

Abstract
Genome-scale human protein–protein interaction networks are critical to understanding cell biology and interpreting genomic data, but challenging to produce experimentally. Through data integration and quality control, we provide a scored human protein–protein interaction network (InWeb_InBioMap, or InWeb_IM) with severalfold more interactions (>500,000) and better functional biological relevance than comparable resources. We illustrate that InWeb_InBioMap enables functional interpretation of >4,700 cancer genomes and genes involved in autism.
Since the turn of the millennium, it has become increasingly feasible to experimentally map large-scale protein–protein interaction networks (that is, hundreds of proteins systematically tested for thousands of interactions in a single study). Despite the unquestionable importance of these efforts in humans, the most recent screens have only produced on the order of <30,000 new direct interactions (for example, refs. 1,2; reviewed in ref. 3), representing only 4–22% of the most conservative estimates of the human interactome4,5.
Integration of protein–protein interaction data between heterogeneous databases and different organisms, and from fundamentally different types of interaction experiments, is not straightforward. Nonetheless, it has been consistently demonstrated that robust computational integration of many different data sets not only improves coverage, but also can lead to very high accuracy when the resulting inferred protein networks are tested experimentally or against repositories of well-established interactions (reviewed in ref. 3). This is in part because the different experimental large-scale methods complement each other so that no single approach captures the full spectrum of stable and transient interactions between proteins relevant to cell biology6. Importantly, the high value of integrated protein networks for the interpretation of vast genomic data sets is illustrated by the onslaught of exome-sequencing projects and genome-wide association studies that have used integrated protein–protein interaction data to reveal non-obvious molecular pathways perturbed by somatic mutations in cancers as well as germline common and rare genomic variation in metabolic, psychiatric and immune-mediated diseases (reviewed in ref. 3 and exemplified in refs. 7–13).
We devised a general computational framework to exploit, leverage and complement ongoing experimental interaction studies and to provide a systematically integrated human protein–protein interaction network for the annotation of genomic and genetic data sets (details can be seen in Online Methods, Supplementary Fig. 1 and Supplementary Table 1). Through thorough quality control of the raw data, we created an integrated human protein–protein interaction network named InWeb_InBioMap (hereafter InWeb_IM).
InWeb_IM consisted of 585,843 interactions (Fig. 1 and Supplementary Table 1) at the time of submission of the paper (625,641 at the time of publication), aggregated from 8 source databases and spanning 87% of reviewed human UniProt IDs. We applied a stringent orthology majority-voting scheme to map interactions from other organisms. Specifically, we use eight different orthology databases and transfer data to human protein pairs only if the majority of these databases agree on the phylogenetic relationship between protein pairs in the model organism and humans (see Online Methods and Supplementary Note 1). Importantly, 57% of the total data come directly from experiments with human proteins, 68% from either mouse or human, and 95% from human, mouse, rat, cow, nematode, fly, or yeast (Fig. 1e).
Figure 1.
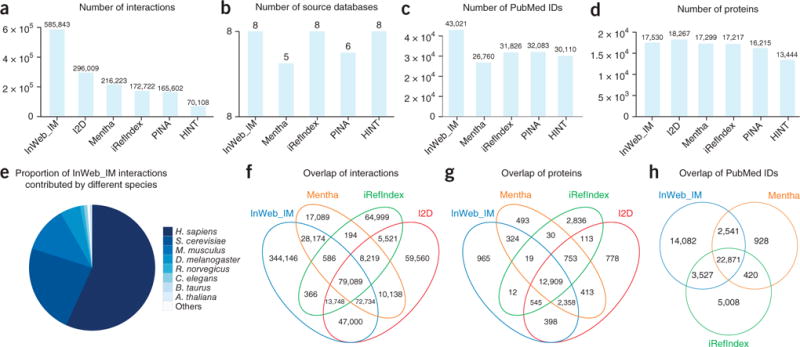
A quantitative comparison of InWeb_IM and five widely used human protein–protein interaction networks. (a–d) Shown are comparisons of the total amount of interactions (a), the number of source databases (b), the number of PubMed identification numbers (c) and the number of proteins (determined using the UniProt reference proteome; d) covered by at least one interaction for each of the six networks. (e) The proportion of InWeb_IM interactions that have been found in humans. (f–h) In the four networks containing the most data, we also measured the overlap of interactions (f), the overlap of proteins covered by at least one interaction (g) and the overlap of publications supporting the interaction data (h) (I2D was excluded in c and h because PubMed identification numbers could not be traced).
