

Abstract
The tunable mechanical and structural properties of protein-based hydrogels make them excellent scaffolds for tissue engineering and repair. Moreover, using protein-based components provides the option to insert sequences associated with the promoting both cellular adhesion to the substrate and overall cell growth. Protein-based hydrogel components are appealing for their structural designability, specific biological functionality, and stimuli-responsiveness. Here we present highlights in the field of protein-based hydrogels for tissue engineering applications including design requirements, components, and gel types.
Structural and Mechanical Properties
To function properly and to be compatible with tissue growth, hydrogels for tissue engineering must meet a number of requirements, both physical and biological. The mechanical and structural properties of the gel - tensile strength, stiffness, elasticity, and so on - must be matched to the specific tissue type [1]. The elastic modulus (the ratio of tensile strength to tensile stress) has been shown to directly influence to cell growth and tissue development [2]. Engler et al. investigated the influence of the elastic modulus on cell growth and development by attaching mesenchymal stem cells (MSCs) to substrates with different elastic moduli (see Figure 1a). MSCs are derived from marrow and can differentiate into various anchorage-dependent cell-types, including neurons, myoblasts and osteoblasts. The substrates were chosen to have moduli which mimicked the brain (0.1–1 kPa), muscle (8–17 kPa), and stiff cross-linked collagen matrices (25–40 kPa) (see Figure 1b) [3]. The experiments showed that cells seeded on gels with brain-like, muscle- like and collagen matrix-like viscoelastic properties showed similar morphologies and RNA transcript profiles to neurons, myoblasts and osteoblasts respectively.
Fig. 1.
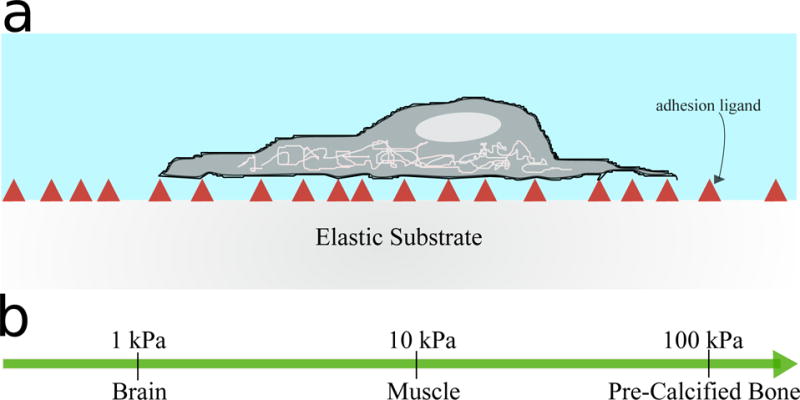
(a) Schematic illustration of cells grown on elastic substrates with varying elastic moduli. Collagen-I was used as the adhesion ligand (red triangles) to promote cell adhesion to gels through via interactions with cellular integrins. (b) Scale showing a range of elastic moduli for different tissue types
Self-Assembly
The desired mechanical and structural properties, such as a cell-type specific elastic modulus, of a protein hydrogel scaffold can be engineered by fine-tuning the self-assembly process. Obas et al. [4] created hydrogels with elastic moduli tunable by the alteration of ionic strength. As scaffolds, they used a 20 amino acid building block β-hairpin molecule (named MAX1), shown in Figure 2a, which consists of alternating valine and lysine residues connected with a tetrapeptide turn promoting sequence. The authors hypothesize that at high ionic strength, the positive charges of the lysine residues are screened and an intramolecular folding event is favored, resulting in a β-hairpin molecule with buried lysine residues and solvent-exposed valine residues. In such a scenario, intermolecular interactions between valine residues on monomers result in aggregation into a β-sheet rich quaternary structure and ultimately hydrogel formation. MAX1 (2% by wt) solutions in the absence of NaCl did not form gels, whereas at concentrations of 20, 150 and 400 mM NaCl, gels formed with elastic moduli of ~100 Pa, ~300 Pa and ~3000 Pa respectively. Thus, increasing ionic strength not only triggers self-assembly but also results in gels with increased rigidity.
Fig. 2.
