

Abstract
The metabolism of malignant cells is profoundly altered in order to maintain their survival and proliferation in adverse microenvironmental conditions. Autophagy is an intracellular recycling process that maintains basal levels of metabolites and biosynthetic intermediates under starvation or other forms of stress, hence serving as an important mechanism for metabolic adaptation in cancer cells. Although it is widely acknowledged that autophagy sustains metabolism in neoplastic cells under duress, many questions remain with regard to the mutual relationship between autophagy and metabolism in cancer. Importantly, autophagy has often been described as a “double-edged sword” that can either impede or promote cancer initiation and progression. Here, we overview such a dual function of autophagy in tumorigenesis and our current understanding of the coordinated regulation of autophagy and cancer cell metabolism in the control of tumor growth, progression, and resistance to therapy.
1. INTRODUCTION
The ability of cells to adapt to stress requires diverse changes in cellular metabolism. One of the principal pathways contributing to this metabolic adaptive response is macroautophagy (commonly termed autophagy), a tightly regulated lysosomal digestion process. Because degradation through autophagy allows recycling of nutrients, autophagy serves as an important survival and fitness pathway induced by a wide array of stresses including nutrient deprivation, growth factor withdrawal, oxidative stress, infection, and hypoxia (Avivar-Valderas et al., 2011; Boya, Reggiori, & Codogno, 2013; Lum et al., 2005; Yin, Kharbanda, & Kufe, 2009). In addition to its role in the stress-induced response, autophagy plays an essential homeostatic function by selectively removing damaged or nonfunctional proteins and organelles. These quality control functions have been demonstrated to be especially crucial in certain cell types: Liver cells are reliant on autophagy for the breakdown of stored metabolites, pancreatic β-cells utilize autophagy to manage high levels of endoplasmic reticulum (ER) stress, and postmitotic neurons require autophagy to remove potentially damaging proteins that cannot be diluted by cell division (Hara et al., 2006; Jung et al., 2008; Komatsu et al., 2005, 2006; Wu et al., 2009).
Given these key functions of autophagy in normal cells and tissues, it is not surprising that disruptions in autophagy have been implicated in numerous human diseases, including neurodegeneration, liver disease, inflammation, type 2 diabetes, and cancer (Debnath, 2011; Levine & Kroemer, 2008; Murrow & Debnath, 2013). While autophagy has been demonstrated to improve disease outcome in many cases by facilitating stress-induced metabolic adaptation or cellular homeostasis, the role of autophagy is more complex in cancer. Autophagy serves as an important tumor suppressor mechanism that impedes cancer initiation; at the same time, autophagy can promote the survival of tumor cells in response to diverse microenvironmental and therapeutic stresses and support anabolic capacity in fast-replicating, metabolically stressed tumor cells (Kimmelman, 2011; Levine & Kroemer, 2008; Rabinowitz & White, 2010). Despite this widely accepted notion that autophagy critically fuels metabolism in tumor cells under duress, many questions remain with regard to the interrelationships between autophagy and metabolism in cancer. This chapter focuses on the coordinated regulation of autophagy and cancer cell metabolism, controlling tumor growth, progression, and resistance.
2. OVERVIEW OF THE AUTOPHAGY MACHINERY
The process of macroautophagy occurs in a series of distinct steps: (1) initiation of the isolation membrane; (2) nucleation; (3) elongation of the double-membrane structure to form the autophagosome; and (4) fusion to the lysosome to form an autolysosome, in which the contents are degraded (Fig. 2.1). Studies in yeast have revealed over 30 autophagy related genes and proteins (ATGs and Atgs respectively) involved in the autophagic trafficking process, many of whose mammalian orthologues have also been identified (Nakatogawa, Suzuki, Kamada, & Ohsumi, 2009). This section provides an overview of the key molecular complexes that comprise the autophagy machinery in mammalian cells—more detailed reviews can be found elsewhere (Klionsky, 2013; Klionsky & Emr, 2000; Yang & Klionsky, 2010).
Figure 2.1. Overview of the autophagy trafficking process.
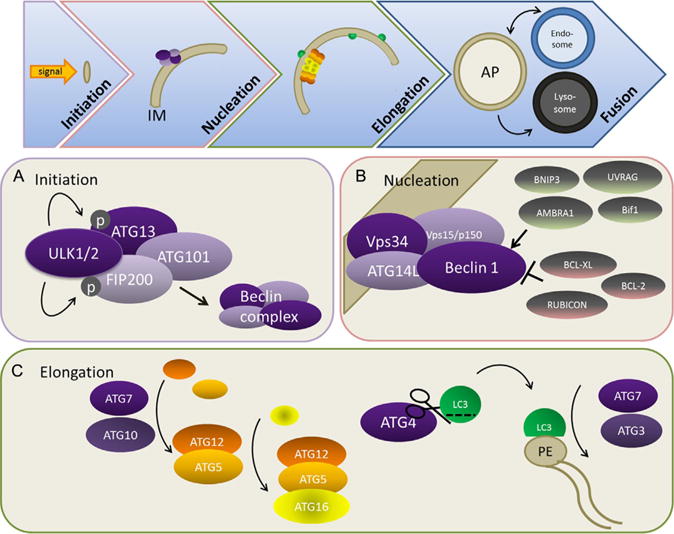
The process of macroautophagy occurs in a series of distinct steps: (1) initiation of the isolation membrane (IM); (2) nucleation; (3) elongation of the double-membrane structure to form the autophagosome (AP); and (4) fusion to endosomes and lysosomes ultimately results in the formation of an autolysosome, in which the contents are degraded. (A) Initiation is mediated by the ULK complex. Activation of ULK activity leads to the phosphorylation of FIP200 and ATG13 and initiates nucleation via interaction with the Beclin 1 complex. (B) Beclin 1/VPS34/ATG14/PIK3R4 (p150) complex interacts with multiple interacting partners that positively and negatively regulate Beclin 1/VPS34 lipid kinase activity, resulting in the fine-tuning of autophagosome nucleation. (C) Elongation requires two ubiquitin-like conjugation pathways that form the ATG12–ATG5/ATG16 complex and phosphatidylethanolamine (PE)-conjugated LC3.
2.1. Initiation and the ULK complex
In mammals, autophagosome initiation requires the ULK complex, which consists of ULK1/2 (orthologous to yeast Atg1) associated with ATG13, FIP200, and ATG101 (Mizushima, 2010; Fig. 2.1A). At least three different ULK proteins are involved in different aspects of autophagy, among which ULK1/2 bear the highest similarity to yeast Atg1. Under nutrient-rich conditions, the ULK complex interacts with mTORC1 and remains inactivated by mTORC1-mediated phosphorylation. However, upon nutrient deprivation, mTORC1 dissociates from the complex resulting in the dephosphorylation of inhibitory sites and concomitant autophosphorylation of activating sites in ULK1/2 (Chan, 2009). The kinase activation of ULK1/2 then leads to the phosphorylation and activation of ATG13 and FIP200 (Jung et al., 2009). The active complex then initiates nucleation by interaction with the Beclin 1/ATG14/VPS34 complex.
2.2. Nucleation and Beclin 1/ATG14/VPS34 complex
The formation of autophagosomes requires the activity of class III phosphatidylinositol 3-kinase (PI3K) VPS34, which is essential for phosphatidylinositol 3-phosphate production during the early stages of phagophore nucleation. VPS34 forms a complex with the yeast Atg6 orthologue Beclin 1, ATG14L, and VPS15/PIK3R4 (p150) (Itakura, Kishi, Inoue, & Mizushima, 2008; Zhong et al., 2009). Various binding partners of Beclin 1 have been identified (Fig. 2.1B), including UV irradiation resistance-associated gene (UVRAG) (Itakura et al., 2008; Liang et al., 2006), ATG14L/Barkor (Matsunaga et al., 2009; Zhong et al., 2009), and AMBRA1 (Fimia et al., 2007), all of which positively regulate Beclin 1 activity. Notably, ATG14L plays a critical role in specifying the site of the VPS34 complex relocation and therefore phagophore nucleation (Matsunaga et al., 2009). UVRAG also interacts with SH3GLB1/Bif-1 (an N-BAR domain protein), which potentially leads to phagophore membrane curvature, and expedites autophagosome–lysosome fusion (Liang et al., 2008; Takahashi et al., 2007). In addition to these positive regulators, other Beclin 1-interacting partners, including BCL-2, BCL-xL, Rubicon (RUN domain and cysteine-rich domain containing, Beclin 1-interacting protein), AKT, and EGFR, are negative regulators of the Beclin 1/VPS34 autophagy-promoting complex (Matsunaga et al., 2009; Pattingre et al., 2005; Wang et al., 2012; Wei et al., 2013; Zhong et al., 2009). Overall, these studies indicate that multiple class III PI3K complexes exist concurrently within the cell, suggesting that these proteins can exquisitely tune the level of autophagy. Notably, several proteins in this complex have tumor-suppressive or antiproliferative effects, which are discussed in detail in the succeeding text.