In InWeb_IM, interactions are given an initial score based on a number of metrics, most notably the reproducibility of the interaction data between different publications (see Online Methods, Supplementary Fig. 1 and Supplementary Note 2). To validate the initial score, we defined a highly trusted (‘gold-standard’) set of protein–protein interactions from pathway databases (see Online Methods and Supplementary Note 2). We ranked the non-pathway-database-derived interactions on the basis of initial score and plotted a curve of the enrichment of the gold-standard interactions as a function of the rank based on the initial score. This analysis suggests that the initial score indeed up-prioritizes the gold-standard interactions, as the curve is well above the diagonal and its slope is steeper than the diagonal for the non-pathway-database-derived interactions that rank in the top 30% (Fig. 2a, Online Methods and Supplementary Note 2). To give the score a probabilistic interpretation, we then calibrated the initial score against the gold-standard interactions to transform it into a lower bound of the true positive rate of interactions with that initial score or better (Fig. 2b, Online Methods and Supplementary Note 2). We confirmed that this final confidence score correlates with an experimentally determined measure of the confidence of interactions between proteins in an independent experiment of 58 human immunoprecipitations14 (Fig. 2c, Online Methods and Supplementary Note 3) with a statistically robust correlation (of 0.39, confidence interval (CI) [0.35, 0.42]). We repeated this analysis for the only other two networks that assign scores to the interactions, and we found a comparable correlation in iRefIndex (0.41, CI [0.38, 0.44]) and a lower correlation in Mentha (0.21, CI [0.15, 0.26]), where both correlations are statistically significant. Together these analyses confirm the reliability of our score and show that it is significantly correlated with an experimentally derived measure of the interaction confidence between proteins.
Figure 2.
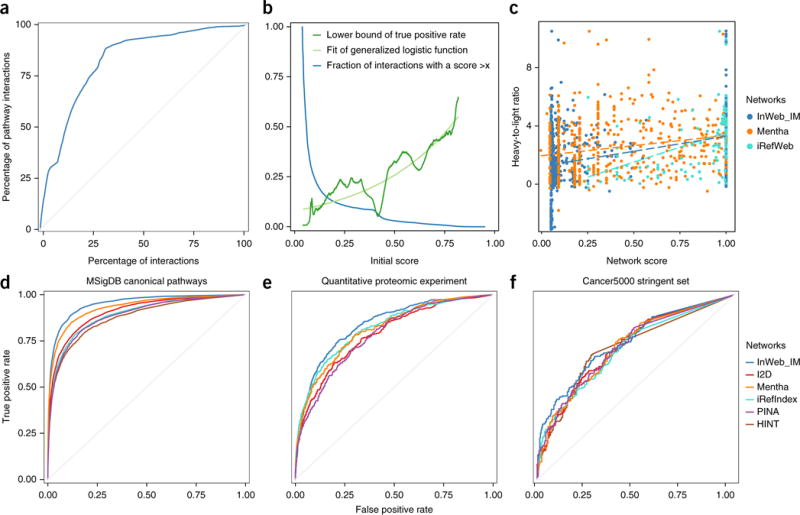
Validating the InWeb_IM score and comparing its biological signal to those of five other networks. (a) A plot of the cumulative fraction of the gold-standard set (pathway-database-derived interactions) as a function of the rank based on the initial score (normalized to percentages) (AUC = 0.83). (b) Calibration of the initial score against the gold-standard interactions (dark green line for initial data points and light green line for the fitted general logistic function; the blue line shows the fraction of interactions with scores higher than the values indicated on the x axis.). (c) Correlation of confidence scores from InWeb_IM, Mentha, and iRefIndex and experimental values of the confidence of binding between proteins (i.e., the heavy-to-light isotope ratios from mass spectrometry data of 58 independent human immunoprecipitations). (d) Cross-validation to test the ability of the six networks to recapitulate pathway relationships between genes in 853 MSigDB canonical pathways through a 30% holdout analysis. InWeb_IM AUC = 0.95; Mentha AUC = 0.93; I2D AUC = 0.91; iRefIndex and PINA AUCs = 0.89; and HINT AUC = 0.88. (e) Agreement between interactions reported in each network with quantitative protein–protein interactions from 58 immunoprecipitations in human cells. InWeb_IM AUC = 0.84; iRefIndex AUC = 0.82; Mentha AUC = 0.81; I2D AUC = 0.79; and PINA AUC = 0.78. (f) Ability of networks to classify 219 cancer genes. InWeb_IM AUC = 0.74; I2D, Mentha, and PINA AUCs = 0.72; and HINT and iRefIndex AUCs = 0.71.