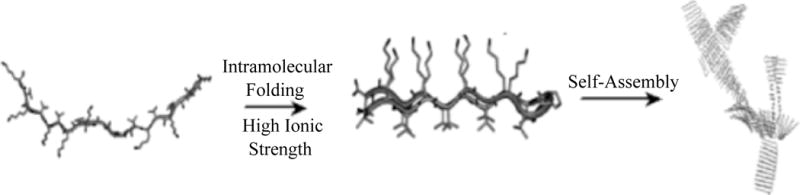
(a) Proposed hairpin structure of the 20 residue long MAX1 peptide. (b) Cartoon representation of intramolecular folding followed by self-assembly in response to increased ionic strength
Self-assembly of α-helical peptides can also lead to hydrogel formation. Banwell et al. designed hydrogel scaffolds comprised of non-covalently cross-linked fibrils formed from short, α-helical peptides [5]. These hydrogelating self-assembling peptides (hSAFs) are formed by two 28-residue peptides designed to assemble into out of register, α-helical coiled-coil fibrils with complementary sticky ends. These α-helical coiled-coils are proposed to assemble longitudinally into fibrils, which then bundle to form mature fibers (Figure 3).
Fig. 3.
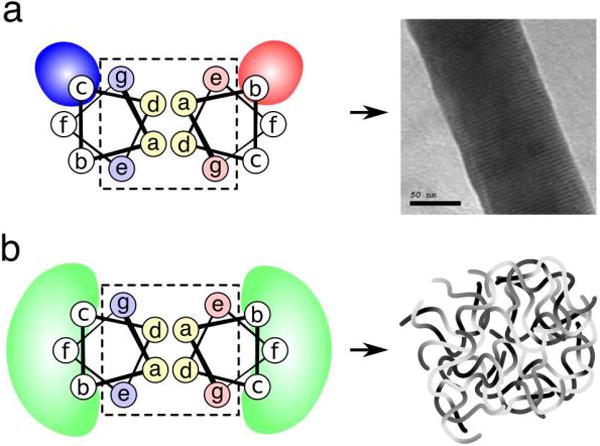
(a) In previous SAF designs, the solvent exposed residues b and c contained specific charged residues, which led to helix alignment and thick fiber formation. (b) In hSAF designs, the b, f, and c positions were changed to combinations of glutamine and alanine with the goal of achieving weaker interactions, leading to small, flexible bundles of fibers
The hSAFs were based on previous designs from the lab based on the coiled-coil heptad repeat abcdefg [6]. The a,d,e, and g positions facilitate dimerization, while b, c, and f are surface-exposed. The b, c, and f positions were mutated to various combinations of alanine and glutamine. From these designs, hSAFAAA and hSAFQQQ both formed hydrogel networks. Using rat adrenal pheochromocyoma (PC12) cells, the therapeutic potential of the hSAFAAA hydrogel network was tested. For these cell biology experiments, a mutation was made at position f replacing an alanine with tryptophan (hSAFAAA-W), allowing for easier quantification of peptide concentration. Serendipitously, this mutation resulted in enhanced stability and gelation at room temperature. PC12 cells were then seeded on hSAFAAA-W hydrogels alone and treated with nerve growth factor. The cells proliferated and showed signs of differentiation into neural cells, indicated by the visualization of neurite projections [5].
Stimuli-responsiveness
In addition to controlling the self-assembly process, the development of “smart” hydrogels that are able to respond to a stimulus can be useful in many applications, such as targeted delivery of cargo. Grove et al. demonstrated the formation of self-assembling hydrogels by taking advantage of an ionic interaction between a protein and peptide [7, 8]. Tetratricopeptide repeat (TPR) domains were concatenated in arrays that formed non-covalent cross-links with their corresponding peptides attached to four-arm star polyethylene glycol (PEG) molecules (Figure 4). The binding interactions forming the junctions of the network were both pH- and ionic strength-dependent, allowing for reversible gelation in response to external stimuli. When placed in solutions containing 500mM NaCl, the hydrogel dissolves, but gelation can be reconstituted through the removal of salt through dialysis [8]. This reversibility can also be demonstrated by alteration of pH [7]. Hydrogelation occurred when components were mixed in a 1:2 stoichiometric ratio (TPR arrays: PEG-peptides). The elastic modulus was measured to be ~270 Pa, which is well above the minimum value necessary (50 Pa –100 Pa) to support mammalian cells in suspension, demonstrating potential in applications for tissue engineering and regeneration [7].
