedure results in fluorescently labeled PCR products with a nucleotide sequence specific to its methylation status. A set of two oligonucleotides is synthetically made with their sequences complementary to the unmethylated and methylated sequence of the labeled PCR products and is attached to the surface of a glass slide. These immobilized oligonucleotides serve as a target for the labeled PCR products and are referred to as probes. The labeled PCR products are incubated with an array of immobilized probes to permit complementary sequences to hybridize to a given probe. A high-resolution camera captures the position of emitted fluorescence. The difference in signal intensities between the paired methylated and unmethylated alleles is used to calculate the percentage of methylation for the sequence associated with the probe. Modified from “Methylation-specific oligonucleotide microarray: A new potential for high-throughput methylation analysis,” by R. S. Gitan, H. Shi, C.-M. Chen, P. S. Yan, & T. H.-M. Huang, Genome Research, 12, p. 159. Copyright 2002 by Cold Springs Harbor Laboratory Press.
Advantages of microarray analysis are that only small amounts of genomic DNA are required, and whole genome coverage is possible. The widespread availability of software programs to analyze and interpret array data renders this method more feasible than large amounts of sequencing output. However, compared to sequencing, microarrays have lower resolution, and the areas of the genome that need to be examined must be known so specific probes can be developed. Designing highly specific probes is critical to the success of this technology. Imperfect base-pairing between the probe and target sequences produce erroneous results (Gitan et al., 2002). In addition, interrogation of areas contained within repetitive sequences is limited by the ability to develop probes that are unique to the specific locus of interest.
Small RNA Expression
The role of RNA as an intermediate carrier of genetic information is well established. Noncoding RNAs were initially believed to consist of ribosomal RNAs and transfer RNAs that are dedicated to protein synthesis. However, the development of specialized methods to detect and quantify small RNAs has led to the discovery of novel species. In the mammalian genome, three classes of small, noncoding RNAs are known to exist (i.e., microRNA [miRNA], endogenous small-interfering RNA [endo-siRNA], and piwi-interacting RNA [piRNA]) (Kim, Han, & Siomi, 2009). These classes of RNAs are believed to regulate gene expression posttranscriptionally by targeting messenger RNAs and silencing transposons through heterochromatin formation. These small RNAs are generally defined by nucleotide length, biogenesis, and their associated proteins (Kim, Han, & Siomi, 2009). Of note, these newly discovered species of noncoding RNAs are conceptualized to function in an epigenetic manner. However, the inclusion of transient gene expression as an epigenetic mechanism is under debate.
While the role of endo-siRNAs in physiologic processes is unknown, current evidence suggests that miRNAs play critical roles in the development of the organism (Ambros, 2003; Chen, Li, Lodish, & Bartel, 2004) and in cell differentiation (Calin et al., 2002; Calin et al., 2004). In addition, miRNAs are associated with oncogenesis (Calin et al., 2002, 2004) and human disease (Perera & Ray, 2007). Evidence suggests that piRNAs suppress gene expression by influencing the activity of methyltransferases (Aravin et al., 2008) and alter DNA methylation patterns in a sequence-specific manner (Watanabe et al., 2011). Additional information on small noncoding RNAs can be found in a recent review by Wery, Kwapisz, and Morillon (2011).
Due to the potential impact of tissue-specific and temporal expression of miRNA genes, highly sensitive methods that are capable of assessing large numbers of genes are required to verify the roles of miRNAs identified using in silico approaches. The most common methods used to measure small noncoding RNA are quantitative real-time polymerase chain reaction (qPCR), microarrays, and sequencing.
Choice of Cell Type for Evaluation of Epigenetic Regulation
All nucleated cells within an organism contain identical copies of DNA. However, different cell types display a variety of structural and functional differences related to differences in gene expression that define their phenotype. Epigenetic mechanisms regulate gene expression transcriptionally and posttranscriptionally. The dynamic cell-type specific changes that occur through epigenetic mechanisms during development and in disease states must be considered when using any of the methods described previously.
Many of the tissues of interest for epigenetic profiling (e.g., cerebral spinal fluid, cardiac muscle) require invasive procedures that limit their accessibility for clinical investigations. Epigenetic profiles in surrogate tissues can be used to assess and monitor epigenetic status in more remote tissues. Peripheral blood contains several types of circulating cells, including peripheral blood mononuclear cells (PBMCs), that are involved in or exposed to molecular signals produced by disease processes in remote tissues. Importantly, peripheral blood samples are easy and inexpensive to obtain.
An assessment of global epigenetic expression within PBMCs may identify a set of genes that are sensitive to the trait/condition of interest. These expression patterns provide an “epigenetic signature” that can be used to measure changes over time. These signatures may identify novel mechanistic pathways involved in the pathophysiology of a condition. PBMCs have been used to evaluate epigenetic changes in a variety of conditions including heart failure (Voellenkle et al., 2010) and transplant rejection (Anglicheau et al., 2009). These studies underscore the central role of neuroimmunomodulation in these disease states. The sensitivity of PBMCs as mediators of inflammatory processes may help investigators to identify additional biomarkers that are capable of determining changes in health status.