We compared InWeb_IM to five similarly constructed and widely used human protein interaction networks (I2D, Mentha, iRefIndex, PINA and HINT15–20) using a number of quantitative and qualitative metrics (Fig. 1a–h; details of other networks are provided in Online Methods and Supplementary Note 4). All networks (including InWeb_IM) were downloaded on the same date (November 2015, as close to the submission of the article as analytically possible). In terms of absolute number of protein–protein interactions, InWeb_IM has twice as many interactions as I2D, the next-largest network, and 2.8 times the median for all networks (Fig. 1a). We draw our data from 34.1% more publications than PINA, the network with the next best coverage in terms of source articles (Fig. 1c). Considering the total number of proteins that are implicated in one or more interactions, InWeb_IM has 4% fewer than I2D, but more than the other four networks (Fig. 1d). While two resources (Mentha and I2D) have been updated since (indeed I2D is now a new resource, IID21), we did not compare to the updated resources as the amount of underlying experimental data did not change significantly (Supplementary Note 4).
To test the biological relevance of InWeb_IM, we implemented a classifier and cross-validation scheme (see http://apps.broadinstitute.org/genets, Online Methods and Supplementary Note 5) that tests the ability of each network to recapitulate known pathway relationships from 853 stringently defined canonical pathways in the Molecular Signatures Database (MSigDB http://software.broadinstitute.org/gsea/msigdb/). We normalized for the number of proteins covered by data to enable the networks to be compared interaction for interaction, meaning that we only looked at the signal of the interactions that did exist in each network and did not penalize networks that were smaller than InWeb_IM for missing data (see Online Methods and Supplementary Note 5). In a 30% holdout analysis, InWeb_IM has an area under the receiver operating characteristics curve (AUC) of 0.95; in comparison, the AUCs for the other networks range from 0.93 to 0.88, with a median of 0.89 (Fig. 2d). If we do not normalize for coverage, but make an absolute comparison of the ability to recapitulate pathway relationships in MSigDB, InWeb_IM has an AUC of 0.86, which is 16% better than for the next best network (the other five networks range in AUC from 0.78 to 0.63; see Supplementary Note 5), as expected of a high-quality network with severalfold the amount of data of the other networks. To further dissect the InWeb_IM data, and to support the quality of both the unique and orthology-transferred subsets of interactions in the network, we repeated the analysis on both of these subsets (Supplementary Note 6), which resulted in high AUCs (0.90 and 0.85, respectively). These analyses confirm that not only the network as a whole, but also the >344,000 unique interactions and the >252,000 interactions stemming from orthology transfer, have a very good biological signal.
In addition to testing the correlation between the confidence scores from InWeb_IM, Mentha and iRefIndex and the experimentally derived confidence scores from the aforementioned 58 pulldowns14, we also tested the overall concordance between data in the different networks and this independent set of human protein–protein interactions (comprising 15,205 interactions; Fig. 2e, Online Methods and Supplementary Note 3). Again, InWeb_IM shows the best agreement with this independent data set (AUC of 0.84) as compared to the other networks (AUCs ranging from 0.82 to 0.78, with a median of 0.80).
Many of the genes emerging from recent cancer sequencing studies do not integrate into well-defined pathways, and it is challenging to functionally interpret the many tumor genomes that are now available. To illustrate the potential for interpretation of massive genomic data sets using InWeb_IM, we combined the protein networks with sequencing data from >4,700 tumor genomes (from 21 tumor types) that identified 219 significantly mutated cancer genes22 to assign these genes to networks (i.e., draft pathways) that are associated with cancer based on the overall burden of mutations seen in the network in question (using an algorithm called network mutation burden, NMB; see Online Methods, Supplementary Note 7 and Supplementary Table 2). This analysis provides contextual information about the molecular groupings of cancer genes; in comparison to the five other networks, InWeb_IM had the best ability to predict cancer genes (AUC = 0.74, as compared to AUCs ranging from 0.72 to 0.71 with a median of 0.72, Fig. 2f).