Fig. 4.
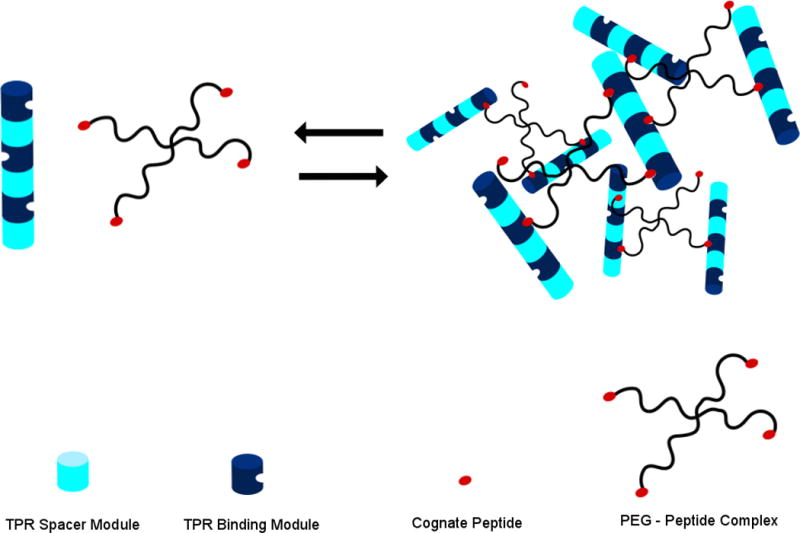
A cartoon showing the reversible formation of a TPR hydrogel network. TPR arrays are shown as blue rods with alternating binding modules (navy) and spacer modules (cyan). Corresponding peptides (red) are displayed on the ends of 4-armed PEG star molecules
Temperature is an alternative stimulus for controlling hydrogelation properties. Glassman et al. developed a thermosensitive hydrogel that is injectable at ambient temperature but exhibits enhanced stiffness and durability at higher, physiological temperatures [9]. This heat-sensitive reinforcement is derived from two separate networks that contribute to the overall properties of the hydrogel. Junctions formed by coiled-coil associations constitute the shear-thinning hydrogel, while the endblocks are comprised of a thermoresponsive polymer called poly(N-isopropylacrylamide) (PNIPAM). The engineered coiled-coil segment was flanked by PNIPAM endblocks (Figure 5). The coiled-coils interact to form a shear-thinning hydrogel, which is reinforced at higher temperatures due to association of the PNIPAM heat-sensitive domains. A shear-thinning hydrogel that transitions to a more rigid network at physiological temperatures offers practical use for tissue repair via injection.
Fig. 5.
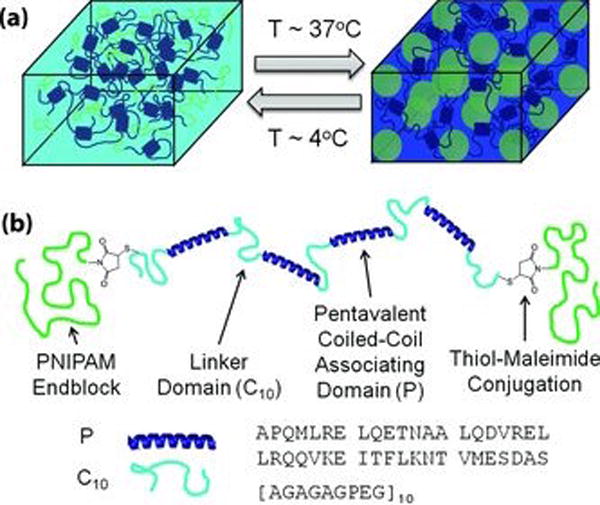
(a) An illustration of the shear-thinning hydrogel (left) which is reinforced to become a stiffer, more rigid network at higher temperatures. (b) A cartoon of the design of the dual-system hydrogel components
Pore Size
Gel scaffold pore size has been hypothesized to both influence nutrient transport to implanted cells and to guide growth in three dimensions. Zeltinger et al. [10] investigated the effects of pore size on the attachment, proliferation, extracellular matrix formation, and cellular metabolism of canine skin and vascular cell types. They generated hydrogel scaffolds with four different pore sizes (<38 μm, 38–63 μm, 63–106 μm, and 106–150 μm), determined by scanning electron microscopy (SEM). Each cell type required a particular pore size for optimal growth; microvascular epithelial cells required a pore size <38μm to form a thin endothelial lining, vascular smooth muscle cells preferred scaffolds >63μm to form uniform tissue, whereas the growth of dermal fibroblasts was independent of pore size. Other experiments have also shown that optimum pore size is cell-type specific and can range from 5μm for neovascularization [6] to greater than 500μm for fibrovascular tissue [7].
