

Abstract
Nuclear receptors regulate gene expression in response to environmental cues, but the molecular events governing the cell-type specificity of nuclear receptors remain poorly understood. Here we outline a role for a non-coding RNA in modulating the cell type-specific actions of LXRs, sterol-activated nuclear receptors that regulate the expression of genes involved in cholesterol homeostasis and that have been causally linked to the pathogenesis of atherosclerosis. We identify the lncRNA MeXis as an amplifier of LXR-dependent transcription of the critical cholesterol efflux gene Abca1. Mice lacking the MeXis gene show reduced Abca1 expression in a tissue-selective manner. Furthermore, loss of MeXis in mouse bone marrow cells alters chromosome architecture at the Abca1 locus, impairs cellular responses to cholesterol overload, and accelerates the development of atherosclerosis. Mechanistic studies reveal that MeXis interacts with and guides promoter binding of the transcriptional coactivator DDX17. The identification of MeXis as a lncRNA modulator of LXR-dependent gene expression expands our understanding of the mechanisms underlying cell-type selective actions of nuclear receptors in physiology and disease.
Introduction
The accumulation of excess cholesterol by macrophages within the arterial wall is a pivotal step in the pathogenesis of atherosclerosis. The ability of macrophages to integrate metabolic and immune signaling in response to environmental cues and lipid excess is therefore an important determinant of disease susceptibility1-2,3. LXRs are ligand-dependent transcription factors that regulate expression of genes involved in macrophage responses to cholesterol, and also modulate inflammatory signaling 4,5. Activation of LXRs promotes reverse cholesterol transport through induction of a cadre of genes, including Abca1, which encodes the plasma membrane transporter ABCA1. This ATP-dependent transporter is critical for HDL generation and its function is compromised in Tangier disease, a syndrome characterized by both HDL deficiency and accelerated atherosclerosis 6,7.
LncRNAs have been shown to function through diverse mechanisms, including exerting direct transcriptional effects in response to environmental cues 8. A number of lncRNAs have been shown to regulate the expression of neighboring genes; however, the mechanisms by which noncoding gene activation serve local regulatory functions remain to be fully clarified 9,10. Although a number of lncRNAs have been shown to have sequence-specific features, recent work highlights the strong contribution of neighboring promoter activity, including the processes of transcriptional initiation and splicing, on gene expression 11. In this work we characterize an LXR-responsive lncRNA that affects transcriptional pathways linked to macrophage cholesterol efflux and atherosclerosis. Our results suggest that cell-type selective action of lncRNAs may contribute to the temporal and spatial gene activation patterns of nuclear receptors.
Results
Regulation of macrophage lncRNAs in response to cholesterol loading
LXRs influence the expression of a large repertoire of genes linked to lipid metabolism in a context-specific fashion. We noted that the gene encoding critical cholesterol efflux mediator ABCA1 was much more highly induced by synthetic LXR in macrophages than in other cell types such as hepatocytes and adipocytes (Supplementary Fig. 1a). To gain insight into the basis for this cell-type selective LXR response, we performed genome-wide transcriptional profiling on mouse peritoneal macrophages treated with or without the synthetic LXR agonist GW3965. The expression of canonical protein-coding LXR target genes was robustly induced by LXR agonist treatment, as expected (Supplementary Fig. 1b). Consistent with established roles of LXR in metabolism, the network of ontology terms for GW3965-induced genes showed robust enrichment for lipid regulatory processes and macrophage-specific pathways (Supplementary Fig. 1c). Interestingly, we also observed that LXR activation induced a limited number of lncRNAs (Supplementary Fig. 1d and Supplementary Table 1). Several of these lncRNAs are located in neighboring genomic regions to protein-coding genes with established roles in mediating LXR effects on metabolism. Gene ontology analysis of protein-coding genes that were nearest to these lncRNAs showed enrichment for cholesterol metabolic processes, consistent with the idea that a subset of these non-coding RNAs may modulate the activity of adjacent protein-coding genes (Supplementary Table 2).
From the list of regulated lncRNAs in Supplementary Table 2, one of the most robustly induced hit that also showed evidence of LXR binding to its gene regulatory regions by LXR ChIP-seq studies 12 was a predicted transcript annotated as AI427809. We named this transcript MeXis (Macrophage-expressed LXR-induced sequence). Notably, the MeXis gene is located in close proximity to the established LXR target genes Abca1 and LeXis 13 (Fig. 1a). Analysis of chromatin signatures from ENCODE indicated that MeXis and Abca1 are distinct genes with separate promoters (Fig. 1a) 14,15. 5’ and 3’ rapid amplification of cDNA ends (RACE) experiments defined the MeXis transcript ends (Supplementary Fig. 2). RNA-copy number analysis showed that MeXis was highly expressed in murine macrophages (Supplementary Fig. 3a) and real-time PCR analysis showed that MeXis and Abca1 expression was induced by LXR (GW3965) and RXR (LG268) agonists in primary macrophages in an LXR-dependent manner (Fig. 1b). MeXis expression was also induced in macrophages by physiologic lipid signals such as oxidized or acetylated LDL (Fig. 1c). In addition, oxysterol agonist of LXR induced MeXis expression in macrophages (Supplementary Fig. 3b). Intriguingly, MeXis showed a distinct pattern of LXR-dependent regulation compared to the lncRNA LeXis, which is expressed in liver but not macrophages (Fig. 1d) 13. MeXis was also expressed in adipose tissue (Supplementary Fig. 3c).
Figure 1. Regulation of the non-coding RNA MeXis by LXR.
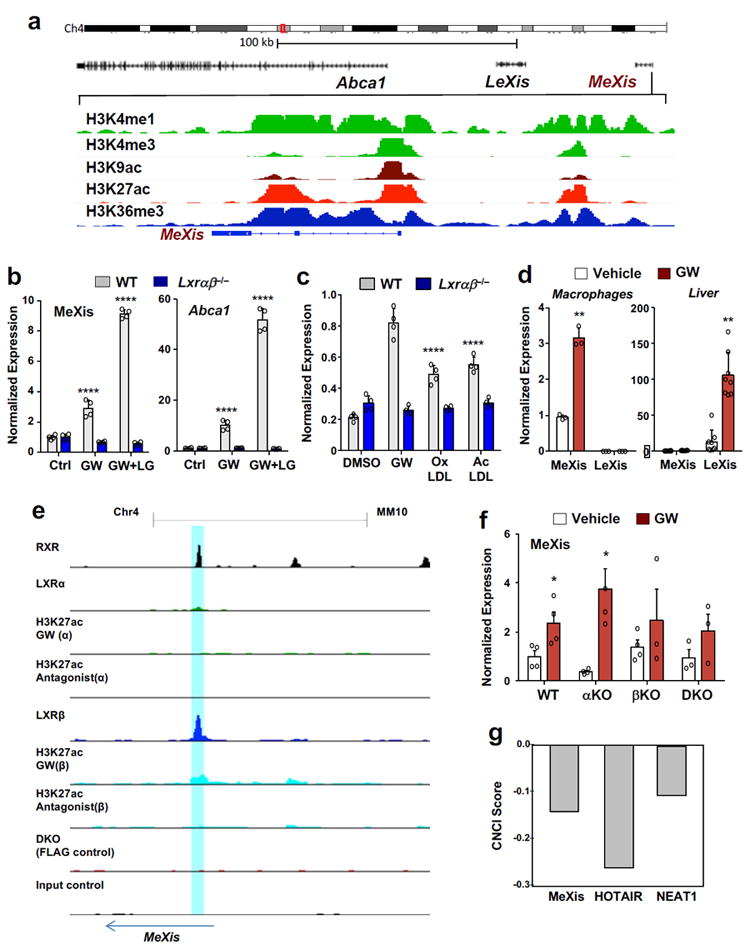
A. Schematic representation of the MeXis gene locus on the Integrative Genome Viewer (IGV) (top) and histone marks from LICR ENCODE data in the immediate region of the MeXis gene (bottom). B. Real-time PCR analysis of MeXis and Abca1 expression in primary mouse macrophages treated with vehicle (Ctrl), GW3965 (GW, 0.5 μM) and/or the RXR ligand LG268 (LG, 50 nM). Results are representative of four independent experiments. Values are means ± SD. **** P<0.0001 by Two-way ANOVA followed by multiple comparisons test (Dunnett’s). C. Real-time PCR analysis of MeXis expression in primary mouse macrophages treated with vehicle (Ctrl), GW3965 (GW, 0.5 μM) , oxidized LDL (oxLDL, 50 μg/ml), or acetylated LDL (acLDL, 50 μg/ml). Results are representative of four independent experiments. Values are means ± SD. **** P<0.0001 by Two-way ANOVA followed by multiple comparisons test (Dunnett’s). D. Real-time PCR analysis of MeXis and LeXis expression in primary mouse macrophages treated with vehicle or GW3965 (GW, 0.5 μM) (n = 3/group) or in liver harvested from WT mice treated with vehicle or GW3965 (40 mg/kg, by gavage) for 3 consecutive days (n = 8/group). Values are means ± SD. E. Chip-Seq analysis of LXR binding at the MeXis gene locus.Chip for LXRα and LXRβ in 3xFLAG-LXRα and 3xFLAG-LXRβ expressing immortalized bone marrow macrophage cell using FLAG, RXR and H3K27ac antibody. Cells treated with LXR agonist (GW3965, 1uM) and antagonist (GW2033, 1uM) shown. Blue shaded bar highlighting binding of LXR /RXR at MeXis.F. Real-time PCR analysis of MeXis expression in primary mouse macrophages from mice of the indicated genotypes treated with vehicle or GW3965 (GW, 0.5 μM). N=(4 for WT, αKO, βKO vehicle and 3 for DKO & βKO GW). Experiment repeated once with similar results. Values are means ± SEM. * P<0.05 by two-sided student’s t-test. G. Prediction of coding potential of the indicated lncRNAs using Coding-Non-Coding Index (CNCI) software. A negative value indicates low coding potential.