2.3. Elongation and the ATG12/ATG8 conjugation systems
The elongation of the phagophore membrane requires two ubiquitin-like conjugation systems. In the first, ATG7 and ATG10 (E1- and E2-like enzymes, respectively) conjugate ATG12 to ATG5. The ATG5–ATG12 complex binds ATG16 and forms a large multimeric complex called the ATG16L complex, which is essential for the elongation of the nascent phagophore (Fig. 2.1C). The second conjugation system involves cleavage of the ubiquitin-like molecule, ATG8, by the protease ATG4 to expose a C-terminal glycine residue required for subsequent activation and conjugation reactions. Several mammalian orthologues of yeast Atg8 have been identified, of which the best characterized is microtubule-associated protein 1 light chain 3 (LC3) alpha (MAP1LC3A) (Weidberg, Shpilka, Shvets, Shinder, & Elazar, 2010). Atg4 also has four mammalian isoforms, although the specificities are entirely known (Li et al., 2011; Shpilka, Weidberg, Pietrokovski, & Elazar, 2011). Ultimately, LC3 is conjugated to the lipid phosphatidylethanolamine (PE) via ATG7 and E2-like ATG3 and is subsequently recruited to both the outer and inner surfaces of the autophagosomal membrane (Fig. 2.1C). LC3 and other Atg8 family members can mediate membrane tethering and hemifusion, which may be important in the fusion of the ends of the phagophore membrane into a closed autophagosome (Weidberg et al., 2011).
In addition, LC3 is an important mediator for selectively targeting cargo for autophagic degradation. Several ubiquitin-binding proteins have been identified as cargo receptors for autophagy substrates (Johansen & Lamark, 2011), including p62/SQSTM1 (Bjørkøy et al., 2005), NBR1 (Kirkin et al., 2009), NDP52 (Thurston, Ryzhakov, Bloor, von Muhlinen, & Randow, 2009), and OPTN (Wild et al., 2011). These cargo receptors contain a well-conserved linear amino acid motif called the LIR (LC3-interacting region) that is necessary for specific targeting to the autophagosome. Interestingly, the LIR consensus sequence has been identified in a number of proteins, suggesting that the repertoire of LC3-interacting proteins acting as cargo receptors for selective autophagy may be expansive. In support of this, a large-scale proteomic study demonstrated that the mammalian ATG8 family has 67 high-confidence interactions with other cellular proteins (Berhends, Sowa, Gygi, & Harper, 2010).
2.4. Fusion
After an autophagosome forms, it fuses with the endosome or lysosome where the engulfed components may be recycled (Fig. 2.1E). Autophagosomes travel along microtubules, pushed by dynein, to lysosomes. Fusion requires ESCRT, SNAREs—specifically syntaxin 17 (Itakura, Kishi-Itakura, & Mizushima, 2012)—VPS family proteins, and RAB7. Fusion to the lysosome is the last step in the degradation of the intracompartmental components, and impaired lysosome function prevents complete autophagic flux. Hence, lysosomotrophic agents such as hydroxychloroquine (HCQ) are used experimentally to inhibit autophagy. These lysosomal inhibitors are proposed to impair autophagosome maturation and flux by altering the pH of the lysosome; nonetheless, it is important to recognize that these compounds impact a broad array of processes other than autophagy.
2.5. Chaperone-mediated autophagy
Although this chapter principally focuses on macroautophagy, it is important to recognize that multiple routes of autophagic degradation exist, including microautophagy and chaperone-mediated autophagy (CMA) (Mizushima, 2007). CMA warrants special attention because of its emerging role in cancer (Kon et al., 2011; Lv et al., 2011; Vakifahmetoglu-Norberg et al., 2013). CMA is a highly selective form of autophagy in which specific proteins are targeted to the lysosome via their interaction with a cytosolic chaperone protein—HSC70—that recognizes and binds to a specific pentapeptide motif, the KFERQ sequence. This interaction leads to binding to the lysosome via a variant of the lysosome-associated membrane protein 2A (LAMP2A), and after some unfolding, the targeted protein is directly delivered into the lysosome for degradation (Fig. 2.2; Cuervo, Terleckyh, Dice, & Knecht, 1994; Dice, Chiang, Spencer, & Backer, 1986; Koga, Martinez-vicente, Macian, Verkhusha, & Cuervo, 2011). Interestingly, CMA can be induced in mammalian cells when macroautophagy is inhibited and vice versa, indicating that a switch in one type of autophagy can compensate for a deficiency in the other (Massey, Kaushik, Sovak, Kiffin, & Cuervo, 2006; Wang et al., 2008).
Figure 2.2. Chaperone-mediated autophagy.
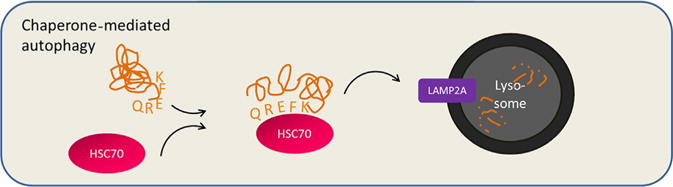
Chaperone-mediated autophagy is an additional route by which proteins are degraded in the lysosome. HSC70 binds to proteins with a KFERQ pentapeptide motif, assists in protein unfolding, and delivers the targeted protein directly to the lysosome for degradation via interaction with lysosome-associated membrane protein 2A (LAMP2A).
3. METABOLIC STIMULI REGULATING AUTOPHAGY
Metabolic stresses often occur in solid tumors and the tumor microenvironment—rapidly multiplying tumor cells and tumors that have yet to initiate angiogenic programs and often cannot maintain nutrient supply and quickly become hypoxic. To forestall senescence or death, tumor cells metabolically reprogram and engage autophagy to survive in the hostile tumor microenvironment (DeBerardinis, Lum, Hatzivassiliou, & Thompson, 2008; Lozy & Karantza, 2012). Metabolites, oxygen concentration, and oncogenes all regulate the initiation of autophagosome formation, and the regulation of autophagy is finely balanced by the integration of all these signals (Fig. 2.3). In this section, we provide an overview of the regulation of autophagy by specific metabolites and metabolic stressors in tumor cells, focusing on cancer-relevant pathways.
Figure 2.3. AMPK and mTORC1 as metabolic regulators of autophagy.
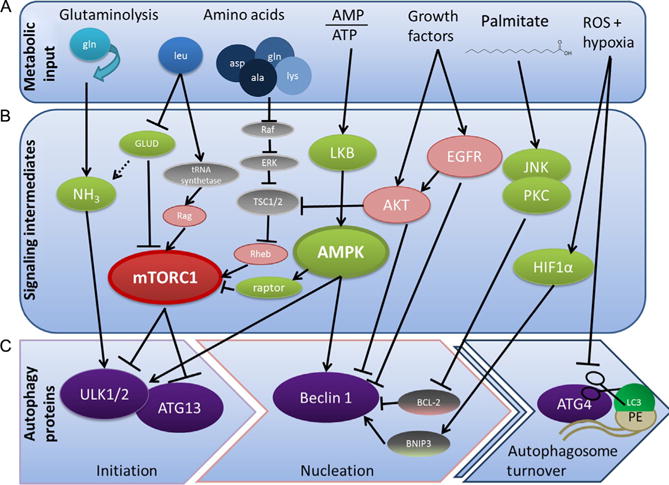
The control of autophagosome formation and turnover is tightly controlled by many upstream metabolic stimuli. Metabolic input (A), such as concentrations of ammonia, general and specific amino acid levels, ATP to ADP ratio, and signals of growth and stress such as growth factors, reactive oxygen species, and palmitate, signals to initiate autophagosome formation and inhibits turnover either through signaling intermediates (B) or by directly inhibiting or activating key autophagy-related proteins (C). AMPK1 and mTOR are the main signaling integrators and modulators of autophagy—they sense glucose and amino acid levels and act on ULK1/2, ATG13, and Beclin 1 to inhibit autophagy in times of plenty and promote autophagy under energy-lean circumstances.
3.1. Nutrient starvation
Autophagy is strongly induced in response to nutrient starvation, which is primarily controlled by mammalian target of rapamycin (mTOR). mTOR was initially identified as a key negative regulator of autophagy in yeast and has been confirmed to function as a major regulator of mammalian autophagy (Kroemer, Mariño, & Levine, 2010). mTOR acts as a master sensor of metabolic state; signals from growth factors, amino acids, oxidative stress, and DNA damage alter mTOR interactions with binding partners, thereby regulating mTOR activity. Active mTORC1 under nutrient conditions modulates the rates of translation, lipid synthesis, and mitochondrial proliferation and phosphorylates ULK1/2 and ATG13 to block autophagy. Under nutrient deprivation, ATG13 and ULK1/2 are dephosphorylated by an unknown phosphatase, leading to autophagosome formation (Jung et al., 2009; Jung, Ro, Cao, Otto, & Kim, 2010; Neufeld, 2010; Zoncu, Efeyan, & Sabatini, 2011).
3.2. Glucose
As noted by Otto Warburg in 1924, cancer cells preferentially utilize glycolysis over oxidative phosphorylation as a source of energy in aerobic conditions. Glycolysis is thought to provide a growth advantage by maintaining intracellular pools of metabolites for anabolism (Vander Heiden, Cantley, & Thompson, 2009). Therefore, cancer cells are more sensitive to low levels of glucose than nontransformed cells. Low glucose levels induce autophagy in a wide variety of mammalian cell types, and this regulation appears to be partially dependent on the activation of AMPK (Williams, Forsberg, Viollet, & Brenman, 2009). AMPK is activated by a high ratio of AMP to ATP (Kroemer et al., 2010). Under conditions of low intracellular energy, activated AMPK induces autophagy both by phosphorylating ULK1, resulting in its activation, and by inhibiting mTORC1 via phosphorylation of Raptor (Kim, Kundu, Viollet, & Guan, 2011; Mihaylova & Shaw, 2011). During glucose deprivation, AMPK-dependent Beclin 1 phosphorylation activates the proautophagy Beclin 1/VPS34 complex (Kim et al., 2013). However, the balance of nutrient availability is crucial for autophagy induction, especially since autophagy is an ATP-consuming process. Under starvation conditions, the addition of glucose (up to a threshold) promotes autophagy via a p38 MAPK-dependent pathway (Moruno-Manchón, Pérez-Jiménez, & Knecht, 2013).