Controlling the pore size in protein-based gels requires the tuning of both component concentration [9,10] and the physical conditions of gelation such as temperature [11] or ionic strength [10]. In some instances, finer control of pore size can be accomplished through the use of a rapid freeze-drying technique [11], which is well-illustrated by the work of Mandal et al. [9] in the fabrication of silk fibroin 3D scaffolds with different pore sizes. The researchers created porous protein scaffolds by pouring regenerated silk solutions (2–6wt %) in Teflon molds. The solutions were then frozen for 24 hours at −20, −80 or −196 °C by keeping the bottom of the mold in contact with the freezer. Uniform directional cooling was accomplished by sealing all sides, except the bottom, with an insulated cover (see Figure 6). The slow and induced cooling from the bottom of the mold results in the uniform and directional formation of pores within the scaffold by ice crystal formation. Pore size decreased directly as a function of temperature and inversely as a function of silk fibroin concentration (see Table 1).
Fig. 6.
Schematic representation of directional ice crystal formation within silk fibroin hydrogel scaffolds. Cylinder representation shows upward ice crystal formation over time and circular cross-sections show inward ice crystal formation over time
Table. 1.
Pore sizes obtained under different silk fibroin concentrations and freezing conditions
| Scaffold temperature (°C) | Silk fibroin concentration (wt %) | |||
|---|---|---|---|---|
| 2 | 4 | 6 | ||
| Pore size range (μm) | ||||
| −20 | 200–250 | 100–150 | 75–100 | |
| −80 | 100–150 | 75–100 | 50–75 | |
| −196 | 80–100 | 60–80 | 50–75 |
Another critical component of hydrogel-based tissue engineering is the ability of the system to mimic the function of the extracellular matrix (ECM) by presenting factors necessary to direct tissue formation and regeneration [16]. This can be done through the incorporation of growth factors, which are signaling molecules that can direct cell proliferation, differentiation, healing, and migration [17]. By controlling pore size, Lindsey et al. designed a hydrogel delivery system for nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) [18] using the self-assembling peptide-based hydrogel MAX8 [19]. MAX8 peptide hydrogels are derived from the previously mentioned MAX1 peptides and have unique shear-thinning properties that are attractive for cargo delivery via injection [29]. Their studies showed that encapsulation of growth factors did not affect the physical properties of MAX8, and the rate of growth factor release was dependent on the growth factor concentration within the gel. Additionally, the pore size of the hydrogel decreased inversely with the concentration of MAX8 peptides, allowing tunable encapsulation and release of molecular cargo.
Degradation
The controlled degradation of hydrogel scaffolds is an important physical parameter in tissue engineering. Hydrogels used for implantation must degrade, at a rate that is compatible with the desired function, into non-toxic products that can be easily cleared from the body, leaving healthy tissue post degradation [18]. Depending on the nature of the gel components, degradation can be primarily the result of enzymatic cleavage, hydrolysis or dissolution. Hydrogels formed by covalent cross-links between components must be degraded by enzymatic cleavage or hydrolysis, whereas gels formed by non-covalent physical interactions between components may be degraded by dissolution [19].
Balakrishnan et al. developed an injectable hydrogel with cross-links produced by a Schiff’s base reaction between the lysine or hydroxylysine side chains of gelatin and the aldehyde on the chemical alginate dealdehyde (ADA) catalyzed by Na2B4O7 (borax) (see Figure 7) [20]. To mix the components with the potential to form the gel in situ, researchers used a double-syringe fibrin glue applicator with one syringe filled with the gelatin solution and the other with oxidized ADA plus borax. Both solutions are pushed into a hypodermic needle where the polymers mix, gel, and cross-link within seconds. The susceptibility of the Schiff’s linkage to hydrolysis, and the degradability of ADA and gelatin by the body make the scaffold fully biodegradable over time.