MeXis is an LXR-responsive lncRNA that influences Abca1 expression
We identified an LXR-response element (LXRE) within the MeXis promoter region that was bound by LXRs in ChIP-seq analysis (Fig. 1e). In contrast to most of LXR target genes which are responsive to both LXRs, the MeXis promoter was bound by LXRβ but not LXRα. Consistent with this result, MeXis expression was induced by LXR activation in WT and Lxra−/− but not Lxrb−/− peritoneal macrophages, whereas Abca1 expression was responsive to both LXRs (Fig. 1f and Supplementary Fig. 3d).To further explore isoform specific regulation of MeXis, we treated immortalized LXR DKO bone-marrow-derived macrophages (BMDMs), as well as LXR DKO BMDMs stably expressing LXRα, with GW3965. Abca1 was induced with LXR activation in LXRα-expressing BMDMs but not in DKO controls, whereas MeXis was not induced in either cell type (Supplementary Fig. 3e). These data further suggest that MeXis is an LXRβ-selective target gene.
Computational scores that distinguish protein-coding from non-coding RNAs predicted a low-coding potential for the MeXis transcript (Fig. 1g). Although the MeXis transcript does contain a number of short potential open-reading frames, we found no evidence of translation and production of a protein product from MeXis using a coupled in vitro transcription-translation assay (Supplementary Table 3 and Supplementary Fig. 4a). Single molecule RNA FISH in immortalized mouse bone-marrow derived macrophages confirmed nuclear localization of MeXis (Supplementary Fig. 4b). MeXis was predominantly located in the insoluble nuclear pellet enriched for chromatin, whereas the protein-coding 36b4 mRNA was predominantly present in cytoplasm (Supplementary Fig. 4c). Even when exogenously expressed at elevated levels, MeXis was predominantly nuclear (Supplementary Fig. 4d). This localization strongly suggests that MeXis most likely acts as a nuclear RNA, rather than being translated into a short protein in the cytoplasm.
Since some lncRNAs have been shown to regulate the expression of adjacent genes 16,17, we hypothesized that loss of MeXis may impact the expression of Abca1. In support of this idea, siRNA-mediated knockdown of either MeXis or RXRα/β in mouse peritoneal macrophages decreased Abca1 transcript levels (Fig. 2a). An antisense oligonucleotide (ASO) targeting MeXis also reduced Abca1 levels (Supplementary Fig. 4e). Reciprocally, stable overexpression of MeXis in macrophages enhanced Abca1 expression and cholesterol efflux capacity to ApoA-I acceptors (Fig. 2b, 2c, Supplementary Fig. 4f).
Figure 2. MeXis regulates Abca1 expression and function.
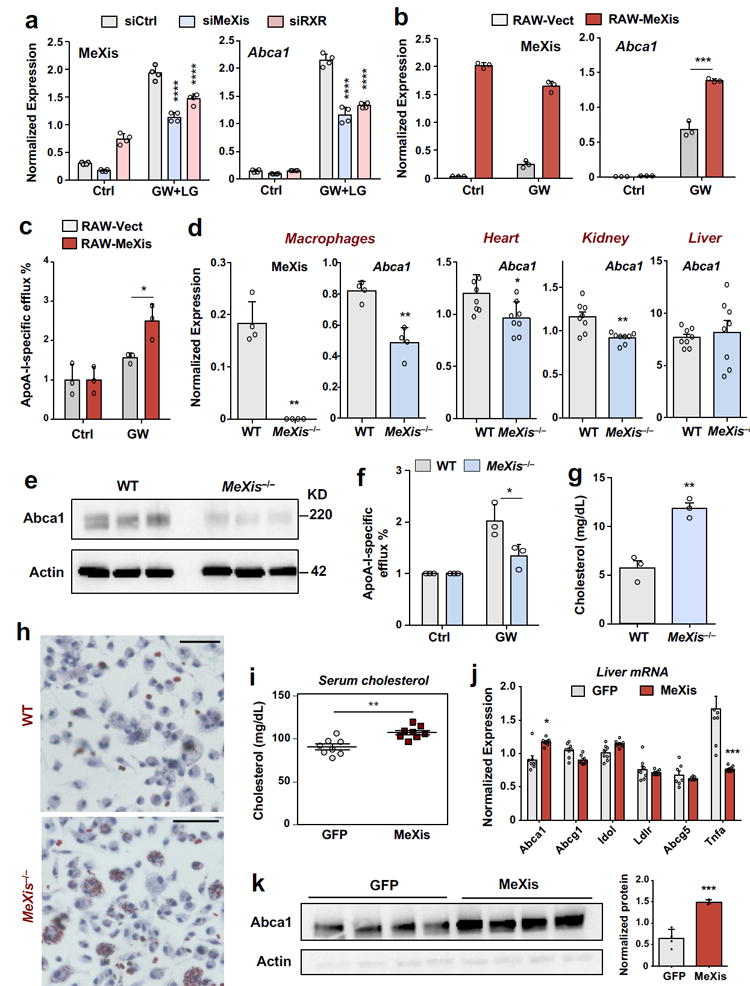
A. Real-time PCR analysis of MeXis and Abca1 expression from primary macrophages treated with the indicated siRNAs (50 nM) followed by either vehicle (Ctrl) or a combination of GW3965 (GW, 0.5 μM) and the RXR ligand LG268 (LG, 50 nM) for 36 h. Results are representative of four independent experiments. Values are means ± SD.**** P<0.0001 by Two-way ANOVA followed by multiple comparisons test (Sidak’s). B. Real-time PCR analysis of MeXis and Abca1 expression 10 days after stable overexpression of control vector (Vect) or MeXis in RAW cells treated with vehicle (Ctrl) or GW3965 (GW, 0.5 μM). Results are representative of three independent experiments. Values are means ± SD.*** P < 0.001 by two-sided student’s t-test. C. Cholesterol efflux in the presence of ApoA-I from RAW macrophages loaded with [3H]cholesterol (1.0 μCi/ml) and treated with the acyl-CoA:cholesterol O-acyltransferase inhibitor (2 μg/ml) and either with DMSO or LXR ligand (1 μM GW3965). ApoA-I-specific efflux represents percent radiolabelled cholesterol efflux in the presence of ApoA-I normalized to DMSO. Experiments were conducted in triplicate. Data are expressed as mean ± SD.* P<0.05 by two-sided student’s t-test.D. Real-time PCR analysis of MeXis and Abca1 expression in primary mouse macrophages (results are representative of four independent experiments; values are means ± SD) and of Abca1 expression in heart, kidney and liver of mice fed a western diet for 3 weeks (N = 8/group; values are means ± SEM).* P<0.05; ** P < 0.01 by two-sided student’s t-test.E. Western blot analysis of Abca1 levels in primary mouse macrophages of WT and MeXis−/− mice treated with GW (0.5 μM for 16 hours). Actin was used as a loading control. The experiment repeated twice with similar results. F. Cholesterol efflux in the presence of ApoA-I or HDL from WT or MeXis−/− macrophages loaded with [3H]cholesterol (1.0 μCi/ml) and treated with the acyl-CoA:cholesterol O-acyltransferase inhibitor (2 μg/ml) and either with DMSO or LXR ligand (1 μM GW3965). Experiments were conducted in triplicate. Data are expressed as mean ± SD. * P<0.05 by two-sided student’s t-test.G. Cholesterol content measured in peritoneal macrophages isolated from mice on western diet for 12 weeks (N = 3/group). ** P < 0.01 by two-sided student’s t-test. H. Oil-red-O staining of peritoneal macrophages isolated from WT or MeXis−/− mice and treated with oxidized LDL (100 μg/ml) for 72 h.The experiment was repeated 3 times with similar results.Scale bars, 50 μm. I. Total serum cholesterol levels in 10-week-old chow-fed male C57BL/6 mice transduced with adenoviral vectors encoding GFP control (Ad-GFP) or MeXis (Ad-MeXis) for 6 days (n = 8 per group). ** P < 0.01 by two-sided student’s t-test. J. GFP and MeXis expression in liver 6 days after transduction of mice with Ad-GFP or Ad-MeXis, respectively (n = 8 per group except Abcg1 n=7 per group).* P<0.05; *** P < 0.001 by two-sided student’s t-test. Data are expressed as mean ± SEM. K. Left, western blot analysis of Abca1 levels in liver from the mice in I (n = 4 per group). Right, quantification of protein levels normalized to actin. Data expressed as mean ± SD. *** P < 0.001 by two-sided student’s t-test.