3.3. Amino acids
Autophagy is inhibited in an mTORC1-dependent manner based on the levels of amino acids in the cytoplasm. Amino acids activate Rag GTPases, which promote translocation of mTORC1 to the lysosomal surface, resulting in mTORC1 activation and inhibition of autophagy via ULK1/2. Intralysosomal amino acid levels also regulate mTORC1 activity in a vacuolar ATPase-dependent manner, which may function as a means of feedback inhibition of the autophagic process (Sancak et al., 2008, 2010; Zoncu, Bar-Peled, et al., 2011). Amino acid levels also alter the signaling of the RAS/RAF1/ERK1/2 pathway, which regulates autophagy induction. High amino acid levels inhibit the activation of RAF1, which prevents ERK1/2-dependent phosphorylation of Gα-interacting protein, resulting in decreased stimulus-induced autophagy in HT-29 intestinal cells (Gozuacik & Kimchi, 2004; Ogier-Denis, Pattingre, El Benna, & Codogno, 2000; Pattingre, Bauvy, & Codogno, 2003). Specific amino acids also have distinct effects on autophagy inhibition. Leucine has the strongest inhibitory effect on autophagy. Leucyl-tRNA synthetase, an intracellular leucine sensor, binds to and regulates Rag GTPase interaction with mTORC1, leading to autophagy inhibition (Han et al., 2012).
3.4. Glutamine
When glucose levels are low, cells commonly shift to glutaminolysis to maintain tricarboxylic acid (TCA) cycle ATP and NADPH production. Ammonia produced during glutaminolysis increases autophagic flux by an mTORC1-independent pathway (Cheong, Lindsten, & Thompson, 2012; Eng, Yu, Lucas, White, & Abraham, 2010). Moreover, leucine levels regulate glutamate dehydrogenase 1 (GLUD1) activity that promotes autophagy by inhibiting mTORC1 activity and modulating reactive oxygen species (ROS) levels (Lorin et al., 2013). Accordingly, the production of ammonia by GLUD1-mediated oxidative deamination of glutamate to alpha ketoglutarate may also regulate autophagy in a similar fashion to ammonia generated from glutaminolysis; however, this intriguing hypothesis requires further testing. Moreover, it is important to recognize that glutaminolysis may not always promote autophagy. Indeed, glutamine and leucine together have been reported to activate mTORC1 and therefore inhibit autophagy in a glutaminolysis-dependent manner (Durán et al., 2012), indicating that the regulation of autophagy by glutamine is sensitive to metabolic context. Glutamine depletion was reported to decrease mRNA levels of Atg5 in wild-type MEFs, supporting the finding that glutaminolysis may promote autophagy (Lin et al., 2012).
3.5. Lipids and free fatty acids
In cancer cells, the impact of altered lipid metabolism on autophagy regulation is not as well defined as that of glucose and glutamine metabolism. Fatty acid synthesis is generally restricted to specific tissues, but is often upregulated in cancers (Santos & Schulze, 2012). Palmitate, the simplest and most abundant fatty acid and the product in fatty acid synthesis, stimulates autophagy in the muscle, liver, neurons, and pancreatic cells. Palmitate-induced autophagy is mediated by JNK1 activity and PKC activity and is independent of mTOR (Komiya et al., 2010; Martino et al., 2012; Tan et al., 2012). However, the induction of autophagy may not increase autophagic flux in pancreatic cells (Las, Serada, Wikstrom, Twig, & Shirihai, 2011), although there are conflicting data about the turnover of long-lived proteins. Further experiments, such as using GFP and mCherry-tagged LC3, will clarify this point. Additionally, the autophagic response to fatty acids may be highly tissue-specific. In hepatocytes, palmitate was found to promote apoptosis instead of autophagy, while oleate—the most abundant monounsaturated fatty acid—was found to promote autophagy via increasing ROS levels (Mei et al., 2011). The synthetic fatty acid 2-hydroxyoleic acid induced ER stress and autophagy in glioma cell lines but not a control fibroblast cell line and resulted in glioma cell differentiation (Marcilla-Etxenike et al., 2012; Terés et al., 2012). How 2-hydroxyoleic acid induces ER stress remains unknown. However, it has been shown that excess lipid storage in nonadipose tissue can cause ER stress, which increases autophagy via MTOR, JNK, and increased transcription of autophagy genes (B’chir et al., 2013; Ogata et al., 2006; Qin, Wang, Tao, & Wang, 2010; Tomohiro & Klionsky, 2007).
3.6. Hypoxia and ROS
Hypoxia and ROS, often found in the poorly vascularized tumor microenvironment, have been shown to increase autophagic flux via several mechanisms. Most directly, ROS inhibit ATG4 autophagosome turnover activity, allowing for the maintenance of lipidated LC3 necessary for autophagosome formation (Scherz-Shouval et al., 2007). Autophagy is upregulated during hypoxia via hypoxia-inducible factor 1α (HIF1α) induction of BNIP3 and BNIP3L, which binds to Beclin 1 to promote autophagy (Bellot et al., 2009). AMPK promotes autophagy independently of HIF in response to severe hypoxia. While BNIP3-regulated autophagy protects cells from death, AMPK-induced autophagy promotes cell death, pointing to the influence of cellular context on the outcome of autophagy (Papandreou, Lim, Laderoute, & Denko, 2008).
ROS-mediated damage also likely controls autophagy. ROS damages DNA, proteins, and organelles (Wellen & Thompson, 2010), and accumulated damage and subsequent metabolic stress activate autophagic programs. In addition to starvation, JNK-mediated autophagy induction is often associated with oxidative stress (Eisenberg-Lerner & Kimchi, 2007; Kang, Zeh, Lotze, & Tang, 2011). Stress-activated JNK results in the phosphorylation of BCL-2, an antiapoptotic protein that binds to and inhibits Beclin 1 (Pattingre et al., 2005), causing the release of Beclin 1 and autophagy induction (Wei, Pattingre, Sinha, Bassik, & Levine, 2008). In another indirect mechanism, low-oxygen concentrations lead to the acidification of the environment, and autophagy is upregulated in response to low pH independent of oxygen concentration (Wojtkowiak et al., 2012).
4. AUTOPHAGY AND TUMOR SUPPRESSION
Scientific evidence supports both tumor-promoting and tumor-suppressive functions for autophagy and the exact role of autophagy during cancer progression depends on tumor type, context, and stage. Here, we discuss the genetic evidence supporting the role of autophagy-related proteins (ATGs) as tumor suppressors and review the potential mechanisms through which autophagy impairs tumor initiation and progression (Fig. 2.4).
Figure 2.4. Tumor-suppressive roles of autophagy.
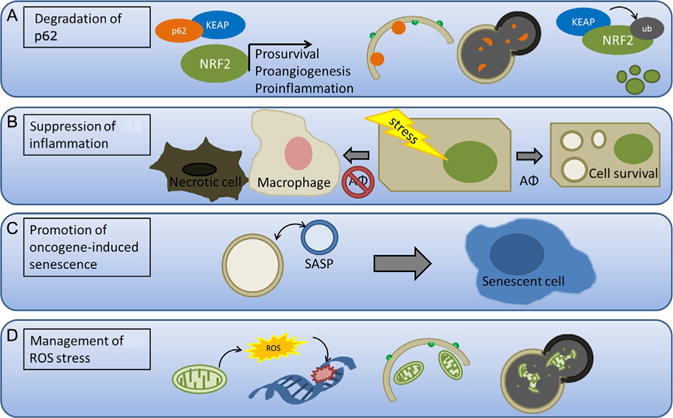
(A) Autophagy prevents p62/NRF2 pathway prosurvival, proangiogenic, and proinflammatory signaling. During hypoxia, p62 binds to and sequesters KEAP, thereby preventing ubiquitination of NRF2. NRF2 can then promote transcription of prosurvival, proangiogenic, and proinflammatory genes that enhance tumor growth. When autophagy is active, p62 is degraded by sequestration within the autophagolysosome, thereby allowing ubiquitination and degradation of NRF2. (B) Autophagy prevents necrosis and inflammation by promoting survival in stressed cells. Tumor cells with decreased autophagy (AΦ) are more prone to necrosis following stress, which recruits macrophages, promotes inflammation, and fuels tumor growth. (C) Autophagy promotes oncogene-induced senescence by enhancing the senescence-associated secretory phenotype (SASP). (D) Autophagy suppresses reactive oxygen species (ROS) accumulation and genomic damage, which helps prevent genomic instability—an important driver of tumorigenesis. The mitochondria produce ROS under normal metabolic conditions and may increase the production of ROS when damaged. Mitophagy is upregulated in response to ROS and clears excess and damaged mitochondria, which then mitigates ROS production.