Conclusions
Epigenetic regulation increases the functional complexity of DNA by providing mechanisms for modifying cellular processes. A comprehensive understanding of health and disease involves an evaluation of both the DNA sequence and epigenetic modulation of gene expression. Several forms of epigenetic regulation exist, and a variety of methods can be employed to characterize them. The choice of method depends on a variety of factors including the number of samples that will be analyzed, the amount of DNA available, the nature of the samples, and the resources available. The progressive development of improved techniques and the refinement of current methods will enable a more comprehensive understanding of additional types of epigenetic regulation and their interactions with genetic variation.
Epigenetic control mechanisms may additively or synergistically interact to precisely respond to internal and external environmental cues. Determining the hierarchical relationships involved in epigenetic regulation and identifying mechanisms that are differentially expressed in different cell types will increase our understanding of gene expression and protein regulation. Comparative studies of epigenetic mechanisms involved in physiologic processes may reveal transcriptional and translational changes in genes that can be used to develop biomarkers and therapeutic targets for specific diseases. In combination with genomics, an improved understanding of epigenetic regulation of gene expression may provide a more comprehensive understanding of human health and the trajectories of various diseases.
Acknowledgments
Kimberly Stephens is supported by an institutional training grant (T32 NR07088) from the National Institute of Nursing Research. Dr. Aouizerat was funded through the National Institutes of Health Roadmap for Medical Research Grant (K12RR023262). Drs. Miaskowski, Aouizerat, and Levine are supported by grants from the National Institutes of Health. Dr. Miaskowski is supported by a grant from the American Cancer Society.
Contributor Information
Kimberly E. Stephens, Department of Physiological Nursing, University of California, San Francisco, 2 Koret Way, Box 0610, San Francisco, CA 94143, (415) 476-6089 – phone, (415) 476-8899 - fax, Kimberly.stephens@ucsf.edu.
Christine A. Miaskowski, Department of Physiological Nursing, University of California, San Francisco, Chris.miaskowski@nursing.ucsf.edu.
Jon D. Levine, Department of Oral and Maxillofacial Surgery, University of California, San Francisco, Jon.levine@ucsf.edu.
Clive R. Pullinger, Department of Physiological Nursing and Cardiovascular Research Institute, University of California, San Francisco, Clive.pullinger@ucsf.edu.
Bradley E. Aouizerat, Department of Physiological Nursing and Institute for Human Genetics, University of California, San Francisco, Bradley.aouizerat@nursing.ucsf.edu.
References
- Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113(6):673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Anglicheau D, Sharma VK, Ding R, Hummel A, Snopkowski C, Dadhania D, Suthanthiran M. MicroRNA expression profiles predictive of human renal allograft status. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(13):5330–5335. doi: 10.1073/pnas.0813121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Hannon GJ. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Molecular Cell. 2008;31(6):785–799. doi: 10.1016/j.molcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nature Reviews Genetics. 2001;2(1):59–67. doi: 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Medical Genomics. 2010;3:33. doi: 10.1186/1755-8794-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes & Development. 2009;23(7):781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nature Structural & Molecular Biology. 2007;14(11):1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes & Development. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 1991;64(6):1123–1134. doi: 10.1016/0092-8674(91)90267-3. [DOI] [PubMed] [Google Scholar]
- Brower V. Epigenetics: Unravelling the cancer code. Nature. 2011;471:S12–S13. doi: 10.1038/471S12a. [DOI] [PubMed] [Google Scholar]
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Croce CM. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(24):15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-Z, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303(5654):83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Research. 1994;22(15):2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements A, Poux AN, Lo W-S, Pillus L, Berger SL, Marmorstein R. Structural basis for histone and phosphohistone binding by the GCN5 histone acetyltransferase. Molecular Cell. 2003;12(2):461–473. doi: 10.1016/s1097-2765(03)00288-0. [DOI] [PubMed] [Google Scholar]
- de Bustos C, Ramos E, Young JM, Tran RK, Menzel U, Langford CF, Trask BJ. Tissue-specific variation in DNA methylation levels along human chromosome 1. Epigenetics & Chromatin. 2009;2(7):1–21. doi: 10.1186/1756-8935-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Wang RY. 5-Methylcytosine in eukaryotic DNA. Science. 1981;212(4501):1350–1357. doi: 10.1126/science.6262918. [DOI] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. Journal of the National Institutes of Cancer. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421(6921):448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438(7071):1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- Friedenson B. The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer. 2007;7:152. doi: 10.1186/1471-2407-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(5):1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. Journal of Molecular Biology. 1987;196(2):261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gitan RS, Shi H, Chen C-M, Yan PS, Huang TH-M. Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome Research. 2002;12(1):158–164. doi: 10.1101/gr.202801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SIS, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301(5634):798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405(6785):486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- Hattman S. DNA-[adenine] methylation in lower eukaryotes. Biochemistry. 2005;70(5):550–558. doi: 10.1007/s10541-005-0148-6. [DOI] [PubMed] [Google Scholar]