MeXis deficiency affects Abca1 expression, cholesterol efflux, and atherogenesis
To better decipher the contributions of MeXis to macrophage metabolism, we generated MeXis-knockout mice (Supplementary Fig. 5a). We used a strategy in which FLP excised the targeting cassette following homologous recombination, such that no extraneous promoter sequences were left behind after recombination. Consistent with our siRNA studies, MeXis−/− peritoneal macrophages showed decreased Abca1 transcript levels (Fig. 2d). Notably, in western-diet fed mice, Abca1 mRNA expression was differentially altered across tissues in MeXis−/− as compared to WT mice (Fig. 2d). For example, liver Abca1 was not significantly different between groups, whereas heart and kidney Abca1 expression was lower in MeXis−/− compared to WT mice (Fig. 2d). We also confirmed that Abca1 protein levels were reduced in MeXis−/− compared to WT macrophages (Fig. 2e). These results suggest that MeXis augments Abca1 expression in a context-specific manner.
We observed minimal changes in expression of other LXR target genes in response to MeXis overexpression, knockdown or knockout (Supplementary Fig. 5b-d), suggesting that MeXis does not regulate all LXR target genes equivalently. Unbiased transcriptomic analysis of WT and MeXis−/− primary peritoneal macrophages showed enrichment for lipid metabolic pathways and inflammatory signaling (Supplementary Table. 4). Consistent with the alteration in Abca1 expression in MeXis−/− macrophages, we observed a decrease in ApoA-I-dependent efflux capacity in peritoneal macrophages from MeXis−/− compared to WT mice (Fig. 2f, Supplementary Fig. 4g). By contrast, loss of MeXis in macrophages was not associated with a change in cholesterol uptake (Supplementary Fig. 4h). Furthermore, compared to WT macrophages, MeXis−/− peritoneal macrophages showed higher cholesterol content in vivo in western-diet fed mice (Fig. 2g). Additionally, morphological “foam cell” formation, as assessed by oil-red O staining, was in MeXis−/− peritoneal macrophages (Fig 2h, Supplementary Fig. 4i). In accordance with the idea that MeXis primarily affects macrophage responses, there were no differences in serum cholesterol or triglyceride levels between MeXis−/− and WT mice, either when fed chow or western diet (Supplementary Fig. 6). Taken together, these results suggest that MeXis is required for maximal Abca1 expression in the face of macrophage cholesterol loading.
To further explore the ability of MeXis to act as an RNA and to confirm its ability to modulate Abca1 expression in vivo, we expressed MeXis in mouse liver using an adenoviral vector. Notably, we observed an increase in serum cholesterol levels—a hallmark feature of enhanced hepatic ABCA1 expression—in mice ectopically expressing MeXis in liver (Fig. 2i). We also confirmed that MeXis expression in liver induced Abca1 mRNA without affecting the expression of other LXR target genes (Fig. 2j). ABCA1 protein levels were correspondingly increased by MeXis expression (Fig. 2k).
Cholesterol efflux capacity is an important determinant of atherosclerotic plaque development and progression. To examine the impact of MeXis on atherosclerosis, we reconstituted the bone marrow of irradiated Ldlr−/− mice with WT or MeXis−/− hematopoetic cells (Fig. 3a). Real-time PCR analysis of bone marrow from recipient mice collected at the time of sacrifice confirmed engraftment (Supplementary Fig. 7a). We found reduced Abca1 expression and enhanced inflammatory gene expression in the bone marrow of mice transplanted with MeXis−/− bone marrow (Supplementary Fig. 7a). En face analysis of atherosclerotic plaque area after 17 weeks of western diet feeding showed markedly increased atherosclerotic burden in mice transplanted with MeXis−/− compared to WT bone marrow (Fig. 3a, 3b). No differences in plasma cholesterol or triglyceride levels were observed between the two groups (Supplementary Fig. 7b). Consistent results were obtained when we assessed atherosclerosis by quantification of oil-red O–stained aortic root sections (Fig 3c, 3d). Histological analysis showed larger lesions in MeXis−/− bone marrow-transplanted compared to WT bone marrow-transplanted mice, as well as increased staining for the macrophage-specific marker CD68 (Fig. 3e and Supplementary Fig. 7c). Laser-capture microdissection of CD68-positive cells revealed decreased expression of Abca1 and MeXis in macrophages within atherosclerotic lesions from MeXis−/− bone marrow-compared to WT bone marrow-transplanted mice (Fig 3f,g). Taken together, these results demonstrate that macrophage MeXis expression is a determinant of susceptibility to atherosclerosis in mice.
Figure 3. Loss of MeXis impairs macrophage Abca1 expression and accelerates atherosclerosis.
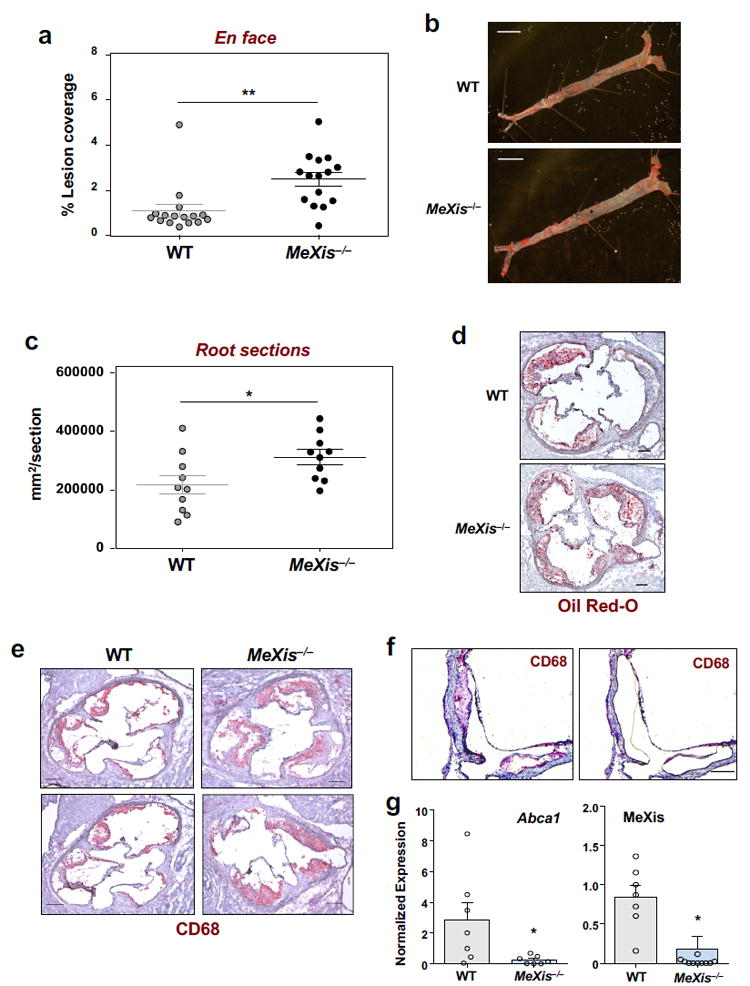
Ldlr−/− mice were transplanted with WT or MeXis−/− bone marrow and maintained on a Western diet for 17 weeks. A. Percentage of aorta surface area with atherosclerotic plaque by en face analysis. Data are mean ± SEM. (N = 16 WT, 15 MeXis). ** P < 0.01 by two-sided student’s t-test. B. Representative photographs (from 16 WT and 15 MeXis−/−) from en face analysis of aortas. Scale bars, 5 mm. C. Quantification of lesion area from oil-red O-stained aortic root sections. Mean ± SEM. (N = 10/ group). * P<0.05 by two-sided student’s t-test. D. Oil Red-O staining of frozen sections from the aortic roots. Representative of 10 per group. Scale bars, 200 μm. E. Representative histology of the aortic root stained with the macrophage marker CD68 and H & E. Representative of 8 per group. Scale bars, 200 μm.F. Representative images of an aortic lesion before and after laser capture microdissection of CD68-positive cells from MeXis−/− mice. Scale bar, 200 μm. G. Abca1 and MeXis expression in laser capture samples as determined by realtime PCR. Samples taken from 6 animals WT and 4 MeXis−/−. Number of samples for Abca1expression 7 per group and MeXis expression 7WT & 11MeXis−/−. Data are mean ± SEM. * P<0.05 by two-sided student’s t-test.