4.1. ATGs as tumor suppressors
Genetic evidence that autophagy can prevent tumor formation first emerged through studies of beclin 1 (Liang et al., 1999), which was found to be monoallelically deleted in 40–75% of cases of sporadic human breast, ovarian, and prostate cancer. Furthermore, mice lacking a single copy of beclin 1 developed spontaneous lymphoma, hepatocellular carcinoma, and lung adenocarcinomas (Qu et al., 2003; Yue, Jin, Yang, Levine, & Heintz, 2003). Notably, the second allele of beclin 1 was not lost in these tumors, further corroborating that beclin 1 functioned as a haploinsufficient tumor suppressor. In addition, multiple Beclin 1-interacting partners have been implicated as tumor suppressors. UVRAG, a Beclin 1-interacting protein that positively regulates autophagy, is allelically deleted in human colon carcinoma (Liang et al., 2006; Liang, Feng, Ku, & Oh, 2007). Moreover, frameshift mutations in the polyadenine tract of the UVRAG gene resulting in decreased autophagy are present in gastric carcinomas (Kim et al., 2008). Mice lacking SH3GLB1/Bif-1, which interacts with Beclin 1 via UVRAG, exhibit a significantly higher rate of spontaneous tumors (Takahashi et al., 2007), and reduced SH3GLB1/Bif-1 expression, which correlates with decreased autophagy, is observed in gastric carcinoma (Lee et al., 2006). Besides the well-characterized oncoprotein BCL-2 interaction with Beclin 1, two other oncoproteins have been more recently shown to interact with Beclin 1 leading to autophagy suppression and oncogenesis. AKT-mediated Beclin 1 serine phosphorylation enhances its interaction with vimentin and decreases autophagy. Depletion of vimentin or expression of a nonphosphorylatable Beclin 1 mutant in AKT-overexpressing cells increases autophagy and inhibits transformation, supporting the hypothesis that autophagy suppresses tumor initiation in AKT-driven tumors (Wang et al., 2012). EGFR-mediated Beclin 1 tyrosine phosphorylation suppresses the formation of the proautophagy Beclin 1/VPS34 complex, which may contribute to tumor progression and chemoresistance in human non-small cell lung cancer xenografts harboring oncogenic EGFR mutations (Wei et al., 2013).
In addition to Beclin 1 and its associated proteins, other ATGs have been implicated as suppressors of spontaneous tumorigenesis. Mice with systemic mosaic deletion of Atg5 and liver-specific Atg7−/− mice develop liver adenomas (Inami et al., 2011; Takamura et al., 2011). Atg4C knockout mice exhibit increased susceptibility to fibrosarcomas in a chemical carcinogen model (Mariño et al., 2007). Mice with hematopoietic stem cell deletion of Atg7 develop an atypical myeloproliferation resembling human myelodysplastic syndrome and acute myeloid leukemia (Mortensen et al., 2011). Frameshift mutations in ATG2B, ATG5, and ATG9B have been reported in gastric and colorectal carcinomas, further suggesting that the components of the core autophagic machinery act as tumor suppressors in human cancers (Kang et al., 2009).
4.2. Autophagy-dependent degradation of p62/SQSTM1
The accumulation of p62/SQSTM1, an autophagy cargo receptor, promotes tumorigenesis: Liver tumor size is reduced in Atg7−/− mice by simultaneous p62 deletion (Takamura et al., 2011), p62 gene targeting reduces anchorage-independent growth of human hepatocellular carcinoma cells (Inami et al., 2011), p62−/− mice fail to develop RAS-induced lung carcinomas (Duran et al., 2008), and p62-null cells have impaired RAS transformation (Guo et al., 2011). In KRAS-driven tumor cells, p62 activates Nrf2 and NF-kB, which stimulate proangiogenic and proinflammatory responses, respectively, thereby contributing to aggressive tumor progression. Thus, increased autophagy enhances p62 degradation, leading to diminished angiogenic and inflammatory responses (Duran et al., 2008; Kim, Hur, et al., 2011; Mathew et al., 2009).
p62/SQSTM1 activation of the Nrf2 pathway in autophagy-deficient cells is especially important in tumor progression (Komatsu et al., 2010). Notably, the Nrf2 pathway, due to inactivating somatic mutations in the E3 ubiquitin ligase Keap1, has been implicated as a survival pathway in non-small cell lung carcinomas (Singh et al., 2006). The transcription factor Nrf2 (nuclear regulatory factor 2) regulates the expression of a wide range of genes that promote angiogenesis and facilitate cell survival. Keap1 ubiquitinates Nrf2 resulting in its degradation under normal conditions. Accumulated p62/SQSTM1 in autophagy-deficient cells directly binds to Keap1, disrupting Keap1-mediated degradation of Nrf2 and promoting aberrant Nrf2-mediated transcription (Komatsu et al., 2010). Thus, aberrant regulation of Nrf2 in autophagy-deficient cells may be an important pathway in tumor cell survival (Fig. 2.3A). Indeed, this pathway has been implicated in the spontaneous tumorigenesis of autophagy-defective liver cells (Inami et al., 2011; Takamura et al., 2011) and in the early growth acceleration of BRAF-driven lung cancers lacking Atg7 (Strohecker et al., 2013).
4.3. Autophagy prevents protumor inflammation and facilitates senescence
Because autophagy promotes tumor cell adaptation and survival during hypoxic and metabolic stress, it may suppress tumor progression by inhibiting necrosis. In solid tumors, necrotic cell death causes macrophage infiltration and proinflammatory cytokine production, and chronic inflammation generally favors cancer growth and progression (DeNardo, Johansson, & Coussens, 2008). Thus, by limiting necrosis, autophagy may actually suppress tumor growth by preventing leukocyte infiltration of the primary tumor site (Fig. 2.3B). Indeed, this ability of autophagy to restrict necrosis prevented macrophage-associated tumor inflammation and inhibited primary tumor growth in apoptosis-resistant cells (Degenhardt et al., 2006). Additionally, autophagy can facilitate the transition to senescence (Fig. 2.3C), which also prevents immune activation due to necrosis, and can lead to the elimination of premalignant cells by senescence-mediated surveillance (Kang, Yevsa, et al., 2011; Narita et al., 2011; Young et al., 2009). Autophagy allows the cancer cells to quietly survive but helps to restrict proliferation by facilitating senescence, thereby overall suppressing tumor growth.
4.4. Autophagy clears dysfunctional mitochondria
Autophagy is an important mechanism for the clearance of damaged mitochondria, a process termed mitophagy. Mitochondrial number may indirectly regulate tumor progression as the mitochondria produce ROS, which can promote tumor progression via damage to proteins or DNA causing chromosomal instability (Ishikawa et al., 2008). In response to ROS, mitophagy is upregulated to remove excess mitochondria and mitigate ROS production (Fig. 2.3D). Increased ROS production from increased metabolic rate can damage the mitochondria, which in turn can increase metabolic stress in the cell. Accordingly, in autophagy-defective cells, metabolic stress induces more DNA damage, increased genomic instability, and increased accumulation of damaged mitochondria than in wild-type control cells (Belaid et al., 2013; Mathew et al., 2009). By clearing damaged mitochondria and controlling intracellular ROS levels, autophagy may exert a tumor suppressor function.
5. TUMOR-PROMOTING FUNCTIONS OF AUTOPHAGY
Although reduced autophagy can promote tumor development, autophagy provides cancer cells with certain selective advantages to cope with stress and promote metabolic adaptation. Hence, a basal level of autophagy appears to be necessary for the optimal survival and fitness of cancer cells. The following section provides an overview of several potential mechanisms by which autophagy may promote tumor progression (Fig. 2.5).
Figure 2.5. Tumor-promoting roles of autophagy.
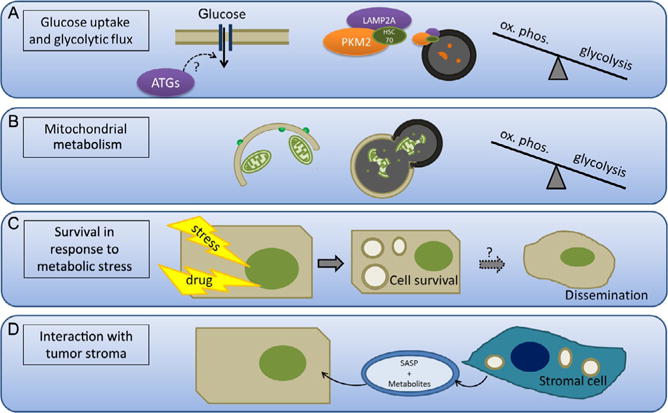
(A) Autophagy promotes glucose uptake and glycolytic flux. Autophagy has been shown to promote glucose uptake in cancer cells, although the mechanism remains to be elucidated. Additionally, increased chaperone-mediated autophagy (CMA) promotes degradation of PKM2, a rate-limiting glycolytic enzyme. Thus, CMA can control the rate of flux through glycolysis and whether glycolytic intermediates are used for energy production or anaplerosis. (B) Autophagy selectively degrades the mitochondria and therefore the machinery necessary for fatty acid oxidation and oxidative phosphorylation. This enhances the shift to glycolysis, which is characteristic of cancer cells. (C) Autophagy promotes survival in response to metabolic stress such as growth factor deprivation, acidic environment, and ER stress by recycling cytoplasmic material in order to maintain the basal energy state and clear damaged, misfolded proteins. This process may also be important for survival during tumor dissemination and metastasis. (D) Autophagy in stromal cells induced by the hypoxic and acidic tumor microenvironment promotes the secretion of metabolites and growth signals via senescence-associated secretion phenotype (SASP) that enhances tumor cell growth.
5.1. Autophagy and metabolic adaptation in cancer
5.1.1 Autophagy and oxidative mitochondrial metabolism
Strong oncogenic insults like RAS activation lead to increased autophagy. In pancreatic ductal adenocarcinoma (PDAC), where activating KRAS mutations are present in greater than 90% of tumors, elevated autophagy is found in both primary PDAC tumors and cell lines. Genetic inhibition of autophagy in PDAC cells potently suppresses proliferation in vitro and elicits robust tumor regression and prolonged survival in pancreatic cancer xenografts and genetic mouse models (Yang et al., 2011). Because RAS activation is marked by profound metabolic alterations that promote energy production and support the biosynthesis of macromolecules needed for rapid proliferation, it has been hypothesized that autophagy maintains key metabolic pathways in RAS-transformed cells. In support, the loss of autophagy during RAS transformation is associated with reduced mitochondrial oxygen consumption and decreased levels of TCA cycle intermediates (Guo et al., 2011; Yang et al., 2011). This requirement for autophagy to maintain oxidative mitochondrial metabolism of RAS-transformed cells indicates that the protumor effects of autophagy are not limited to survival functions in response to external stresses. Rather, autophagy contributes to the metabolic fitness of the entire tumor population. Remarkably, this requirement for autophagy may be oncogene-dependent, as autophagy has been demonstrated to restrict, rather than promote, proliferation driven by oncogenic PI3K in a three-dimensional mammary culture model (Chen, Eritja, Lock, & Debnath, 2013). As RAS is one of the few oncogenes that stimulate—rather than suppress—autophagy, it will be interesting to determine whether this requirement for autophagy is conserved in other oncogenic contexts.