- Hayatsu H, Wataya Y, Kai K, Iida S. Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. Biochemistry. 1970;9(14):2858–2865. doi: 10.1021/bi00816a016. [DOI] [PubMed] [Google Scholar]
- Higgs DR, Vernimmen D, Hughes J, Gibbons R. Using genomics to study how chromatin influences gene expression. Annual Review of Genomics and Human Genetics. 2007;8:299–325. doi: 10.1146/annurev.genom.8.080706.092323. [DOI] [PubMed] [Google Scholar]
- Hu Y, Kireev I, Plutz M, Ashourian N, Belmont AS. Large-scale chromatin structure of inducible genes: transcription on a condensed, linear template. The Journal of Cell Biology. 2009;185(1):87–100. doi: 10.1083/jcb.200809196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295(5562):2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jin S-G, Jiang C-L, Rauch T, Li H, Pfeifer GP. MBD3L2 interacts with MBD3 and components of the NuRD complex and can oppose MBD2-MeCP1-mediated methylation silencing. The Journal of Biological Chemistry. 2005;280(13):12700–12709. doi: 10.1074/jbc.M413492200. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet I, Schlesinger Y, Farkash S, Rand E, Hecht M, Segal E, Simon I. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nature Genetics. 2006;38(2):149–153. doi: 10.1038/ng1719. [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nature Reviews Molecular Cell Biology. 2009;10(2):126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kuo KC, McCune RA, Gehrke CW, Midgett R, Ehrlich M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Research. 1980;8(20):4763–4776. doi: 10.1093/nar/8.20.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nature Reviews Genetics. 2010;11(3):191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- Li L-C, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, Schrump DS. Epigenomic alterations and gene expression profiles in respiratory epithelial exposed to cigarette smoke condensate. Oncogene. 2010;29:3650–3664. doi: 10.1038/onc.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger Karolin, Hansen JC. Nucleosome and chromatin fiber dynamics. Current Opinion in Structural Biology. 2005;15(2):188–196. doi: 10.1016/j.sbi.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code. Current Opinion in Genetics & Development. 2005;15(2):163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Mersfelder EL, Parthun MR. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Research. 2006;34(9):2653–2662. doi: 10.1093/nar/gkl338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nature Genetics. 2005;37(10):1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- Nguyen A, Rauch TA, Pfeifer GP, Hu VW. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB Journal. 2010;24:3036–3051. doi: 10.1096/fj.10-154484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C, Vercelli D. Gene-environment interactions in human disease: nuisance or opportunity? Trends in Genetics. 2011;27:107–115. doi: 10.1016/j.tig.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LP, Turner BM. Immunoprecipitation of native chromatin: NChIP. Methods. 2003;31(1):76–82. doi: 10.1016/s1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nature Reviews Genetics. 2009;10(10):669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RJ, Ray A. MicroRNAs in the search for understanding human diseases. BioDrugs. 2007;21(2):97–104. doi: 10.2165/00063030-200721020-00004. [DOI] [PubMed] [Google Scholar]
- Ramsahoye BH. Measurement of genome wide DNA methylation by reversed-phase high-performance liquid chromatography. Methods. 2002;27(2):156–161. doi: 10.1016/s1046-2023(02)00069-5. [DOI] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation and human disease. Nature Reviews Genetics. 2005;6(8):597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nature Medicine. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- Singal R, Ginder GD. DNA methylation. Blood. 1999;93(12):4059–4070. [PubMed] [Google Scholar]
- Snyder LR, Dolan JW. High-Performance Gradient Elution: The Practical Application of the Linear-Solvent-Strength Model. New York: Wiley-Interscience; 2006. [Google Scholar]
- Tazi J, Bird A. Alternative chromatin structure at CpG islands. Cell. 1990;60(6):909–920. doi: 10.1016/0092-8674(90)90339-g. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Delrow J, Henikoff S. Chromatin profiling using targeted DNA adenine methyltransferase. Nature Genetics. 2001;27(3):304–308. doi: 10.1038/85871. [DOI] [PubMed] [Google Scholar]
- Viet CT, Schmidt BL. Methylation array analysis of preoperative and postoperative saliva DNA in oral cancer patients. Cancer Epidemiology, Biomarkers & Prevention. 2008;17:3603–3611. doi: 10.1158/1055-9965.EPI-08-0507. [DOI] [PubMed] [Google Scholar]
- Voellenkle C, van Rooij J, Cappuzzello C, Greco S, Arcelli D, Di Vito L, Martelli F. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiological Genomics. 2010;42(3):420–426. doi: 10.1152/physiolgenomics.00211.2009. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tomizawa SI, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, Sasaki H. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science. 2011;332(6031):848–852. doi: 10.1126/science.1203919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver ICG, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Meaney MJ. Epigenetic programming by maternal behavior. Nature Neuroscience. 2004;7(8):847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Wery M, Kwapisz M, Morillon A. Noncoding RNAs in gene regulation. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2011 doi: 10.1002/wsbm.148. [DOI] [PubMed] [Google Scholar]
- Wilkins JF. Genomic imprinting and methylation: epigenetic canalization and conflict. Trends in Genetics. 2005;21(6):356–365. doi: 10.1016/j.tig.2005.04.005. [DOI] [PubMed] [Google Scholar]