Fig. 7.
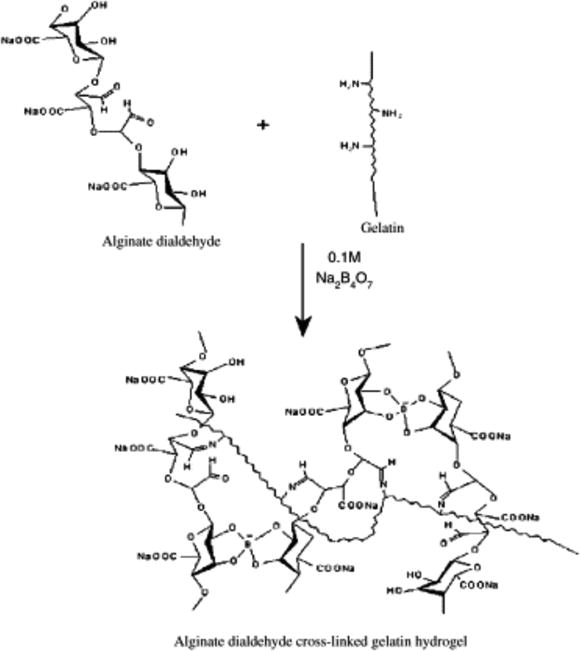
ADA cross-linking with gelatin in the presence of borax (Na2B4O7)
Hydrogels designed to undergo degradation by enzymatic cleavage are well-illustrated by the work of Galler et al. with a multidomain peptide (MDP) hydrogel [21]. MDPs are amphiphilic peptide systems with amino acids arranged in a block motif that self-assemble into nanofibers. The core interactions of supramolecular assembly are driven by the “B” block, which consists of alternating hydrophobic (leucine, L) and hydrophilic (serine, S) residues (see Table 2). In aqueous environments, the side chains present on opposite sides of the peptide backbone and two peptides can come together at the hydrophobic face to create a dimeric “sandwich” stabilized by the packing of the leucine residues (Figure 8) [22]. The dimers are proposed to interact through anti-parallel β-sheet hydrogen bonding oriented down the fiber axis (Figure 8). The flanking “A” block has charged amino acids, lysine (K), that create water solubility as well as an electrostatic repulsion that works against fiber formation when unshielded, thus can be manipulated to control fiber assembly and length [23]. The addition of multivalent ions can be used to screen these charges and allows fiber elongation beyond dimerization and eventual gel formation.
Table. 2.
Names and sequences of peptides studied
| MDP# | Sequence |
|---|---|
| 1 | KK-SLSLSLSLSLSL—KK |
| 2 | KK-SLSLSLSLSLSL—KKGRGDS |
| 3 | KK-SLSL SLSLSL-KK |
Fig. 8.
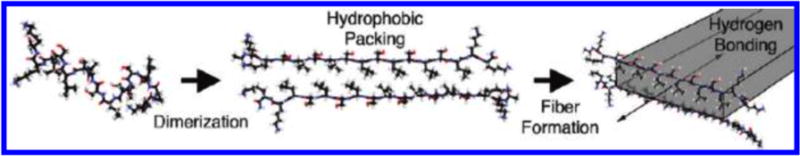
Cartoon illustration of MDP fiber formation
To encode stimuli-responsiveness into this design, the researchers created a peptide arranged in an ABA block motif containing the matrix metalloproteinase 2 (MMP-2) leucine-arginine-glycine consensus cleavage motif, see peptide MDP3 in Table 2 [24]. Gelation of the peptide was induced by the addition of phosphate buffered saline. Gels containing the MMP-2 cleavage sequence, (i.e. made using peptide MDP3), degraded upon treatment with MMP-2 whereas gels without the cleavage sequence, MDPs 1 and 2, did not. MDP2 included the well-known amino acid cell adhesion motif RGD to increase the hydrogel compatibility for cell culture. RGD sequences have been shown increase cell spreading, proliferation and migration within hydrogel scaffolds [25].
Another example of enzymatically degradable hydrogels is demonstrated by the work of Sun et al. using SpyTag/SpyCatcher technology [26]. SpyTag and SpyCatcher are peptide and protein components, respectively, that originated from splitting a fibronectin-binding domain of a bacterial adhesin and engineering it into separate parts [27]. Upon mixing, SpyCatcher and SpyTag spontaneously form an isopeptide linkage between the sidechains of a lysine on SpyCatcher and an aspartate on SpyTag. This has been shown by Zhang et al. to be a powerful tool for covalent attachment and the creation of interesting protein topologies [28].