5.1.2 Glucose metabolism
Many tumors preferentially use aerobic glycolysis, which allows for the accumulation of metabolic intermediates required for anabolism (Hsu & Sabatini, 2008). CMA and selective macroautophagy both play important roles in regulating the shift to aerobic glycolysis in cancer cells. CMA is upregulated in diverse tumor types and is necessary for tumor growth and metastasis in lung cancer cells, and inhibition of CMA decreases the rate of glycolysis characteristic of tumor growth (Kon et al., 2011). More specifically, CMA controls the levels of the metabolic enzyme PKM2 (Fig. 2.4A), which is often upregulated in many tumor types and particularly glioblastoma. The PKM2 isoform of pyruvate kinase is slower at metabolically converting phosphoenolpyruvate to pyruvate than the M1 isoform; this causes glycolytic intermediates to accumulate and drives tumor cell proliferation and growth by promoting key biosynthetic side reactions in the glycolytic pathway. CMA can selectively degrade PKM2, thereby regulating the levels of the metabolic intermediates, glucose-6-phosphate and fructose-1,6-bisphosphate, and the levels of ATP (Lv et al., 2011). Recently, PKM2-specific deletion was shown to have increased mammary tumor formation driven by Brca-1 deletion (Israelsen et al., 2013), consistent with the notion that cancer cells prefer low pyruvate kinase activity. Therefore, the degradation of PKM2 by CMA may promote tumor progression.
The number of mitochondria present also regulates the shift to anaerobic metabolism. BRAF-driven melanoma cells decrease the rate of mitochondrial biogenesis in order to shift from oxidative phosphorylation to glycolysis (Haq et al., 2013; Ho et al., 2012; Vazquez et al., 2013). If mitophagy is aberrantly activated, decreased numbers of mitochondria shift the cells to glycolysis in a similar mechanism to BRAF regulation of mitochondrial biogenesis (Fig. 2.4B). RCAN1-1L, whose expression is increased in response to oxidative stress, can open the MPT pore and decrease ATP levels. This inhibits mTOR signaling via AMPK, resulting in increased mitophagy and a shift to glycolysis (Ermak et al., 2012). In addition to shifting the metabolic pathways to preferentially use glucose, autophagy also facilitates glucose uptake (Fig. 2.4A) and glycolytic flux in RAS-transformed cells, which is required for adhesion-independent proliferation (Lock & Debnath, 2011; Lock et al., 2011).
5.1.3 Amino acids
In addition to glucose, amino acids are necessary for cancer cell growth. In yeast, autophagic breakdown of proteins during starvation generates cytosolic amino acid pools crucial for survival (Onodera & Ohsumi, 2005). Amino acids feed into cataplerotic pathways and can be used to maintain biosynthetic capacity in rapidly dividing cancer cells. Glutamine, the most abundant amino acid in mammalian cells, is important in cancer progression as a metabolic intermediate (DeBerardinis et al., 2008; Gaglio et al., 2011). As glycolytic rates increase, tumor cells rely increasingly on glutamine to replenish the TCA cycle and maintain ATP production (Burgess, 2013). In pancreatic cancer, glutamine feeds into glutaminolysis, utilizing steps in the TCA cycle to generate NADPH, maintain the cellular redox state, and provide metabolites for anaplerosis (Son et al., 2013). In wild-type MEFs, loss of autophagy was also found to decrease the levels of intracellular glutamine and also mimic the metabolic changes associated with glutamine depletion, indicating that autophagy normally helps to maintain intracellular stores of glutamine. However, in the same study, glutamine deprivation did not increase the levels of autophagy, and the Atg5 mRNA level decreased (Lin et al., 2012). Therefore, how autophagy may increase specific amino acids during deprivation remains to be defined.
5.1.4 Lipids
Lipid metabolism is altered in cancer—tumor cells reactivate de novo lipid synthesis, ATP-citrate lyase is required for transformation in vitro, cholesterol synthesis in prostate cancer is increased, and fatty acid oxidation is an important source of energy for prostate cancer cells (Santos & Schulze, 2012). Autophagy in the specific form of lipophagy is important for the degradation of lipid droplets in the adipose tissue (Singh & Cuervo, 2012), and autophagy regulates lipid metabolism in hepatocytes as triglyceride hydrolysis is impaired in Atg5−/− cells (Singh et al., 2009). Whether these processes affect tumor lipid metabolism requires further study.
Additionally, autophagy impacts lipid metabolism by altering the mitochondrial number. Atg7 deleted, p53 mutant cells in a KRAS-driven NSCLC model have intracellular lipid accumulation because of increased dysfunctional mitochondria that compromises fatty acid oxidation, suggesting that autophagy is crucial to maintain lipid metabolism in KRAS and p53 mutant cells. This prevents the efficient growth of tumor cells and turns them into lipid cysts instead of tumors (Guo et al., 2013).
5.2. Autophagy promotes cell survival under metabolic stress
As discussed earlier in the text, autophagy is strongly activated under periods of oxidative and metabolic stress, and depending on the extent and severity of stress, autophagy serves to prolong cell survival in the primary tumor and possibly also during tumor dissemination and metastasis (Fig. 2.4C). In melanoma cells driven by oncogenic Ras or MEK, the removal of leucine does not induce autophagy to the same extent as nontransformed, immortalized melanocytes. The aberrant activation of mTOR via Ras prevents autophagy induction and the cells are sensitized to apoptosis, presumably because translation continues although the lost leucine is not recycled intracellularly (Sheen, Zoncu, Kim, & Sabatini, 2011). Following growth factor withdrawal, autophagy is essential for maintaining cell survival in apoptosis-deficient hematopoietic cells and can sustain viability for several weeks. IL3-deprived cells become less glycolytic and use autophagy as a catabolic process to maintain mitochondrial respiration and levels of ATP (Lum et al., 2005). Increased autophagy regulated by the PI3K/AKT/mTOR pathway prolongs cancer cell survival under acidic environment stress produced by glycolysis (Wojtkowiak et al., 2012). Autophagy also prevents ER stress-induced cell death during protein overproduction (e.g., induced by oncogenes such as Myc) by clearing excess and misfolded proteins (Tomohiro & Klionsky, 2007). Indeed, Myc-driven tumors have increased cell growth, ER stress, and metabolic rate, and autophagy inhibition enhances therapy-induced apoptosis in a Myc-driven model of lymphoma (Amaravadi, Yu, & Lum, 2007; Dang, 1999; Miller, Thomas, Islam, Muench, & Sedoris, 2012).
5.3. Autophagy in the tumor stroma
Autophagy prolongs tumor cell survival under stressful conditions. It should be noted that the acidic, hypoxic, or nutrient-starved environment also induces autophagy in the surrounding stromal cells, which promotes tumor growth (Fig. 2.4D). Serum-deprived mesenchymal stem cells induce autophagy and support MCF7 growth in xenograft models by secreting growth factors and antiapoptotic factors (Sanchez et al., 2011). While autophagy-induced senescence in cancer cells limits growth, autophagy-induced senescence in the tumor stroma may promote cancer by enhancing the senescence-associated secretory phenotype (SASP) and promoting the secretion of growth factors and cytokines that enhance tumor progression (Capparelli, Chiavarina, et al., 2012; Capparelli, Guido, et al., 2012; Capparelli, Whitaker-Menezes, et al., 2012; Maes, Rubio, Garg, & Agostinis, 2013).
In addition to modulating secretion in senescent fibroblasts, autophagy in cancer-associated fibroblasts (CAFs) may directly fuel cancer cell metabolism. Autophagic senescent CAFs release metabolites such as glutamine, ketone bodies, and glycolytic intermediates that may promote tumor growth and metastasis. These studies raise the possibility that autophagy in the tumor stroma is important for the continued growth of the tumor (Ko et al., 2011; Martinez-Outschoorn et al., 2010; Salem et al., 2012).
5.4. Autophagy inhibition in cancer therapy
The increased dependence of tumors on altered metabolism is an attractive therapeutic target. In addition to targeting metabolic enzymes, targeting autophagy may provide a similar benefit. However, such an approach is complicated by the multifaceted role of autophagy in tumor formation and progression (Cheong, Lu, Lindsten, & Thompson, 2012). Increased autophagy has been observed in tumor cells following numerous anticancer treatments and is proposed to represent a common adaptive stress response that enables tumor cells to survive these therapeutic insults (Fig. 2.4C). This has motivated significant interest in combining autophagy inhibition with other agents to synergistically eliminate cancer cells. Readers are referred to several reviews for additional information (Amaravadi et al., 2011; Eisenberg-Lerner & Kimchi, 2009; Høyer-hansen & Jäättelä, 2008).