MeXis interacts with DDX17 and modulates Abca1 transcription
eRNA expression can serve as a surrogate marker of enhancer site activity and transcriptional activation 18. Enhancer elements surrounding Abca1 in macrophages have been defined previously (Supplementary Fig. 8) 19. Interestingly, MeXis−/− macrophages showed decreased eRNA expression from Abca1 enhancers in response to LXR activation, as compared to WT cells (Fig. 4a). To assess whether MeXis can affect transcription of the Abca1 gene in trans, we crossed Abca1Flox/Flox mice with heterozygous MeXis−/+ mice. This strategy enabled us to measure effects of MeXis deficiency specifically on the Abca1 transcript that is trans to the mutant MeXis allele. We found that a reduction in MeXis expression reduced Abca1 expression in trans (Fig. 4b, Supplementary Fig. 9a).
Figure 4. MeXis alters chromosome architecture at the Abca1 locus.
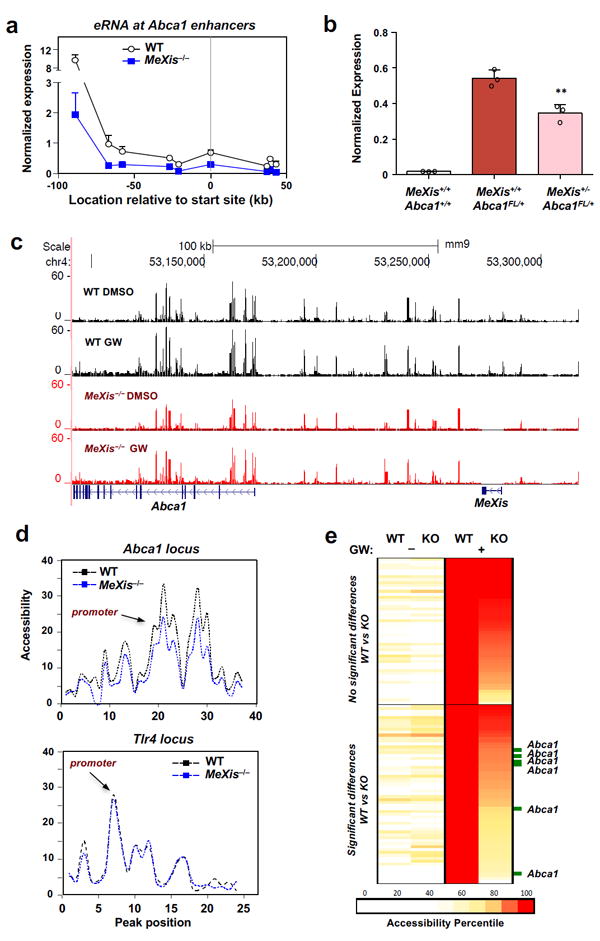
A. Gene expression of enhancer RNAs (short noncoding RNAs transcribed from enhancer elements)at the Abca1 locus in primary mouse macrophages of WT or MeXis−/− mice treated with GW3965 (1 μM). The x-axis indicates the location at which gene expression was measured relative to the Abca1 transcription start site. N= 2WT, 3 MeXis−/−. Mean ± SD. B. Expression from the Abca1FL allele in primary macrophages from mice of the indicated genotypes (n=3/group). Mean ± SD.** P < 0.01 by two-sided student’s t-test. C. Genome browser view of ATAC-seq data from primary mouse macrophages of WT or MeXis−/− mice treated with DMSO control or GW3965 (1 μM) for 3 h. Reads were from 4 individual samples per group. D. ATAC seq analysis showing accessibility at peaks around the Abca1 and Tlr4 genepromoters in WT or MeXis−/− macrophages with GW stimulation. Peak position arbitrary numbers the accessibility peaks shown in C at the Abca1 locus in relationship to promoter TSS. E. Heat map of accessibility regions in WT and MeXis−/− macrophages with or without GW3965 treatment.Top of panel shows genome-wide accessibility sites significantly induced by GW3965 in both WT &MeXis−/− macrophages. Bottom panel shows accessibility sites significantly induced by GW3965 only WT macrophages.
These observations led us to hypothesize that MeXis may influence chromatin dynamics at the Abca1 locus, thereby modulating its transcription. Consistent with this idea, ATAC-seq performed on peritoneal macrophages showed blunted accessibility at multiple sites within the Abca1 gene locus in the setting of MeXis deficiency (Fig. 4c). Quantification of accessible sites neighboring the Abca1 promoter revealed decreased accessibility in MeXis−/− macrophages. By contrast, we found no difference in accessibility at the Tlr4 locus (Fig. 4d), indicating selectivity of the effect of MeXis deficiency for certain chromatin regions. Genome-wide normalization revealed that the LXR target gene activation signature was largely preserved in MeXis-deficient macrophages; however, multiple sites within the Abca1 locus showed differential accessibility between WT and MeXis−/− macrophages (Fig. 4e and Supplementary Fig. 9b). By contrast, the accessibility of very few sites was substantially altered at other LXR target gene loci (Supplementary Fig. 9c). Unbiased genome-wide analysis of differentially-accessible chromatin sites between WT and MeXis−/− macrophages showed enrichment for lipid transport and related processes (Supplementary Fig. 9d).
To further investigate the mechanism of MeXis action, we used an unbiased lncRNA:chromatin affinity capture technique 20 to pull down MeXis from macrophage extracts and identify interacting proteins (Supplementary Fig.10a). Analysis of the MeXis interactome by mass spectrometry identified DDX17, an established nuclear receptor coactivator21,22, as a potential interacting partner (Supplementary Fig.10b).We confirmed a robust interaction between MeXis and DDX17 in RNA immunoprecipitation studies in mouse macrophages, either with or without the use of a cross-linking agent (Fig. 5a). The prior characterization of DDX17 as a transcriptional coactivator led us to hypothesize that DDX17 may serve this function at the Abca1 locus. In line with this idea, ChIP-PCR analysis revealed that DDX17 was enriched at LXR binding sites in Abca1 enhancer regions in macrophages (Fig 5b). Moreover, DDX17 binding was substantially reduced as a consequence of MeXis deletion (Fig. 5b). Interestingly, the pattern of LXR binding was also altered as a consequence of loss of MeXis, with reduced occupancy at the Abca1 promoter, but enhanced binding at an intronic site (Fig. 5b). These results suggest that MeXis facilitates the coactivator actions of DDX17 to enhance LXR-mediated Abca1 expression. To definitively determine if MeXis is recruited to the Abca1 gene locus, we used ChIRP-qPCR. Using this technique, we found that MeXis bound to at a number of sites at Abca1 that show differential accessibility between MeXis−/− and WT macrophages (Fig. 5c)
Figure 5. Identification of DDX17 as a binding partner of MeXis.