To further explore the biological possibilities of the InWeb_IM data, we integrated it with tissue-specific expression quantitative trait locus information from the Genotype Tissue Expression Project (GTEx, http://www.gtexportal.org/home/) to derive 27 protein–protein interaction networks for which the corresponding genes are under tissue-specific regulation. On average the tissue-specific networks derived from InWeb_IM have 2–3 times more data than analogous networks derived from the other five resources (Supplementary Note 8 and Supplementary Figs. 2–4). One example is brain, where InWeb_IM has severalfold more interaction data connecting brain-regulated genes at the protein level than the next largest network (I2D; Supplementary Figs. 2 and 4). This observation suggests that InWeb_IM could help discover new pathway relationships in neuropsychiatric diseases. We tested the ability of each network to annotate and interpret 65 autism genes from a recent study23 using NMB, and observed that InWeb_IM is the only network that can assign these autism genes into statistically significant networks with each other (Supplementary Note 9 and Supplementary Fig. 5).
While other excellent functional genomics networks and databases exist, such as STRING24, ConsensusPathDB25 and HumanNet26, with InWeb_IM we aimed to develop a comprehensive and transparent scored experimental protein–protein interaction network as a tool for genome interpretation (Supplementary Notes 10–12). We believe that the unique features of InWeb_IM make it a versatile resource to interpret and augment the very large genomic data sets that are now being produced as part of the ongoing genomic revolution. Interim versions of InWeb_IM27 have been used to interpret genetic data from neurological7, cardiovascular8, immunological9,10 and metabolic diseases11, and cancers12. Previous evolutions of the data have also been used as part of the 1000 Genomes Project to annotate population-scale genetic variation13.
InWeb_IM is available from http://www.lagelab.org/resources/ and http://www.intomics.com/inbio/map. We have a strong commitment to maintaining and updating the resource quarterly. Moreover, we make the data accessible from graphical user interfaces at both http://www.intomics.com/inbio/map and http://apps.broadinstitute.org/genets#InWeb_InBiomap so that it can be interactively explored by any individual researcher who wishes to study the interactions of proteins of interest. We provide a roadmap for future updates and an overview of file formats and ways to query the InWeb_IM data in Supplementary Note 13 and Supplementary Figure 6.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
General design strategy of computational framework to generate InWeb_InBioMap
Details of our computational framework and network statistics are available in Supplementary Figure 1 and Supplementary Table 1. We extracted data from eight heterogeneous protein–protein interaction resources (using only data on physical interactions between proteins or data from protein complexes) covering data from 68,160 independent publications (where independent means articles indexed with a unique PubMed [PMID] identification number). These data span 4,910,949 redundant protein–protein interactions and 191,336 protein identifiers (covering all accession mapping systems used by the different databases) and stem from 1,493 organisms. Orthology transfer of interaction data is not straightforward, and for this reason we used eight different orthology databases with stringent settings (see below) and only transferred the interactions if at least four of these databases agreed on the orthology relationship (see Supplementary Note 1 for an analysis supporting this design). After thorough quality control of the raw data (including filtering to be sure only experimentally measured or curated protein–protein interactions were included in workflow, see more details below) InWeb_InBioMap was generated.
Raw interaction data
Raw and partially overlapping interaction data sets were obtained from eight source databases: BIND28, BioGRID29, DIP30, IntAct31, MatrixDB32, NetPath33, Reactome34, WikiPathways35, along with key information about the individual interaction, such as protein IDs, species, interaction type and PubMed identification numbers for the paper reporting the interaction (where we consider publications independent if they are indexed by a different PMID). Interactions that were annotated as genetic interactions, colocalizations, or neighboring reactions were ignored, and from the pathway databases (NetPath, Reactome and WikiPathways) we exclusively extracted the small subset of data that describes direct protein–protein interactions or data on experimentally resolved protein complexes. Proteins IDs for all eight databases were mapped to accession identifiers from UniProt36, and PubMed identification numbers were used to identify experiments. The authors would like to acknowledge, first, that InWeb_IM builds on the invaluable foundation laid by the experimental proteomics communities that provide the raw data and, second, the many high quality protein–protein interaction databases that laboriously extract non-structured data on the physical interactions of proteins from the 25+ million articles in PubMed with a very low error rate37. In fact, the curation practices of most protein–protein interaction databases we use are robust, and error rates have been proven to be low (on the order of ~6%37).