By encoding SpyTag and SpyCatcher into specific parts of ELP sequences, Sun et al. designed components intended to form covalent linkages leading to hydrogelation. The first construct, called AAA, was comprised of three SpyTag sequences separating by two ELP sequences (Figure 9). The second construct, referred to as BB, consisted of an ELP sequence flanked by two SpyCatchers (Figure 9). The BB construct also contained an internal integrin-binding RGD sequence and an MMP-1 cleavage site embedded in the ELP region, with the goals of cellular adhesion and potential of enzymatic degradation [26]. To demonstrate potential in tissue engineering applications, Sun et al. showed that the hydrogels could encapsulate mouse 3T3 fibroblasts. After growth in the matrix for 24 hours, the majority of cells were still alive and displayed “elongated morphologies”, suggesting that cellular adhesion and matrix remodeling had occurred [26]. This design illustrates one major advantage of protein-based hydrogels, which is that functionality can be encoded into the sequence in a straightforward way.
Fig. 9.
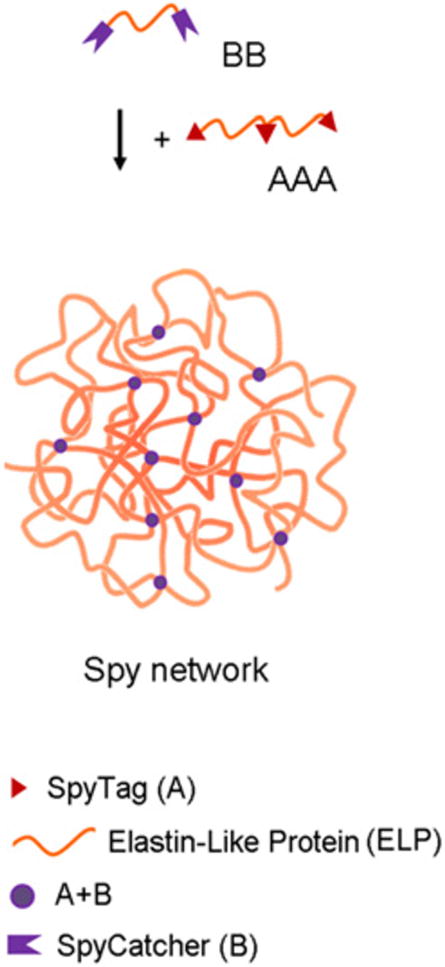
A cartoon illustrating the placement of SpyTag and SpyCatcher on the two constructs labeled AAA and BB. Covalent linkages via isopeptide bonding form the hydrogel network. Gelation occurred within 5 min of mixing AAA and BB at 10 wt % in a 2:3 molar ratio at room temperature
Biocompatibility
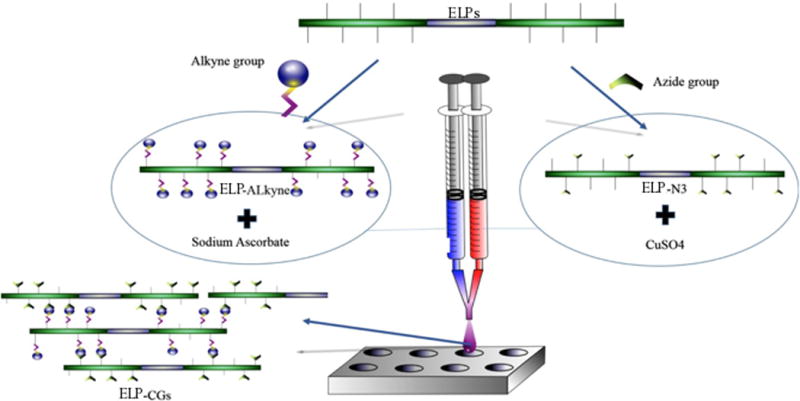
Hydrogel biocompatibility and low immunogenicity are essential for in vivo applications. Researchers must either choose components with known biocompatibility or engineer components to be more compatible. For protein-based hydrogels, site-specific mutagenesis and/or truncation can be used by researchers to remove immunogenic epitopes, while maintaining the maintenance gelation properties [29]. The ease of manipulation of protein-based materials in this fashion to fine-tune their properties is a unique and attractive feature. Some proteins, such as elastin-like polymers (ELPs), naturally exhibit low immunogenicity [30] and can confer this biocompatibility onto tissue engineering scaffolds. Testera et al. used ELPs as protein-based polymer building blocks for a tissue engineering hydrogel scaffold [31]. Using click chemistry, ELPs were decorated at lysine residues with either alkyne or azide groups to create two gel components (see Figure 10). Mixing of the two components resulted in cross-linking between azide and alkyne groups, forming hydrogels that were able to maintain long-term viability with respect to human primary cell types in both surface and 3D cultures, thus showing the importance of hydrogel scaffold biodegradability to viability.