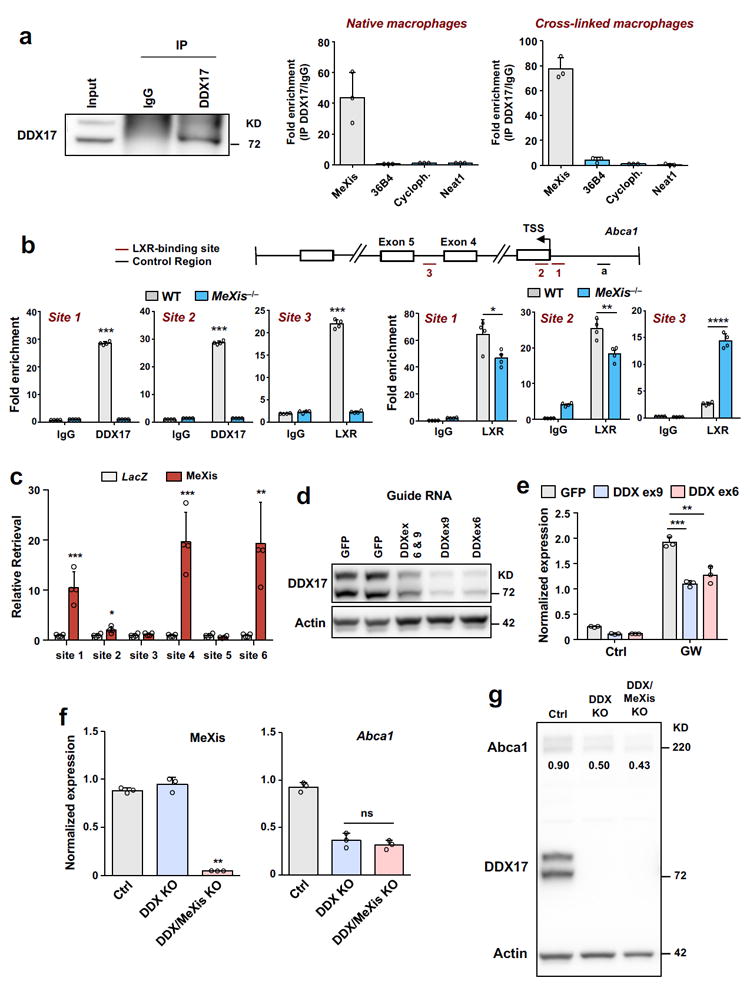
A. RNA immunoprecipitation analysis of DDX17. Following immunoprecipitation using IgG control or DDX17 antibody from mouse peritoneal macrophages that were untreated or treated with the cross-linking agent formaldehyde,expression of MeXis, 36B4, cyclophilin (cyloph) and Neat1 was determined by qPCR analysis. N=3/group. Data are mean ± SD. B. Recruitment of DDX17 and LXR at the Abca1 gene locus in mouse macrophages from WT or MeXis−/− mice as determined by ChIP qPCR analysis. Site 1, 2, and 3 are regions containing LXR binding elements known to bound LXR from Chip studies. Data are expressed as percent input retrieved normalized to an upstream control site (region a). N=4/group. Data are mean ± SD. * P<0.05; ** P < 0.01; **** P < 0.0001 by two-sided student’s t-test. C. ChIRP-qPCR analysis of a series of ATAC-seq sites at the Abca1 locus. Sites 1-6 are accessibility sites shown in figure 4e induced with LXR activation in WT but not MeXis−/− macrophages (n=4/group). Chirp probes designed against LacZ or MeXis. Mean ± SD. * P<0.05; ** P < 0.01; *** P < 0.001 by two-sided student’s t-test. D. Western blot analysis of DDX17 levels in lentivirus-transduced immortalized iBMDM (pool of selection-positive cells). Lentiviruses contained either a GFP control or the indicated guide RNAs targeting the DDX17 locus. Actin was used as a loading control.Representative of two independent western blots. E. Abca1 expression in iBMDMs transduced with the indicated lentiviruses and treated with DMSO (Ctrl) or GW3965 (1 μM) for 12 hours (n= 3 per group). Data are mean ± SD. ** P < 0.01; *** P < 0.001 by Two-way ANOVA followed by two-sided student’s t-test. F. MeXis or Abca1 expression in iBMDMs (n= 3 per group). Ctrl is GFP, DDX is DDX17KO, DDXMeXKO is DDX17/MeXis double knockout. Data are mean ± SD. NS= Not significant at P < 0.05 by two-sided student’s t-test. G. Western blot analysis of Abca1 and DDX17 levels in iBMDMs from F.Numbers in blot are quantitative Abca1 protein normalized to control (actin).
To further define the importance of DDX17 in Abca1 regulation, we generated DDX17-deficient immortalized bone marrow-derived macrophages utilizing CRISPR-Cas editing (Supplementary Fig.10c). Deletion of DDX17 using two different excision strategies reduced Abca1 levels at baseline and in response to LXR activation (Fig. 5d, e). These results strongly suggest that DDX17 is required for maximal Abca1 expression in macrophages. To analyze the epistatic relationship between DDX17 and MeXis, we generated DDX17/MeXis double-knockout macrophages using CRISPR-Cas. Deletion of MeXis in the setting of DDX17 deficiency failed to further reduce Abca1 transcript or protein levels, as compared to DDX17 single knockout (Fig. 5f-g). Taken together, these results demonstrate that DDX17 contributes to MeXis-dependent regulation of Abca1.
Finally, we assessed whether the LXR-MeXis-Abca1 pathway is operational in human cells. Genome-batch conversion between human and mouse genome builds revealed the genomic region surrounding the MeXis/Abca1 locus showed a degree of conservation between species (Supplementary Fig.11). A noncoding human RNA transcript in this region, identified as TCONS00016111, showed some sequence conservation with MeXis (Fig. 6a and Supplementary Fig.11). Intriguingly, TCONS00016111 expression was induced in response to LXR activation in human THP-1 macrophages (Fig 6b). Moreover, ASOs targeting TCONS00016111 reduced ABCA1 transcript levels and ApoA-I-specific cholesterol efflux (Fig. 6c-d). Furthermore, lentiviral transduction of MeXis into MeXis−/− or THP-1 macrophages increased ABCA1 expression (Fig. 6e, f).
Figure 6. Functional conservation of the LXR-MeXis axis in humans.
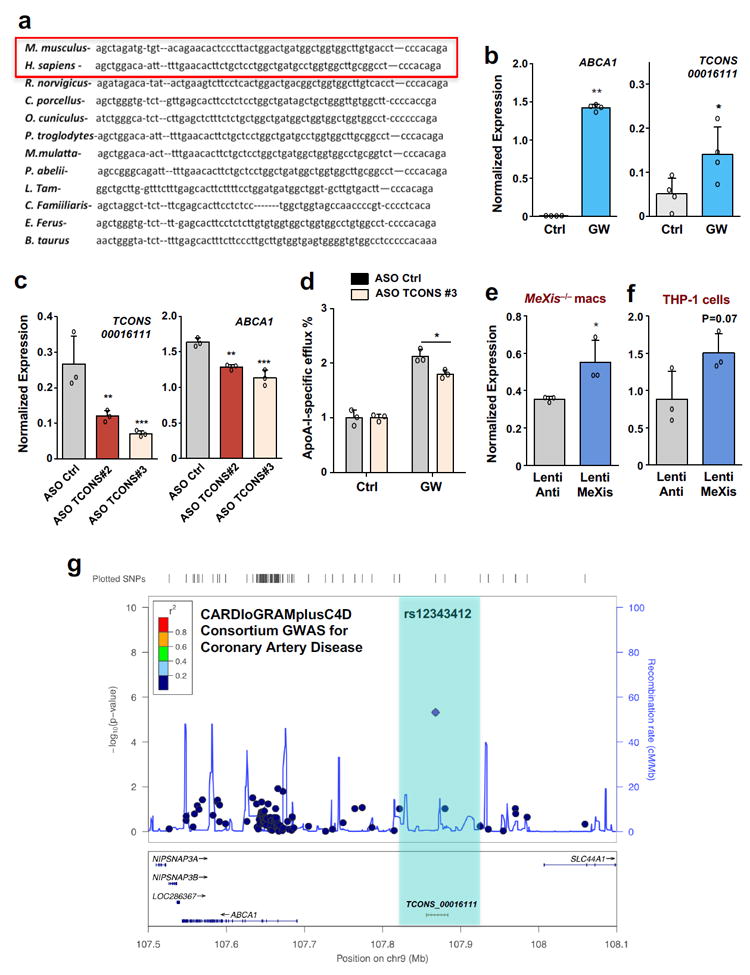
A. Sequence similarity across species at the MeXis gene locus. B. ABCA1 and TCONS00016111 expression in differentiated THP-1 monocytes treated with DMSO (Ctrl) or GW3965 (0.5 μM) for 16 hours (N=4 per group). Mean ± SD. * P < 0.05 by two-sided student’s t-test. C. ABCA1 and TCONS00016111 expression in differentiated THP-1 cells treated with the indicated ASOs (50 nM) and GW3965 (0.5 μM) (N=3 per group). Data are mean ± SD. * P < 0.05; ** P < 0.01; *** P < 0.001 by one-way ANOVA followed by multiple comparisons test (Dunnett’s).D. Cholesterol efflux in the presence of ApoA-I of THP-1 cells treated with the indicated ASO, loaded with [3H]cholesterol (1.0 μCi/ml), and treated with acyl-CoA:cholesterol O-acyltransferase inhibitor (2 μg/ml) and with either DMSO or LXR ligand (1 μM GW3965) (n=3/group). Data are mean ± SD. * P < 0.05 by two-sided student’s t-test. E. Abca1 gene expression in MeXis−/− BMDM macrophages treated with control (antisense MeXis) or MeXis lentivirus (n=3 per group). Data are mean ± SD. * P < 0.05 by two-sided student’s t-test. F. Abca1 expression in THP-1 cells treated with control or MeXis lentivirus (n=3 per group).Mean ± Two-sided student’s t-test. Not significant at P<0.05. 95% Confidence Interval (-0.1025 to 1.338). G. Regional association plot of variants at TCONS-0016111 and the risk of coronary artery disease in humans from the CARDIoGRAMplusC4D Consortium.