Orthology transfer of raw data
Orthology mapping is far from trivial, as different methods rely on different orthology definitions, sequence homology thresholds and handling of paralogs and orthologs. Orthology transfer of interactions in InWeb_IM was built on a voting scheme across eight orthology resources eggNOG38, Ensembl39, HomoloGene40, Inparanoid41, Gene42, OrthoDB43, KEGG44, and HOGENOM45, where interactions are orthology transferred if four or more databases agree in the orthology assignment (which was determined to give the best biological signal of the resulting network; see Supplementary Note 1). Among these inferred interactions we kept only those that were between proteins from the reviewed part of UniProt.
For the orthology transfer scheme to work, we rely on the organism annotations of data from the underlying databases is accurate. To test this accuracy, we randomly extracted 20 human interactions and read the articles from which the data came (Supplementary Note 12). All 20 of the interactions were true positives, and for 19 of 20 interactions the databases also had annotated the proteins with correct organism identifiers. In 1 of 20 interactions the experiments were executed with the murine orthologs of the human proteins and annotated as human in the database, so the interaction was true in the mouse and not a false positive, but an error in the annotation (in terms of organism) of the protein identifiers. This analysis shows that 20 of 20 interactions (100%) were true and 19 of 20 (95%) were annotated with the correct organism identifiers, supporting the high quality of the InWeb_IM data and the source database annotations.
Calculating confidence scores for the interactions
For each inferred interaction we kept track of the number of publications corresponding to the underlying evidence. Each of these publications contributed to the confidence score for the inferred interaction based on the total number of interactions from the publication; publications describing few interactions contributed more than publications describing many interactions because small-scale experiment are more reliable than interactions from a large screening27. In addition, the confidence score was adjusted based on the local topology of the network around the interaction, punishing interactions between proteins with many non-shared neighbors. Finally, using a gold-standard set of known high- confidence pathway interactions, the confidence scores were re-calibrated so that a score for an interaction can be interpreted as a lower bound on the probability for the interaction being a true positive. More details can be found in Supplementary Note 2.
Qualitative and quantitative comparison to other resources
We compared the number of interactions, source databases, supporting publications, and proteins within I2D, Mentha, iRefIndex, PINA, and HINT15–20 to InWeb_IM. All proteins are indexed using UniProt accessions, which are extracted directly from all networks, and we mapped UniProt to gene symbols using HGNC-provided conversion table for functional analyses. Details can be found in Supplementary Note 4.
Correlating the InWeb_IM confidence score to quantitatively measure protein–protein interactions from an independent experiment
We made a linear correlation of the InWeb_IM confidence scores to experimentally derived quantitative interaction confidences (measured as the heavy-to-light isotope ratios from the mass spectrometry data of 58 independent human immunoprecipitations using the stable isotope labeling in cell culture [SILAC] method). More details about this metric, the design choice of our experiment, and the 58 immunoprecipitations can be found in Supplementary Note 3.
Quantifying the ability of InWeb_IM to recapitulate pathway relationships
We used an algorithm called Quack (http://apps.broadinstitute.org/genets) to test how well each network can learn structures for 853 stringently defined pathways catalogued in MSigDB normalized for the amount of interactions covered by data in the network being tested. When predicting genes in, for example, the WNT pathway in the 30% holdout analysis, the positive data points were proteins assigned to the WNT pathway MSigDB and the negatives were sampled from the rest of the network. If ten proteins in a pathway (for example, the WNT pathway) were covered by data in InWeb_IM, but only five of the WNT pathway proteins were covered by data in another network, a true positive rate of 100% for InWeb_IM would mean identifying ten out of ten WNT proteins in InWeb, but a true positive rate of 100% for the other network would mean identifying five out of five of the WNT proteins. In this way we are able to determine the biological signal interaction-for-interaction in each network. If we do not normalize for network size, but make an absolute comparison of the ability to recapitulate pathway relationships in MSigDB, InWeb_IM has an AUC of 0.86, which is 16% better than the next best network (the other five networks range in AUC from 0.78 to 0.63). Details can be found in Supplementary Note 5.