Fig. 10.
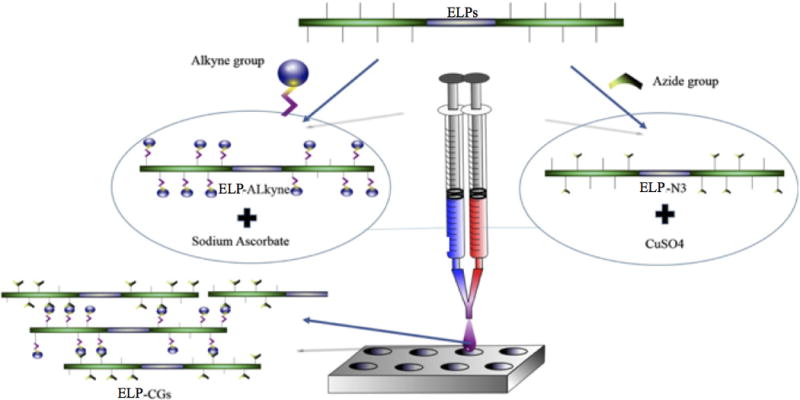
Schematic showing ELP click gels as a two component system
Conclusion
Protein-based hydrogels are excellent candidates tissue engineering and repair scaffolds because of the ease of designability. Here, we have shown that researchers have been able to use sequence-to-structure information to take advantage of the self-assembly process as well as create hydrogel scaffolds with tunable elastic moduli, stimuli-responsiveness, varying pore sizes and increased biocompatibility. As the field of protein engineering continues to develop and create new tools, protein-based hydrogel systems will continue to develop as an innovative tool for tissue engineering.
References
- 1.Whang K, Healy KE, Elenz DR, et al. Engineering bone regeneration with bioabsorbable scaffolds with novel microarchitecture. Tissue Eng. 1999;5:35–51. doi: 10.1089/ten.1999.5.35. [DOI] [PubMed] [Google Scholar]
- 2.Georges PC, Miller WJ, et al. Matrices with compliance comparable to that of brain tissue select neuronal over glial growth in mixed cortical cultures. Biophys J. 2006;90(8):3012–3018. doi: 10.1529/biophysj.105.073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engler AJ, Sen S, Sweeny HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 4.Obas B, Kreysinger J, Rajagopal K, et al. Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus. Macromolecules. 2004;37(19):7331–7337. [Google Scholar]
- 5.Banwell EF, et al. Rational design and application of responsive α-helical peptide hydrogels. Nature Materials. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandya MJ, et al. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry. 2000;39:8728–8734. doi: 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]
- 7.Grove TZ, et al. Stimuli-responsive smart gels realized via modular protein design. J Am Chem Soc. 2010;132(40):14024–14026. doi: 10.1021/ja106619w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grove TZ, et al. A modular approach to the design of protein-based smart gels. Biopolymers. 2012;97(7):508–517. doi: 10.1002/bip.22033. [DOI] [PubMed] [Google Scholar]
- 9.Glassman MJ, et al. Reinforcement of shear thinning protein hydrogels by responsive block copolymer self-assembly. Advanced Functional Materials. 2013;23:1182–1193. doi: 10.1002/adfm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mygind T, Stiehler M, Baatrup A, et al. Mesenchymal stem cell ingrowth and differentiation on coralline hydroxyapatite scaffolds. Biomaterials. 2007;28(6):1036–1047. doi: 10.1016/j.biomaterials.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Zeltinger J, Sherwood JK, Graham DA, et al. Effect of pore size and void fraction on cellular adhesion, proliferation, and matrix deposition. Tissue Eng. 2001;7(5):557–572. doi: 10.1089/107632701753213183. [DOI] [PubMed] [Google Scholar]
- 12.Brauker JH, Carr-Brendel VE, Martinson LA, et al. Neovascularization of synthetic membranes directed by membrane architecture. J Biomed Mater Res. 1995;29(12):1517–1524. doi: 10.1002/jbm.820291208. [DOI] [PubMed] [Google Scholar]
- 13.Mandal BB, Kundu SC. Cell proliferation and migration in silk fibroin 3D scaffolds. Biomaterials. 2009;30(15):2956–2965. doi: 10.1016/j.biomaterials.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Kim U, Park J, Li C, et al. Structure and properties of silk hydrogels. Biomacromolecules. 2004;5:786–792. doi: 10.1021/bm0345460. [DOI] [PubMed] [Google Scholar]