Finally, GWAS from the CARDIOGRAM Plus consortium23 identified a moderately significant association between a SNP overlapping the TCONS00016111 transcript and human coronary artery disease (P=4.78E–6) (Fig. 6g). These results support the notion that an LXR-lncRNA-Abca1 axis is operational in humans and may have relevance for human disease.
Discussion
Previous work has identified important roles for eRNAs in regulating gene expression programs 24. Our study expands the repertoire of noncoding RNA-mediated gene activation by showing that a lncRNA can help specify nuclear receptor regulatory circuits. Our results suggest that induction of MeXis expression in response to activation of LXRs augments Abca1 expression and macrophage cholesterol efflux in a context-specific manner. It seems likely that MeXis may contribute to cell type-specific regulation of Abca1 expression by LXRs. Our work, however, does not exclude the possibility that MeXis may have targets other than Abca1.
Our findings are consistent with previous reports that intergenic lncRNAs and their genomically-adjacent protein-coding genes tend to exhibit similar spatiotemporal expression profiles 25,26. Recent work suggests that at least some of the local effects of lncRNAs are the results of promoter activity and the act of transcription rather than lncRNA-specific features11. Although our results do not exclude the possibility that cis regulatory elements at the MeXis locus exert an influence on Abca1 transcription, our data support that transcript-dependent actions also contribute to Abca1 regulation.
This study also reveals an unexpected to role for a lncRNA in cardiovascular disease. Control of macrophage gene expression by the LXR pathway is causally linked to the pathogenesis of atherosclerosis. Identification of MeXis fills a gap in our understanding of pathways that control cellular responses to cholesterol overload and atherosclerosis.It is tempting to speculate that targeting the LXR-MeXis-Abca1 axis may enhance macrophage reverse cholesterol transport to treat or prevent atherosclerotic disease while bypassing undesirable side effects of LXR activation in other tissues.
Materials & Methods
Reagents, Plasmids, and Gene Expression
GW3965 was synthesized as previously described27. LG268 was from Ligand Pharmaceuticals. Oxysterols were purchased from Sigma and used as described28. Simvastatin sodium salt was from Calbiochem. Ligands were dissolved in dimethyl sulfoxide before use in cell culture. MeXis was amplified from RNA purified from GW3695-treated primary mouse peritoneal macrophages using KOD polymerase (Millipore), forward 5’GTCTGAAAAGGAAGTTGAAGAAGA3’ and reverse 5’ AAGGAATCTAGTAAATTTTAATACTAA3’ primers. Primers were designed to provide flanking attB sequences and a SacI site at the immediate 3’ end. For details of oligonucleotide sequences please see supplementary file number 12. The fragments were then cloned into pDONR221 using the Gateway system and the minimal SV40 polyadenylation sequence was inserted at the SacI site. ON-Targetplus siRNAs (Catalog number R-050345) were used with Dharmafect 4 reagent per the manufacturer’s recommendations for knockdown studies (Dharmacon). For gene expression analysis, RNA was isolated using TRIzol reagent (Invitrogen) and analyzed by real-time PCR using an Applied Biosystems 7900HT sequence detector or Applied Biosystems Quant Studio 6 Flex. Results are normalized to 36B4 or cyclophilin. The following antibodies were used for immunohistochemistry: CD68 (MCA1957GA, AbD) 1:400 with secondary antibody biotin-SP-conjugated AffiniPure goat anti-rat IgG (H+L) (Jackson Laboratories). Details of antibodies and full-length blots are provided in supplementary file 13 and 14. In brief, for immunoblot analysis, the following antibodies were used: ABCA1 (Novus) 1:1,000, and actin (Sigma) 1:10,000.. For ChIP analysis, we used the LXR antibody previously described29; DDX17 antibody was generated by Douglas Black, UCLA30. plentiCRISPR v2 was used for lentivrius production; the guide RNA was inserted into Bsmb1-digested plasmid and the plasmid was ligated with T4 DNA ligase. Guide insertions were verified via sequencing.For lentiviral overexpression studies, MeXis was cloned into the pSLIK-Zeo vector system 31 and modified for lncRNA expression by replacement of the sequence between NcoI and MfeI sites in the pEN_TT entry vector with a minimal SV40 polyadenylation signal. The MeXis sequence was inserted into the NcoI site immediately upstream of the polyadenylation signal. Lentiviruses were packaged and purified as described 32. For retroviral and adenoviral expression, the MeXis sequence was transferred from the pEN-TTpA entry vector to pBABE and pAd/CMV/V5-DEST, respectively, using the Gateway cloning system (Invitrogen Life Technologies).
Animals and diets
Mice were housed in a temperature-controlled room under a 12-hour light/12-hour dark cycle and under pathogen-free conditions. Experiments used 12 week- old male mice unless otherwise specified. Age-matched male Ldlr−/− mice on C57BL/6 background were purchased from Jackson Laboratories (catalog number 2207). Mice lacking LXRα and/or LXRβ were originally provided by David Mangelsdorf, University of Texas Southwestern Medical Center, Dallas, Texas, USA33 and were backcrossed to on C57BL/6 background for more than 10 generations. MeXis global knockout mice on a C57BL/6 background were generated at UCDavis Knockout Mouse Project (KOMP) using the strategy outlined in supplemental figure 5. Abca1flox/flox mice on a C57BL/6 background were obtained from John Parks, Wake Forest University34. Mice were fed a chow diet except as indicated, where mice were placed on a Western diet (21 percent fat, 0.21 percent cholesterol; D12079B; Research Diets Inc.). We measured cholesterol and triglycerides as previously described13. At the time of sacrifice, tissues and blood were collected by cardiac-puncture and immediately frozen in liquid nitrogen and stored at – 80°. For adenoviral infections, age-matched male mice were injected with 2.0 × 109 PFU by tail-vein injection and were sacrificed six days later following a six-hour fast. Tissue was processed for isolation of RNA as above. All animal experiments were approved by the UCLA Institutional Animal Care and Research Advisory Committee and performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Cell culture
Primary peritoneal macrophages were isolated four days after thioglycollate injection and prepared as described27. Mouse primary hepatocytes were isolated as previously described and cultured in William’s E medium with 5 percent FBS27. Peritoneal cells were incubated in 0.5-10 percent FBS in DMEM, with or without 5 μM simvastatin and 100 μM mevalonic acid (EMD Biosciences). Five to eight hours later, cells were pretreated with DMSO or appropriate ligand overnight. The in vitro translation assay was performed using the TnT Coupled Transcription/Translation System (PROMEGA) according to the manufacturer’s protocol.THP-1 cells from ATCC were treated were treated with custom siRNA from Dharmacon (50 nM) and harvested 36 h later. Modified gapmer ASOs were obtained from IDT and used with Dharmafect 4 or Attractene transection reagent at a final concentration of 50 nM. THP-1 cells were differentiated with phorbol 12-myristate 13-acetate (PMA) at 10 ng/ml followed by GW3965 treatment. Immortalization of bone marrow-derived macrophages (iBMDMs)to generate stable cell lines was performed as previously described34. These stable cell lines were obtained via puromycin selection of lentrivirus-treated cells starting from a single clonal population. Gene expression or western blot results are shown for a pool of selected cells unless otherwise noted. All cell lines were tested for mycoplasma contamination. Subcellular RNA fractions were obtained according to the protocol of Bhatt et al.31 and as we previously described 13,35.