Genomic annotation of cancer genes from 21 tumor types
We tested how well known cancer genes can be classified as cancer driver candidates by inferring significance through the aggregated mutation burden of first-order interactors at the protein level using an algorithm Network Mutation Burden (NMB, which is described in detail in http://biorxiv.org/content/early/2015/08/25/025445). We used the Cancer5000 stringent set of genes (n = 219) defined by Lawrence et al.22 as the true positive set and 293 genes that had a FDR = 1 across all 21 tumor types in this paper as negatives. As a negative control for cryptic confounders we randomly selected 87 genes and reran the analysis, which gave a null signal in all networks as expected. More details can be found in Supplementary Note 7.
Statistical analysis
For each of InWeb_IM, Mentha, and iRefIndex, we calculated Pearson correlation coefficient between the interaction scores in the network and the isotope heavy-to-light ratios, and reported 95% confidence interval associated with the correlation coefficient using sample size as the total number of interactions present in both the network and the experimental data set (InWeb_IM n = 1,892; Mentha n = 1,070; iRefIndex n = 2,214). We calculated the area under the ROC curve (AUC) using standard pROC package version 1.8 of the R programming language.
Concordance between the six networks and an independent data set of human protein–protein interactions
To evaluate the concordance between an independent set of human protein–protein interactions and the six protein–protein interaction networks, we used both network architectural metrics and quantitative proteomic metrics from the experimental data to test how well predicted physical interactions amongst 58 baits14 agree with the interactions reported in each of the networks. Specifically, for each resource, we take interactions present in the network as the gold-standard set of known interactions, and constructed a Random Forest model where we used the median-adjusted (by-bait) heavy-to-light ratio, along with Jaccard metric and edge-betweenness centrality, to predict whether each of the 15,205 potential physical interactions are known gold-standard interactions. After training the model on 50% of the data, we computed the AUC on the remaining 50% of the data (where interactions detected in the experiment are used as positive data points in the analysis and negative data points are interactions not found in the experiment) as a measure of how well interaction data from each network correspond to the quantitative proteomic experiment result. HINT was not included in this analysis because of low overlap between HINT and the experimental data set (less than 1.5% of all of the experimentally derived interactions). More details about the 58 pulldown experiments can be found in ref. 14 and Supplementary Note 3.
Code and data availability
Source data for Figure 2 is available in the online version of this article. InWeb_IM and the code for network mutation burden (NMB) are available from http://www.lagelab.org/resources. InWeb_IM is also available from http://www.intomics.com/inbio/map. Moreover, we make the data accessible from graphical user interfaces at both http://www.intomics.com/inbio/map and http://apps.broadinstitute.org/genets#InWeb_InBiomap so that it can be interactively explored by any individual researcher who wishes to study the interactions of proteins of interest. InWeb_IM is updated quarterly, and we provide a roadmap for future updates and an overview of file formats and ways to query the InWeb_IM data in Supplementary Notes 10 and 13. Data on the 58 pulldowns is available from ref. 14. The GTEx data is available from http://www.gtexportal.org/home/.