- 15.Ren L, Tsuru K, Hayakawa S, Osaka A. Novel approach to fabricate porous gelatin-siloxane hybrids for bone tissue engineering. Biomaterials. 2002;23(24):4765–4773. doi: 10.1016/s0142-9612(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 16.Kangwon L, et al. Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. J R Soc Interface. 2011;8:153–170. doi: 10.1098/rsif.2010.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu J, Marchant RE. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev Med Devices. 2011;8(5):607–626. doi: 10.1586/erd.11.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen S, Bano MC, Cima LG, et al. Design of synthetic polymeric structures for cell transplantation and tissue engineering. Clin Mater. 1993;13:3–10. doi: 10.1016/0267-6605(93)90082-i. [DOI] [PubMed] [Google Scholar]
- 19.Nicodemus GD, Bryant SJ. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng. 2008;14(2):149–165. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balakrishnan B, Jayakrishnan A. Self-cross-linking biopolymers as injectable in situ forming biodegradable scaffolds. Biomaterials. 2005;26(18):3942–3951. doi: 10.1016/j.biomaterials.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Galler KG, Aulisa L, Regan KR, et al. Self-assembling multidomain peptide hydrogels: designed susceptibility to enzymatic cleavage allows enhanced cell migration and spreading. J Am Chem Soc. 2010;132:3217–3223. doi: 10.1021/ja910481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aulisa L, Dong H, Hartgerink JD. Self-assembly of multidomain peptides: sequence variation allows control over cross-linking and viscoelasticity. Biomacromolecules. 2009;10:2694–2698. doi: 10.1021/bm900634x. [DOI] [PubMed] [Google Scholar]
- 23.Galler KM, Hartgerink JD, Cavender AC, et al. A customized self-assembling peptide hydrogel for dental pulp tissue engineering. Tissue Eng. 2012;18:178–184. doi: 10.1089/ten.tea.2011.0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 25.Jeschke B, Meyer J, Jonczyk A, et al. RGD-peptides for tissue engineering of articular cartilage. Biomaterials. 2002;22(16):3455–3463. doi: 10.1016/s0142-9612(02)00052-2. [DOI] [PubMed] [Google Scholar]
- 26.Sun F, et al. Synthesis of bioactive protein hydrogels by genetically encoded SpyTag-SpyCatcher chemistry. Proc Natl Acad Sci U S A. 2014;111(31):11269–11274. doi: 10.1073/pnas.1401291111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zakeri B, et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A. 2012;109(12):E690–697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang WB, et al. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. J Am Chem Soc. 2013;135(37):13988–13997. doi: 10.1021/ja4076452. [DOI] [PubMed] [Google Scholar]
- 29.Liu Z, Zhou H, Wang W, et al. A novel method for synthetic vaccine construction based on protein assembly. Sci Rep. 2014;4:7266. doi: 10.1038/srep07266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Cabello JC, Martin L, Alonso M, et al. Recombinamers as advanced materials for the post-oil age. Polymer. 2009;50(22):5159–5169. [Google Scholar]
- 31.Testera AM, Girotti A, Gonzalez de Torre I, et al. Biocompatible elastin-like click gels: design, synthesis and characterization. J Mater Sci. 2015;26:105. doi: 10.1007/s10856-015-5435-1. [DOI] [PubMed] [Google Scholar]