Cholesterol efflux and uptake
Assays were performed as previously described 27. Briefly, peritoneal macrophages from WT or MeXis−/− mice were labeled with [3H]cholesterol (1.0 μCi/ml) (Perkin Elmer) in the presence of acyl-CoA:cholesterol O-acyltransferase inhibitor (2 μg/ml)followed by treatment with DMSO or LXR(1 μM GW3965). After equilibration of the cholesterol pools and washing, cells were incubated in DMEM containing 0.2% BSA in the absence or presence of apoA-I (Meridian Life Sciences) (15 μg/ml) or HDL purchased from Lee Biosolutions (Catalog number 361-10-0.01) (50 μg/ml) for 6 h. The data are presented as percent apoA-I- or HDL-specific efflux. For uptake assays, resident mouse peritoneal mouse macrophages were obtained from age-matched WT and MeXis−/− mice. Peritoneal macrophages were suspended in starvation media (1% Lipoprotein Deficient Serum (LPDS), simvastatin, mevalonic acid) and incubated at 37° C for 16 hours. Cells were then treated with DiI-acetyl-LDL (Invitrogen) at concentrations of 0 μg/ml, 50 μg/ml and 100 μg/ml and incubated at 37 ° C for an additional 4 h. After the 4 h incubation period, cells were washed three times with PBS containing bovine serum albumin (2mg/ml), harvested, and lysed in RIPA buffer. To measure the uptake of DiI-acetyl-LDL by peritoneal macrophages, cell lysates were analyzed for fluorescence using a Clario Star plate reader with an excitation of 554 nm and an emission of 571 nm. Values for fluorescent intensity were normalized to total protein concentration and displayed relative to the untreated group.
Atherosclerosis analysis
Immunohistochemistry of sections and preparation and staining of frozen and paraffin-embedded sections from aortas were performed as described previously 36. Atherosclerosis in the aortic roots and the descending aortas (en face) were quantified by computer-assisted image analysis as described 37. Atherosclerotic lesions at the aortic valve were analyzed as described 27. Laser capture microdissection was used as previously outlined 38, except that an LMD7000 Laser Microdissection System (Leica) was used at the UCLA Advanced Microscope CNSI core lab. The Arcturus PicoPure RNA Isolation Kit (Applied Biosystems) was used for RNA processing and amplification.
RACE
The 5’ and 3’ ends of the MeXis transcript were defined using mouse peritoneal macrophage RNA and the FirstChoice RLM-RACE kit (Ambion) according to manufacturer’s protocol, with modifications. Briefly, for 5’ RACE, degraded mRNA 5’ ends were dephosphorylated with CIP and then full-length mRNA was decapped with TAP. Following 5’RACE adapter ligation, reverse transcription was performed using the SuperScriptIII First-Strand Synthesis system (Invitrogen) and MeXis-specific primers. For 3’ RACE, RNA was reverse transcribed using SuperScriptIII First-Strand Synthesis system (Invitrogen) and adapter-linked oligo dTs. The resulting cDNA was amplified by nested PCR across a 55-65 °C melting temperature gradient using KOD polymerase (Millipore), with the inner primers containing attB sequences. Aliquots of reactions were inspected on 1% agarose gels for product size and abundance. Products of selected PCR reactions were purified using the NucleoSpin Gel and PCR Cleanup kit (Clontech) and were inserted into pDONR221 by Gateway cloning procedures. Cloned fragments were sequenced and then aligned to the mouse genome with the BLAST analysis tool. RNA fractionation assays were done as previously described 13.
RNA Sequencing
RNA sequencing libraries were constructed with the TruSeq RNA Sample Prep Kits (Illumina) on RNA isolated from peritoneal macrophages treated with or without GW3965. Samples were indexed with adapters and submitted for paired-end 2 × 100-bp sequencing using an Illumina HiSeq2000 instrument. RNA-seq reads were aligned with TopHatv2.0.2 to the mouse genome, version mm939. Transcripts were assessed and quantities were determined by Cufflinks v2.0.2, using a GTF file based on Ensembl mouse NCBI37. Comparisons of expression levels were made using FPKM values using Cuffdiff from the Cufflinks package 40. In order to analyze lncRNA expression in more depth, we built a comprehensive, non-redundant mouse gene database by merging the Gencode and Noncode annotations. Reads were aligned to the mouse genome (GRC38/mm10 assembly) using STAR41. Gene-level count summaries were estimated with HTSeq. We employed a comprehensive, non-redundant mm10 genome annotation that incorporates protein and lncRNA genes from Gencode and lncRNA annotations from Noncode v4 42. Coding potential scores for each transcript in the merged annotation were estimated with CPAT 43. In addition, we used CNCI as another method to classify coding potential of sequences of interest 44. Gene-level expression estimates in units of FPKMs were computed in-house as above. For downstream analysis, the following entries were masked: 1) genes with no counts, 2) genes with low (<50bp) nucleotide sequence uniqueness, and 3) Gencode biotypes other than protein coding or lncRNA. The final genome annotation we employed for downstream analyses comprised 50,608 genes (21098 protein-coding, 29510 lncRNA). Expression fold changes after GW treatment were estimated from FPKM ratios. Enrichment analyses on the set of up-regulated coding and non-coding genes included pathways analyses with MetaScape and cis-regulatory region enrichment with default parameters in GREAT 45.
Lipid analysis
Cellular lipid content was obtained using a Folch extraction. Briefly, chloroform extracts were dried under nitrogen and solubilized in water. Tissue and serum cholesterol and triglycerides were determined using a commercially available enzymatic kit (Wako). Mice were fasted for at least 6 hours prior to blood collection and sacrifice.
ChIP
ChIP studies were performed as described elsewhere 13. Briefly primary mouse peritoneal macrophages (approximately 40 million per sample) were cross-linked using a final formaldehyde concentration of 1% at room temperature for 10 minutes. The reaction was quenched with the addition of glycine. Sonicated chromatin isolated with lysis buffer (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100)was incubated overnight at 4°C with control IgG or 2.5 ug per ml of lysate of anti-LXR (previously generated by JaKobsson et al from Karolinska Institute)29, or anti-DDX17 antibody (gift of Douglas Black, UCLA). Protein A Dynabeads (Invitrogen) were added for 4 hours. Washing, reverse crosslinking and sample elution were performed as previously outlined 13. Chip-seq for LXR isoforms was done using Flag-tagged LXRα or LXRβ in iBMDM cell lines34. Macrophages (12 × 106) were crosslinked using a double fixation protocol with 2 mM disuccinimidyl glutarate for 30 min and 1% methanol-free ultrapure formaldehyde for 10 min before quenching with 2 M glycine. Cells were lysed with RIPA buffer and, after chromatin shearing by sonication using a Bioruptor (Diagenode) apparatus, incubated overnight with protein G magnetic Dynabeads (Invitrogen) coupled with 3 μg of either anti-FLAG M2 (SIGMA #F3165) or anti-H3K27ac (Abcam #ab4729) antibodies. Immunoprecipitated DNA was purified using Qiagen purification kit (Qiagen #28144). For high-throughput sequencing, a minimum of 10 ng of DNA was obtained by pooling DNA from 10 independent ChIP preparations (for FLAG-LXR sequencing) or 6 different ChIP preparations (for H3K27ac sequencing). DNA was then used for library preparation and subsequent Illumina HiSeq sequencing by the Centre de Regulació Genomica (CRG, Barcelona, Spain) genome facility. The primary data have been deposited to GEO, accession number GSE104027.