Notably, certain targeted therapies that enhance autophagy in vitro may benefit from combined autophagy inhibition. Autophagy is upregulated in response to erlotinib in NSCLC cell lines and combined treatment with chloroquine, an antimalarial that inhibits autophagy, enhances erlotinib sensitivity (Li, Lam, Mak, Zheng, & Ho, 2013). Similarly, gastrointestinal stromal tumors exhibit enhanced autophagy in response to imatinib, which lessens the therapeutic benefit. Combined inhibition of autophagy with imatinib treatment increased the number of cells undergoing apoptosis, both in vitro and in vivo, and reduced the outgrowth of resistant cells (Gupta et al., 2010). Moreover, upon treatment with the VEGF-neutralizing antibody bevacizumab, increased autophagy due to hypoxia promotes tumor cell survival and resistance to this antiangiogenic therapy (Hu et al., 2012). In contrast, inhibition of erlotinib-induced autophagy in human NSCLC xenografts in vivo by inducible expression of a Beclin 1 tyrosine phosphomimetic mutant resulted in partial chemoresistance (Wei et al., 2013), suggesting that the effects of autophagy inhibition may vary depending upon the autophagy step targeted, in vitro versus in vivo studies, or due to other differences in tumor type or experimental systems.
In the previously mentioned examples, autophagy is targeted due to its induction in response to therapy, but autophagy inhibition can also synergize with therapies that do not normally promote autophagic flux. For example, combining autophagy inhibition with immunotherapy could increase efficacy. Hypoxia-induced autophagy prevents lung cancer cells from cytolytic T-cell mediated cell death, but inhibition of autophagy combined with immunotherapy may provide a powerful and tumor-specific therapy (Noman et al., 2011). Another synergistic approach involves targeting the proteasome pathway and autophagy in tumor cells that are prone to ER stress. Autophagy inhibitors in combination with proteasome inhibitors increase suppression of proliferation and induce apoptosis in hepatocellular carcinoma (Hui et al., 2012). Additional studies in multiple myeloma cells also show the same increased sensitivity to the combination of proteasome inhibitors and autophagy inhibitors in vitro (Kawaguchi et al., 2011).
Importantly, one should recognize that many studies of autophagy inhibition as anticancer therapy have employed the lysosomal inhibitor HCQ. Hence, an important caveat for these experiments is that the cytotoxic effects of HCQ and similar agents are likely to involve processes other than autophagy. To date, the precise contributions of autophagy inhibition toward the efficacy of these antimalarials remain uncertain. Moreover, compensatory pathways, such as CMA, may influence the efficacy of autophagy inhibition as a therapeutic approach. For example, autophagy inhibition in combination with the HDACi vorinostat in a sensitive T-cell lymphoma cell line results in decreased cell death, but the resulting vorinostat-resistant sub-clones become partially resensitized by the inhibition of CMA (Dupéré-Richer et al., 2013).
While it remains controversial whether autophagy can mediate cell death, several studies demonstrate that genetic knockdown of autophagy blocks tumor cell death induced by oncogenic RAS (Elgendy, Sheridan, Brumatti, & Martin, 2011) or by various chemotherapeutic agents (Janku, McConkey, Hong, & Kurzrock, 2011; Notte, Leclere, & Michiels, 2011). Furthermore, acute inhibition of autophagy can limit chemotherapy responses in vivo by preventing autophagy-dependent anticancer immune responses (Michaud et al., 2011). Thus, additional studies are needed to further clarify the contexts in which autophagy inhibition will be beneficial in the treatment of cancer, but as these studies have shown, autophagy inhibition as a clinical therapy will not be straightforward.
6. CONCLUSION
Autophagy and metabolism in cancer cells are inexorably linked. The cross regulation of these processes acts to buffer cancer cells from the environmental and internal stresses caused by excessive proliferation. As more targeted therapies are being designed and tested, unintended consequences on autophagy, both positive and negative, must be considered in order to predict and combat side effects and resistance mechanisms. Therefore, further understanding of how autophagy contributes to cancer cell metabolism will provide insight into how to better treat cancers.
Acknowledgments
J. D. is supported by the NIH (CA126792), the DOD Breast Cancer Research Program (W81XWH-11-1-0130 and W81XWH-12-1-0505), and the Samuel Waxman Cancer Research Foundation. B. L. is supported by the NIH (CA109618) and CPRIT (RP120718-P1).
References
- Amaravadi RK, Lippincott-schwartz J, Yin X, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clinical Cancer Research. 2011;17(4):654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi R, Yu D, Lum J. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. The Journal of Clinical Investigation. 2007;117(2):326–336. doi: 10.1172/JCI28833. http://dx.doi.org/10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Molecular and Cellular Biology. 2011;31(17):3616–3629. doi: 10.1128/MCB.05164-11. http://dx.doi.org/10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Research. 2013;41(16):7683–7699. doi: 10.1093/nar/gkt563. http://dx.doi.org/10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis and genomic stability. Cancer Research. 2013;73(14):4311–4322. doi: 10.1158/0008-5472.CAN-12-4142. http://dx.doi.org/10.1158/0008-5472.CAN-12-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Molecular and Cellular Biology. 2009;29(10):2570–2581. doi: 10.1128/MCB.00166-09. http://dx.doi.org/10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berhends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466(7302):68–76. doi: 10.1038/nature09204. http://dx.doi.org/10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of Cell Biology. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. http://dx.doi.org/10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nature Cell Biology. 2013;15(7):713–720. doi: 10.1038/ncb2788. http://dx.doi.org/10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DJ. Metabolism: Glutamine connections. Nature Reviews Cancer. 2013;13(5):293. doi: 10.1038/nrc3515. http://dx.doi.org/10.1038/nrc3515. [DOI] [PubMed] [Google Scholar]
- Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11(19):3599–3610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparelli C, Guido C, Whitaker-menezes D, Bonuccelli G, Balliet R, Timothy G, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11(12):2285–2302. doi: 10.4161/cc.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11(12):2272–2284. doi: 10.4161/cc.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Science Signaling. 2009;2(84):pe51. doi: 10.1126/scisignal.284pe51. http://dx.doi.org/10.1126/scisignal.284pe51. [DOI] [PubMed] [Google Scholar]
- Chen N, Eritja N, Lock R, Debnath J. Autophagy restricts proliferation driven by oncogenic phosphatidylinositol 3-kinase in three-dimensional culture. Oncogene. 2013;32(20):2543–2554. doi: 10.1038/onc.2012.277. http://dx.doi.org/10.1038/onc.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lindsten T, Thompson CB. Autophagy and ammonia. Autophagy. 2012;8(1):122–123. doi: 10.4161/auto.8.1.18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nature Biotechnology. 2012;30(7):671–678. doi: 10.1038/nbt.2285. http://dx.doi.org/10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Terleckyh SR, Dice JF, Knecht E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. The Journal of Biological Chemistry. 1994;269(42):26374–26380. [PubMed] [Google Scholar]
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Molecular and Cellular Biology. 1999;19(1):1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metabolism. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002. http://dx.doi.org/10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Debnath J. The multifaceted roles of autophagy in tumors-implications for breast cancer. Journal of Mammary Gland Biology and Neoplasia. 2011;16(3):173–187. doi: 10.1007/s10911-011-9223-3. http://dx.doi.org/10.1007/s10911-011-9223-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. http://dx.doi.org/10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Reviews. 2008;27:11–18. doi: 10.1007/s10555-007-9100-0. [DOI] [PubMed] [Google Scholar]
- Dice JF, Chiang H, Spencer EP, Backer JM. Regulation of catabolism of microinjected ribonuclease A. The Journal of Biological Chemistry. 1986;261(15):6853–6859. [PubMed] [Google Scholar]
- Dupéré-Richer D, Kinal M, Ménasché V, Nielsen TH, Del Rincon S, Pettersson F, et al. Vorinostat-induced autophagy switches from a death-promoting to a cyto-protective signal to drive acquired resistance. Cell Death & Disease. 2013;4:e486. doi: 10.1038/cddis.2012.210. http://dx.doi.org/10.1038/cddis.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–354. doi: 10.1016/j.ccr.2008.02.001. http://dx.doi.org/10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis activates Rag-mTORC1 signaling. Molecular Cell. 2012;47(3):349–358. doi: 10.1016/j.molcel.2012.05.043. http://dx.doi.org/10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Kimchi A. DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death and Differentiation. 2007;14(11):1908–1915. doi: 10.1038/sj.cdd.4402212. http://dx.doi.org/10.1038/sj.cdd.4402212. [DOI] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14(4):376–391. doi: 10.1007/s10495-008-0307-5. http://dx.doi.org/10.1007/s10495-008-0307-5. [DOI] [PubMed] [Google Scholar]
- Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Molecular Cell. 2011;42(1):23–35. doi: 10.1016/j.molcel.2011.02.009. http://dx.doi.org/10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Science Signaling. 2010;3(119):ra31. doi: 10.1126/scisignal.2000911. http://dx.doi.org/10.1126/scisignal.2000911. [DOI] [PubMed] [Google Scholar]
- Ermak G, Sojitra S, Yin F, Cadenas E, Cuervo AM, Davies KJ. Chronic expression of RCAN1-1L protein induces mitochondrial autophagy and metabolic shift from oxidative phosphorylation to glycolysis in neuronal cells. The Journal of Biological Chemistry. 2012;287(17):14088–14098. doi: 10.1074/jbc.M111.305342. http://dx.doi.org/10.1074/jbc.M111.305342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121–1125. doi: 10.1038/nature05925. http://dx.doi.org/10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Molecular Systems Biology. 2011;7(523):523. doi: 10.1038/msb.2011.56. http://dx.doi.org/10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23(16):2891–2906. doi: 10.1038/sj.onc.1207521. http://dx.doi.org/10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes & Development. 2011;25(5):460–470. doi: 10.1101/gad.2016311. http://dx.doi.org/10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes & Development. 2013;27(13):1447–1461. doi: 10.1101/gad.219642.113. http://dx.doi.org/10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Roy S, Lazar AJF, Wang WL, McAuliffe JC, Reynoso D, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST) Proceedings of the National Academy of Sciences of the United States of America. 2010;107(32):14333–14338. doi: 10.1073/pnas.1000248107. http://dx.doi.org/10.1073/pnas.1000248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149(2):410–424. doi: 10.1016/j.cell.2012.02.044. http://dx.doi.org/10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23(3):302–315. doi: 10.1016/j.ccr.2013.02.003. http://dx.doi.org/10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724. http://dx.doi.org/10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Ho J, de Moura MB, Lin Y, Vincent G, Thorne S, Duncan LM, et al. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Molecular Cancer. 2012;11(1):76. doi: 10.1186/1476-4598-11-76. http://dx.doi.org/10.1186/1476-4598-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høyer-hansen M, Jäättelä M. Autophagy: An emerging target for cancer therapy. Autophagy. 2008;4(5):574–580. doi: 10.4161/auto.5921. [DOI] [PubMed] [Google Scholar]
- Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134(5):703–707. doi: 10.1016/j.cell.2008.08.021. http://dx.doi.org/10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Research. 2012;72(7):1773–1783. doi: 10.1158/0008-5472.CAN-11-3831. http://dx.doi.org/10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui B, Shi YH, Ding ZB, Zhou J, Gu CY, Peng YF, et al. Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer. 2012;118(22):5560–5571. doi: 10.1002/cncr.27586. http://dx.doi.org/10.1002/cncr.27586. [DOI] [PubMed] [Google Scholar]
- Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. The Journal of Cell Biology. 2011;193(2):275–284. doi: 10.1083/jcb.201102031. http://dx.doi.org/10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320(5876):661–664. doi: 10.1126/science.1156906. http://dx.doi.org/10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155(2):397–409. doi: 10.1016/j.cell.2013.09.025. http://dx.doi.org/10.1016/j.cell.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Molecular Biology of the Cell. 2008 Dec;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. http://dx.doi.org/10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–1269. doi: 10.1016/j.cell.2012.11.001. http://dx.doi.org/10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Janku F, McConkey D, Hong D, Kurzrock R. Autophagy as a target for anticancer therapy. Nature Reviews. Clinical Oncology. 2011;8(9):528–539. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. doi: 10.4161/auto.7.3.14487. http://dx.doi.org/10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metabolism. 2008;8(4):318–324. doi: 10.1016/j.cmet.2008.08.013. http://dx.doi.org/10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro S, Kim Y, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular Biology of the Cell. 2009;20(April):1992–2003. doi: 10.1091/mbc.E08-12-1249. http://dx.doi.org/10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Letters. 2010;584(7):1287–1295. doi: 10.1016/j.febslet.2010.01.017. http://dx.doi.org/10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, et al. Frame-shift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. The Journal of Pathology. 2009;217:702–706. doi: 10.1002/path.2509. http://dx.doi.org/10.1002/path.2509. [DOI] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. doi: 10.1038/nature10599. http://dx.doi.org/10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death and Differentiation. 2011;18(4):571–580. doi: 10.1038/cdd.2010.191. http://dx.doi.org/10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T, Miyazawa K, Moriya S, Ohtomo T, Che XF, Naito M, et al. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: Crosstalk among proteasome, autophagy-lysosome and ER stress. International Journal of Oncology. 2011;38(3):643–654. doi: 10.3892/ijo.2010.882. http://dx.doi.org/10.3892/ijo.2010.882. [DOI] [PubMed] [Google Scholar]
- Kim TH, Hur E, Kang SJ, Kim JA, Thapa D, Lee YM, et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Research. 2011;71(6):2260–2275. doi: 10.1158/0008-5472.CAN-10-3007. http://dx.doi.org/10.1158/0008-5472.CAN-10-3007. [DOI] [PubMed] [Google Scholar]
- Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Human Pathology. 2008;39(7):1059–1063. doi: 10.1016/j.humpath.2007.11.013. http://dx.doi.org/10.1016/j.humpath.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303. doi: 10.1016/j.cell.2012.12.016. http://dx.doi.org/10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13(2):132–141. doi: 10.1038/ncb2152. http://dx.doi.org/10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes & Development. 2011;25(19):1999–2010. doi: 10.1101/gad.17558811. http://dx.doi.org/10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Molecular Cell. 2009;33(4):505–516. doi: 10.1016/j.molcel.2009.01.020. http://dx.doi.org/10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Klionsky D. An overview of autophagy: Morphology, mechanism and regulation. Antioxidants & Redox Signaling. 2013;20(3):460–473. doi: 10.1089/ars.2013.5371. http://dx.doi.org/10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr S. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells. Cancer Biology & Therapy. 2011;12(12):1085–1097. doi: 10.4161/cbt.12.12.18671. http://dx.doi.org/10.4161/cbt.12.2.18671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Martinez-vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nature Communications. 2011;2(386) doi: 10.1038/ncomms1393. http://dx.doi.org/10.1038/ncomms1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biology. 2010;12(3):213–223. doi: 10.1038/ncb2021. http://dx.doi.org/10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. http://dx.doi.org/10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of Cell Biology. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. http://dx.doi.org/10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya K, Uchida T, Ueno T, Koike M, Abe H, Hirose T, et al. Free fatty acids stimulate autophagy in pancreatic β-cells via JNK pathway. Biochemical and Biophysical Research Communications. 2010;401(4):561–567. doi: 10.1016/j.bbrc.2010.09.101. http://dx.doi.org/10.1016/j.bbrc.2010.09.101. [DOI] [PubMed] [Google Scholar]
- Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, et al. Chaperone-mediated autophagy is required for tumor growth. Science Translational Medicine. 2011;3(109):109ra117. doi: 10.1126/scitranslmed.3003182. http://dx.doi.org/10.1126/scitranslmed.3003182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Molecular Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. http://dx.doi.org/10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in β-cells. The Journal of Biological Chemistry. 2011;286(49):42534–42544. doi: 10.1074/jbc.M111.242412. http://dx.doi.org/10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Jeong EG, Soung YH, Nam SW, Lee JY, Yoo NJ, et al. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology. 2006;38(4):312–315. doi: 10.1080/00313020600820880. http://dx.doi.org/10.1080/00313020600820880. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. http://dx.doi.org/10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hou Y, Wang J, Chen X, Shao ZM, Yin XM. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. The Journal of Biological Chemistry. 2011;286(9):7327–7338. doi: 10.1074/jbc.M110.199059. http://dx.doi.org/10.1074/jbc.M110.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Lam SK, Mak JCW, Zheng CY, Ho JCM. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer. 2013;81(3):354–361. doi: 10.1016/j.lungcan.2013.05.012. http://dx.doi.org/10.1016/j.lungcan.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature Cell Biology. 2006;8(7):688–699. doi: 10.1038/ncb1426. http://dx.doi.org/10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Oh B. UVRAG: A new player in autophagy and tumor cell growth. Autophagy. 2007;3(1):69–71. doi: 10.4161/auto.3437. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi: 10.1038/45257. http://dx.doi.org/10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Liang C, Lee J, Inn K, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature Cell Biology. 2008;10(7):776–787. doi: 10.1038/ncb1740. http://dx.doi.org/10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Chen Y, Kensicki E, Li AY, Kong M, Li Y, et al. Resetting glutamine-dependent metabolism and oxygen consumption. Autophagy. 2012;8(10):1477–1493. doi: 10.4161/auto.21228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Debnath J. Ras, autophagy and glycolysis. Cell Cycle. 2011;10(10):1516–1517. doi: 10.4161/cc.10.10.15434. http://dx.doi.org/10.4161/cc.10.10.15434. [DOI] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Molecular Biology of the Cell. 2011;22(2):165–178. doi: 10.1091/mbc.E10-06-0500. http://dx.doi.org/10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorin S, Tol MJ, Bauvy C, Strijland A, Poüs C, Verhoeven AJ, et al. Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy. 2013;9(6):850–860. doi: 10.4161/auto.24083. http://dx.doi.org/10.4161/auto.24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozy F, Karantza V. Autophagy and cancer cell metabolism. Seminars in Cell & Developmental Biology. 2012;23(4):395–401. doi: 10.1016/j.semcdb.2012.01.005. http://dx.doi.org/10.1016/j.semcdb.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–248. doi: 10.1016/j.cell.2004.11.046. http://dx.doi.org/10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Molecular Cell. 2011;42(6):719–730. doi: 10.1016/j.molcel.2011.04.025. http://dx.doi.org/10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends in Molecular Medicine. 2013;19:428–446. doi: 10.1016/j.molmed.2013.04.005. http://dx.doi.org/10.1016/j.molmed.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Marcilla-Etxenike A, Martín ML, Noguera-Salvà MA, García-Verdugo JM, Soriano-Navarro M, Dey I, et al. 2-Hydroxyoleic acid induces ER stress and autophagy in various human glioma cell lines. PLoS One. 2012;7(10):e48235. doi: 10.1371/journal.pone.0048235. http://dx.doi.org/10.1371/journal.pone.0048235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Ot-ín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. The Journal of Biological Chemistry. 2007;282(25):18573–18583. doi: 10.1074/jbc.M701194200. http://dx.doi.org/10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Casey T, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9(17):3515–3533. doi: 10.4161/cc.9.17.12928. http://dx.doi.org/10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino L, Masini M, Novelli M, Beffy P, Bugliani M, Marselli L, et al. Palmitate activates autophagy in INS-1E β-cells and in isolated rat and human pancreatic islets. PLoS One. 2012;7(5):e36188. doi: 10.1371/journal.pone.0036188. http://dx.doi.org/10.1371/journal.pone.0036188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(15):5805–5810. doi: 10.1073/pnas.0507436103. http://dx.doi.org/10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. doi: 10.1016/j.cell.2009.03.048. http://dx.doi.org/10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature Cell Biology. 2009;11(4):385–396. doi: 10.1038/ncb1846. http://dx.doi.org/10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- Mei S, Ni H, Manley S, Bockus A, Kassel KM, Luyendyk JP, et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. The Journal of Pharmacology and Experimental Therapeutics. 2011;339(2):487–498. doi: 10.1124/jpet.111.184341. http://dx.doi.org/10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud M, Martins I, Sukkurwala A, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature Cell Biology. 2011;13(9):1016–1023. doi: 10.1038/ncb2329. http://dx.doi.org/10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clinical Cancer Research. 2012;18(20):5546–5553. doi: 10.1158/1078-0432.CCR-12-0977. http://dx.doi.org/10.1158/1078-0432.CCR-12-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: Process and function. Genes & Development. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. http://dx.doi.org/10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Current Opinion in Cell Biology. 2010;22(2):132–139. doi: 10.1016/j.ceb.2009.12.004. http://dx.doi.org/10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. The Journal of Experimental Medicine. 2011;208(3):455–467. doi: 10.1084/jem.20101145. http://dx.doi.org/10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moruno-Manchón JF, Pérez-Jiménez E, Knecht E. Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. The Biochemical Journal. 2013;449(2):497–506. doi: 10.1042/BJ20121122. http://dx.doi.org/10.1042/BJ20121122. [DOI] [PubMed] [Google Scholar]
- Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annual Review of Pathology. 2013;8:105–137. doi: 10.1146/annurev-pathol-020712-163918. http://dx.doi.org/10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nature Reviews Molecular Cell Biology. 2009;10(7):458–467. doi: 10.1038/nrm2708. http://dx.doi.org/10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332(6032):966–970. doi: 10.1126/science.1205407. http://dx.doi.org/10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld T. TOR-dependent control of autophagy: Biting the hand that feeds. Current Opinion in Cell Biology. 2010;22(2):157–168. doi: 10.1016/j.ceb.2009.11.005. http://dx.doi.org/10.1016/j.ceb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Research. 2011;71(18):5976–5986. doi: 10.1158/0008-5472.CAN-11-1094. http://dx.doi.org/10.1158/0008-5472.CAN-11-1094. [DOI] [PubMed] [Google Scholar]
- Notte A, Leclere L, Michiels C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochemical Pharmacology. 2011;82(5):427–434. doi: 10.1016/j.bcp.2011.06.015. http://dx.doi.org/10.1016/j.bcp.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and Cellular Biology. 2006;26(24):9220–9231. doi: 10.1128/MCB.01453-06. http://dx.doi.org/10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. The Journal of Biological Chemistry. 2000;275(50):39090–39095. doi: 10.1074/jbc.M006198200. http://dx.doi.org/10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- Onodera J, Ohsumi Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. The Journal of Biological Chemistry. 2005;280(36):31582–31586. doi: 10.1074/jbc.M506736200. http://dx.doi.org/10.1074/jbc.M506736200. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death and Differentiation. 2008;15(10):1572–1581. doi: 10.1038/cdd.2008.84. http://dx.doi.org/10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. The Journal of Biological Chemistry. 2003;278(19):16667–16674. doi: 10.1074/jbc.M210998200. http://dx.doi.org/10.1074/jbc.M210998200. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. http://dx.doi.org/10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6(2):239–247. doi: 10.4161/auto.6.2.11062. Retrieved from, http://www.ncbi.nlm.nih.gov/pubmed/20104019. [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of Clinical Investigation. 2003;112(12):1809–1820. doi: 10.1172/JCI20039. http://dx.doi.org/10.1172/JCI200320039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. doi: 10.1126/science.1193497. http://dx.doi.org/10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem AF, Whitaker-menezes D, Lin Z, Martinez-outschoorn UE, Tanowitz HB, Al-zoubi MS, et al. Two-compartment tumor metabolism: Autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012;11(13):2545–2556. doi: 10.4161/cc.20920. http://dx.doi.org/10.4161/cc.20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. http://dx.doi.org/10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. doi: 10.1126/science.1157535. http://dx.doi.org/10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez CG, Penfornis P, Oskowitz AZ, Boonjindasup AG, Cai DZ, Dhule SS, et al. Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis. 2011;32(7):964–972. doi: 10.1093/carcin/bgr029. http://dx.doi.org/10.1093/car-cin/bgr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CR, Schulze A. Lipid metabolism in cancer. The FEBS Journal. 2012;279(15):2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. http://dx.doi.org/10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. The EMBO Journal. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. http://dx.doi.org/10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen JH, Zoncu R, Kim D, Sabatini DM. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell. 2011;19(5):613–628. doi: 10.1016/j.ccr.2011.03.012. http://dx.doi.org/10.1016/j.ccr.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biology. 2011;12(7):226. doi: 10.1186/gb-2011-12-7-226. http://dx.doi.org/10.1186/gb-2011-12-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Cuervo AM. Lipophagy: Connecting autophagy and lipid metabolism. International Journal of Cell Biology. 2012;2012:282041. doi: 10.1155/2012/282041. http://dx.doi.org/10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976. http://dx.doi.org/10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Medicine. 2006;3(10):e420. doi: 10.1371/journal.pmed.0030420. http://dx.doi.org/10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. http://dx.doi.org/10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BRAFV600E-driven lung tumors. Cancer Discovery. 2013;3(11):1272–1285. doi: 10.1158/2159-8290.CD-13-0397. http://dx.doi.org/10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature Cell Biology. 2007;9(10):1142–1151. doi: 10.1038/ncb1634. http://dx.doi.org/10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes & Development. 2011;25(8):795–800. doi: 10.1101/gad.2016211. http://dx.doi.org/10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SH, Shui G, Zhou J, Li JJ, Bay BH, Wenk MR, et al. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin) The Journal of Biological Chemistry. 2012;287(18):14364–14376. doi: 10.1074/jbc.M111.294157. http://dx.doi.org/10.1074/jbc.M111.294157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terés S, Lladó V, Higuera M, Barceló-Coblijn G, Martin ML, Noguera-Salvà MA, et al. 2-Hydroxyoleate, a nontoxic membrane binding anticancer drug, induces glioma cell differentiation and autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(22):8489–8494. doi: 10.1073/pnas.1118349109. http://dx.doi.org/10.1073/pnas.1118349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TLM, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nature Immunology. 2009;10(11):1215–1221. doi: 10.1038/ni.1800. http://dx.doi.org/10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- Tomohiro Y, Klionsky DJ. Endoplasmic reticulum stress: A new pathway to induce autophagy. Autophagy. 2007;3(2):160–162. doi: 10.4161/auto.3653. [DOI] [PubMed] [Google Scholar]
- Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, et al. Chaperone-mediated autophagy degrades mutant p53. Genes & Development. 2013;27(15):1718–1730. doi: 10.1101/gad.220897.113. http://dx.doi.org/10.1101/gad.220897.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. http://dx.doi.org/10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23(3):287–301. doi: 10.1016/j.ccr.2012.11.020. http://dx.doi.org/10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T, et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. The Journal of Biological Chemistry. 2008;283(8):4766–4777. doi: 10.1074/jbc.M706666200. http://dx.doi.org/10.1074/jbc.M706666200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956–959. doi: 10.1126/science.1225967. http://dx.doi.org/10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Molecular Cell. 2008;30(6):678–688. doi: 10.1016/j.molcel.2008.06.001. http://dx.doi.org/10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, et al. EGFR-mediated beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1–16. doi: 10.1016/j.cell.2013.08.015. http://dx.doi.org/10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Developmental Cell. 2011;20(4):444–454. doi: 10.1016/j.devcel.2011.02.006. http://dx.doi.org/10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Weidberg H, Shpilka T, Shvets E, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. The EMBO Journal. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. http://dx.doi.org/10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB. Cellular metabolic stress: Considering how cells respond to nutrient excess. Molecular Cell. 2010;40(2):323–332. doi: 10.1016/j.molcel.2010.10.004. http://dx.doi.org/10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. doi: 10.1126/science.1205405. http://dx.doi.org/10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Forsberg LJ, Viollet B, Brenman JE. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy. 2009;5(8):1155–1165. doi: 10.4161/auto.5.8.10090. Retrieved from, http://www.ncbi.nlm.nih.gov/pubmed/19844161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtkowiak JW, Rothberg JM, Kumar V, Schramm KJ, Haller E, Proemsey JB, et al. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Research. 2012;72(16):3938–3947. doi: 10.1158/0008-5472.CAN-11-3881. http://dx.doi.org/10.1158/0008-5472.CAN-11-3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. 2009;1(4):425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky D. Mammalian autophagy: Core molecular machinery and signaling regulation. Current Opinion in Cell Biology. 2010;22(2):124–131. doi: 10.1016/j.ceb.2009.11.014. http://dx.doi.org/10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes & Development. 2011;25:717–729. doi: 10.1101/gad.2016111. http://dx.doi.org/10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin LI, Kharbanda S, Kufe D. MUC1 oncoprotein promotes autophagy in a survival response to glucose deprivation. International Journal of Oncology. 2009;34:1691–1699. doi: 10.3892/ijo_00000300. http://dx.doi.org/10.3892/ijo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes & Development. 2009;23:798–803. doi: 10.1101/gad.519709. http://dx.doi.org/10.1101/gad.519709.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(25):15077–15082. doi: 10.1073/pnas.2436255100. http://dx.doi.org/10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature Cell Biology. 2009;11(4):468–476. doi: 10.1038/ncb1854. http://dx.doi.org/10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334(6056):678–683. doi: 10.1126/science.1207056. http://dx.doi.org/10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: From growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology. 2011;12(1):21–35. doi: 10.1038/nrm3025. http://dx.doi.org/10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]