Supplementary Material
Acknowledgments
The authors would like to thank L. Wich for developing the graphical user interface for InWeb_IM at https://www.intomics.com/inbio/map. K.L. and H.H. are supported by grant 1P01HD068250, and H.H. is supported by a Fund for Medical Discovery Award from the Executive Committee on Research of Massachusetts General Hospital (project period 02/01/2015-01/31/2016). T.L., H.H., J.M. and K.L. are supported by a grant from the Stanley Center at the Broad Institute of MIT and Harvard (PI: K.L.), a Broadnext10 Grant from the Broad Institute of MIT and Harvard (PI: K.L.), grant 1R01MH109903 from the NIMH (PI: K.L.) and a grant from the Lundbeck Foundation (PI: K.L.). S.B. acknowledges funding from the Novo Nordisk Foundation (grant agreement NNF14CC0001).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
R.W., R.B.H. and T.S.J. developed the computational framework and continue to maintain InWeb_IM. T.L. led and executed the benchmarking analyses and network comparisons with input from R.W., R.B.H., H.H. and J.M. and supervision by K.L., T.L., R.W., R.B.H., H.H., J.M., G.S., C.T.W., O.R., K.R., H.H.S., S.B., T.S.J. and K.L. analyzed data. T.L., R.W., R.B.H., T.S. and K.L. wrote manuscript with input from all authors. K.L. initiated, designed, and led the study.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Huttlin EL, et al. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hein MY, et al. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 3.Lage K. Biochim Biophys Acta. 2014;1842:1971–1980. doi: 10.1016/j.bbadis.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venkatesan K, et al. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stumpf MP, et al. Proc Natl Acad Sci USA. 2008;105:6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen LJ, Bork P. Science. 2008;322:56–57. doi: 10.1126/science.1164801. [DOI] [PubMed] [Google Scholar]
- 7.Neale BM, et al. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lundby A, et al. Nat. Methods. 2014;11:868–874. doi: 10.1038/nmeth.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jostins L, et al. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada Y, et al. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris AP, et al. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zack TI, et al. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khurana E, et al. Science. 2013;342:1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbluh J, et al. Cell Syst. 2016;3:302–316. doi: 10.1016/j.cels.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown KR, Jurisica I. Bioinformatics. 2005;21:2076–2082. doi: 10.1093/bioinformatics/bti273. [DOI] [PubMed] [Google Scholar]
- 16.Brown KR, Jurisica I. Genome Biol. 2007;8:R95. doi: 10.1186/gb-2007-8-5-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calderone A, Castagnoli L, Cesareni G. Nat Methods. 2013;10:690–691. doi: 10.1038/nmeth.2561. [DOI] [PubMed] [Google Scholar]
- 18.Razick S, Magklaras G, Donaldson IM. BMC Bioinformatics. 2008;9:405. doi: 10.1186/1471-2105-9-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowley MJ, et al. Nucleic Acids Res. 2012;40:D862–D865. doi: 10.1093/nar/gkr967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das J, Yu H. BMC Syst Biol. 2012;6:92. doi: 10.1186/1752-0509-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotlyar M, Pastrello C, Sheahan N, Jurisica I. Nucleic Acids Res. 2016;44:D536–D541. doi: 10.1093/nar/gkv1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence MS, et al. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanders SJ, et al. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szklarczyk D, et al. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamburov A, Stelzl U, Lehrach H, Herwig R. Nucleic Acids Res. 2013;41:D793–D800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM. Genome Res. 2011;21:1109–1121. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lage K, et al. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 28.Bader GD, Betel D, Hogue CW. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stark C, et al. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xenarios I, et al. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orchard S, et al. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Launay G, Salza R, Multedo D, Thierry-Mieg N, Ricard-Blum S. Nucleic Acids Res. 2015;43:D321–D327. doi: 10.1093/nar/gku1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kandasamy K, et al. Genome Biol. 2010;11:R3. doi: 10.1186/gb-2010-11-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Croft D, et al. Nucleic Acids Res. 2014 also available from. [Google Scholar]
- 35.Kutmon M, et al. Nucleic Acids Res. 2016;44:D488–D494. doi: 10.1093/nar/gkv1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.UniProt Consortium. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salwinski L, et al. Nat Methods. 2009;6:860–861. doi: 10.1038/nmeth1209-860. [DOI] [PubMed] [Google Scholar]
- 38.Powell S, et al. Nucleic Acids Res. 2014;42:D231–D239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunningham F, et al. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.NCBI Resource Coordinators. Nucleic Acids Res. 2015;43:D6–D17. doi: 10.1093/nar/gku1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonnhammer EL, Östlund G. Nucleic Acids Res. 2015;43:D234–D239. doi: 10.1093/nar/gku1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown GR, et al. Nucleic Acids Res. 2015;43:D36–D42. doi: 10.1093/nar/gku1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kriventseva EV, et al. Nucleic Acids Res. 2015;43:D250–D256. doi: 10.1093/nar/gku1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penel S, et al. BMC Bioinformatics. 2009;10(Suppl. 6):S3. doi: 10.1186/1471-2105-10-S6-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.