Fluorescent RNA FISH
Custom design RNAscope probes against MeXis were prepared and obtained from Advanced Cell Diagnostics (Catalog number 495011). MeXis was visualized in immortalized mouse bone-marrow derived macrophages using an RNAscope assay and the Multiplex Fluorescent Reagent Kit V2, following the manufacturer’s recommended protocol with use of a HybEZ oven (Advanced Cell Diagnostics); however, we used a protease dilution of 1:5 instead of 1:30.
RNA immunoprecipitation
We followed the protocol outlined by Tsai and colleagues46. Briefly cellular extracts from native or cross-linked (1% formaldehyde) primary peritoneal macrophages were treated with DNASE I followed by incubation with DDX17 antibody or IgG control overnight. Complexes were captured using Dynabead Protein G (Life technologies) and RNA was eluted using the RNeasy micro kit (Qiagen).
Microarrays
cDNA microarray analysis was performed for primary peritoneal macrophages treated with GW3695. Transcriptional profiling was performed at the University of California, Los Angeles, TCGB core facility using the Agilent SurePrint G3 Gene Expression array. Data were analyzed using GeneSpring software (Agilent Technologies) and DAVID Functional Analysis Tools 47. Data available at GSE107977.
ChIRP
Chirp was performed as described20 with a few modifications. First, cross-linking of RAW264.7 cells from ATCC treated with GW3965 was done using 3% formaldehyde. Second, we used a longer probe design algorithm (~50 bp instead of recommended 20 bp) that optimized the signal-to-noise ratio and MeXis retrieval, as determined using pilot experiments comparing shorter and longer probe sets performed with RNAase and DNAase controls. For ChIRP-MS, the final protein elution was done in a solution of 50 mM triethyl ammonium bicarbonate, 12 mM sodium lauryl sarcosine, and 0.5% sodium deoxycholate.
ATAC-Seq
Peritoneal macrophages were isolated and treated for 3 hours with or without GW3965. Using four replicates per condition, libraries were prepared using the Nextera Tn5 Transposase kit (Illumina) as described48 with slight modifications. Libraries were single-end sequenced (50bp) on an Illumina HiSeq 2000 instrument. Reads were mapped to the mouse genome (NCBI37/mm9) using Bowtie2, and were removed from the subsequent analysis if they were duplicated, mapped to mitochondrial genome, or aligned to unmapped contiguous sequences. Peak calling was performed using MACS2 using parameters callpeak --nomodel -g mm --keep-dup all -q .01 --llocal 10000. Overlapping peaks were merged together and used as probes for quantifying reads. The reads were converted to reads per million (RPKM) by dividing by the total number of reads within a peak divided by the peak length per million mapped reads. The average RPKM from four replicates was used to quantify the accessibility across all called peaks. Significance was determined by the DESeq2 package in R Bioconductor49. P-values were adjusted using the Benjamini-Hochberg procedure of multiple hypothesis testing 50.
Statistical analysis
A non-paired student t-test was used to determine statistical significance, defined at P-value < 0.05. For multiple group experiments, ANOVA was used followed by multiple group analysis. Unless otherwise noted, error bars represent standard deviations. Except as noted in figure legends experiments were independently performed twice. Sample size is based on statistical analysis of variance and prior experience with similar in vivo studies.
Supplementary Material
Acknowledgments
We thank members of the Tontonoz, Smale, Black, and Nagy laboratories and the UCLA Atherosclerosis Research Unit for technical assistance and useful discussions. This work was support by NIH grants HL030568, HL066088, HL128822, Burroughs Wellcome Fund Career Award for Medical Scientists and UCLA Cardiovascular Discovery Fund (Lauren B. Leichtman and Arthur E. Levine Investigator Award).
Footnotes
Data Availability and Accession Code Availability Statement:
The source data used in the manuscript can be accessed using the following accession numbers: GSE98910 (RNA-seq) , GSE97207 (ATAC-seq), GSE104027(ChIP-Seq), microarray (GSE107977).
Author Contributions:
T.S. and P.T. conceived and designed the study, guided the interpretation of the results and the preparation of the manuscript. P.T. supervised the study and T.S. managed the daily experiments. X.W. performed most mouse experiments and data analysis including atherosclerosis study. T.S, M.J., T.G., K.Q., Z.Z., J.S., D.S., P.R., J.S., C.H., A.I., X.L. participated in various in vivo and in vitro macrophage experiments and data analysis. Bioinformatic data analysis performed by D.C. and A.E. B.T., X.L. and S.S. assisted with ChIP & ATAC-seq experiments including data analysis. J.W. performed the mass spectrometry analysis. B.D. and L.N. assisted with ChIP and performed experiments defining enhancer landscape at Abca1. A.C. performed ChIP-seq for LXR. M.C. and A.L. provided GWAS data and technical guidance with atherosclerosis analysis.T.S. and P.T. edited the manuscript with input from all authors. All authors discussed the results and approved the final version of the manuscript.
Competing Financial Interest Statement:
The authors declare no competing financial interest.
References
- 1.Rohatgi A, et al. HDL cholesterol efflux capacity and incident cardiovascular events. The New England journal of medicine. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de la Llera-Moya M, et al. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libby P, Ridker PM, Hansson GK Leducq Transatlantic Network on, A. Inflammation in atherosclerosis: from pathophysiology to practice. Journal of the American College of Cardiology. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao Q, et al. The central role of EED in the orchestration of polycomb group complexes. Nature communications. 2014;5:3127. doi: 10.1038/ncomms4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. The Journal of clinical investigation. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodzioch M, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nature genetics. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 7.Rust S, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nature genetics. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 8.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annual review of biochemistry. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotzin JJ, et al. The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature. 2016;537:239–243. doi: 10.1038/nature19346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engreitz JM, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539:452–455. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, et al. Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. eLife. 2017;6 doi: 10.7554/eLife.28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sallam T, et al. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature. 2016;534:124–128. doi: 10.1038/nature17674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. P Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nature reviews Genetics. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 17.Huarte M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng JH, Pan DZ, Tsai ZT, Tsai HK. Genome-wide analysis of enhancer RNA in gene regulation across 12 mouse tissues. Scientific reports. 2015;5:12648. doi: 10.1038/srep12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daniel B, et al. The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes & development. 2014;28:1562–1577. doi: 10.1101/gad.242685.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu C, et al. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161:404–416. doi: 10.1016/j.cell.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auboeuf D, Honig A, Berget SM, O’Malley BW. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science. 2002;298:416–419. doi: 10.1126/science.1073734. [DOI] [PubMed] [Google Scholar]
- 22.Wortham NC, et al. The DEAD-box protein p72 regulates ERalpha-/oestrogen-dependent transcription and cell growth, and is associated with improved survival in ERalpha-positive breast cancer. Oncogene. 2009;28:4053–4064. doi: 10.1038/onc.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikpay M, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nature genetics. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khalil AM, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sallam T et al. The macrophage LBP gene is an LXR target that promotes macrophage survival and atherosclerosis. Journal of lipid research. 2014;55:1120–1130. doi: 10.1194/jlr.M047548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rong X, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell metabolism. 2013;18:685–697. doi: 10.1016/j.cmet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jakobsson T, et al. GPS2 is required for cholesterol efflux by triggering histone demethylation, LXR recruitment, and coregulator assembly at the ABCG1 locus. Molecular cell. 2009;34:510–518. doi: 10.1016/j.molcel.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 30.Wongpalee SP, et al. Large-scale remodeling of a repressed exon ribonucleoprotein to an exon definition complex active for splicing. eLife. 2016;5 doi: 10.7554/eLife.19743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin KJ, et al. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13759–13764. doi: 10.1073/pnas.0606179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nature neuroscience. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 33.Peet DJ, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 34.Ito A, et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife. 2015;4:e08009. doi: 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhatt DM, et al. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell. 2012;150:279–290. doi: 10.1016/j.cell.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradley MN, et al. Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. The Journal of clinical investigation. 2007;117:2337–2346. doi: 10.1172/JCI31909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tangirala RK, Rubin EM, Palinski W. Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. Journal of lipid research. 1995;36:2320–2328. [PubMed] [Google Scholar]
- 38.Feig JE, Fisher EA. Laser capture microdissection for analysis of macrophage gene expression from atherosclerotic lesions. Methods in molecular biology. 2013;1027:123–135. doi: 10.1007/978-1-60327-369-5_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Y, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic acids research. 2016;44:D203–208. doi: 10.1093/nar/gkv1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koeth RA, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature medicine. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun L, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic acids research. 2013;41:e166. doi: 10.1093/nar/gkt646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai MC, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 48.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.