

Abstract
Background
The European Randomized Study of Screening for Prostate Cancer (ERSPC) showed that prostate-specific antigen (PSA) screening significantly reduced prostate cancer mortality (rate ratio (RR)=0.79, 95%CI 0.69–0.91). The U.S. Prostate, Lung, Colorectal, and Ovarian (PLCO) trial showed no such reduction but had a wide confidence interval (RR for prostate cancer mortality=1.09, 95%CI 0.87–1.36). Standard meta-analyses are unable to account for key differences between trials that can impact the estimated effects of screening and the trials' point estimates.
Methods
We calibrated two micro-simulation models to individual-level incidence and mortality data from 238,936 men participating in the trials. A cure parameter for the underlying efficacy of screening was estimated by the models separately for each trial. We changed step-by-step major known differences in trial settings, including enrollment and attendance patterns, screening intervals, PSA thresholds, biopsy receipt, control arm contamination and primary treatment, to reflect a more ideal protocol situation and differences between the trials.
Results
Using the cure parameter estimated for the ERSPC, the models projected 19-21% and 6-8% prostate cancer mortality reductions in the ERSPC and PLCO settings, respectively. Using this cure parameter, the models projected a 37-43% reduction under annual screening with 100% attendance and biopsy compliance and no contamination. The cure parameter estimated for the PLCO was 0.
Conclusions
The observed cancer mortality reduction in screening trials is highly sensitive to trial protocol and practice settings. Accounting for these differences, the efficacy of PSA screening in the PLCO setting is not necessarily inconsistent with ERSPC results.
Keywords: PSA screening, prostate cancer, mortality reduction, modelling
Introduction
The European Randomized Study of Screening for Prostate Cancer (ERSPC)1-3 showed a significant prostate cancer mortality reduction of 21% for the PSA screening arm, while the US-based Prostate, Lung, Colorectal, and Ovarian (PLCO) cancer screening trial did not show a difference in prostate cancer mortality between arms but had wide confidence intervals (prostate cancer mortality rate ratio 1.09, 95%CI 0.87-1.36).4 A number of explanations for these seemingly inconsistent results have been debated.5-8
Selective trial populations, different protocols and practice settings, including differences in pre-trial screening, receipt of biopsies, control arm contamination, and primary treatments, may have influenced the trial results.
The ERSPC trial was conducted in seven centers in Europe with 162,243 men aged 55-69 at randomization. PSA testing was not common at the start of the trial and the estimated contamination in the control arm was lower than 15%.7 Most centers used a screening interval of 4 years and a PSA threshold of 3.0 ng/ml for biopsy referral. Approximately 86% of the positive screens were followed by a biopsy.1 The PLCO trial was conducted in 76,693 men aged 55-74 among whom prior screening was already common. At least 45% of the participants had at least one PSA test before randomization. 4 In addition, control arm participants were screened on average 2.7 times during the 6-year intervention phase of the trial.9 Annual screening was used and the threshold for a positive PSA test was 4.0 ng/ml. Since in this trial, the biopsies were performed outside the study, only about 35% of participants with a positive screen received a biopsy.10 Both trials involved variable use of digital rectal exam (DRE) testing.
Because of these differences, the results of the trials are not directly comparable. In standard meta-analyses, the results were simply pooled 11-13, suggesting that PSA screening has little effect on prostate cancer mortality. Possible reasons for the apparent lack of consistency between the trials have not been evaluated formally to determine their quantitative impact on observed mortality reductions.
The aim of this study is to estimate the impact of trial population, protocols, contamination, and practice settings on the observed prostate cancer mortality reduction. We use two independently designed natural history models, which are informed using individual-level data from both trials, to systematically investigate the impact of these characteristics on the estimated efficacy of PSA screening.
Methods
Data
Individual data from both trials were obtained on: age at randomization, trial arm, screening center, screening test dates and results, occurrence of biopsy, prostate cancer incidence, mode of detection (screen or interval cancer), clinical TNM-stage and Gleason score at diagnosis, primary treatment and date and cause of death. The median follow-up was 11 years for ERSPC2 and 13 years for PLCO.4
Modeling the trials
Two multistate disease course models of the Cancer Intervention and Surveillance Modeling Network (CISNET), the Erasmus MC-MIcrosimulation Screening Analysis (Erasmus-MISCAN) model and the Fred Hutchinson Cancer Research Center (FHCRC) model, were used to simulate the trials. The models were independently developed to describe the natural history of prostate cancer and to investigate prostate cancer progression, screening sensitivity, detection, and improvement in prognosis given screening and primary treatment. The two models have been described extensively14-17 (https://resources.cisnet.cancer.gov/registry). In short, in the Erasmus-MISCAN model, disease progresses through a sequence of states defined by stage and grade. In each state, there is a probability of clinical detection and, dependent on the screen sensitivity and attendance, a probability of screen detection.17,18 In the FHCRC model, PSA growth is externally estimated using results of serial PSA tests from the Prostate Cancer Prevention Trial. The risk of onset of a preclinical screen-detectable tumor increases with age and the risks of progression to metastasis and to disease detection in the absence of screening increases with PSA levels.15 Detailed descriptions of the models are provided in Supplementary Material 1.
Calibration
Each model was calibrated to the ERSPC and PLCO trials separately. Disease progression rates (for the Erasmus-MISCAN model also the PSA test sensitivity) were calibrated against the incidence and stage distributions of clinically-detected cancers in both control arms and the screen-detected and interval cancers in the screened arms (Supplementary Material 2). We used enrollment patterns, screen attendance, and receipt of biopsies by age and PSA-level to model the number of screens and biopsies in the screened arms of the trials (Table 1). Screening before, during and after the intervention period (contamination) in the PLCO was simulated using a model described previously.19 Briefly, we assumed that before the trial participants followed screening patterns previously reconstructed for the US population20, which they also followed after the 6-year intervention phase. We assumed control arm participants had a 20% higher intensity of screening than the general US population during the 6-year invention period to match the estimated average 2.7 screens in this period.9 For the ERSPC, we assumed a contamination rate of 5% of US population screening patterns, leading to a comparable number of screened men as estimated in several centers.21-23
Table 1.
Inputs of the models for each trial. Most inputs are age-, stage-, and/or center-specific. The average value is presented for comparison between the arms and trials.
| ERSPC | PLCO | |||
|---|---|---|---|---|
| Screen arm | Control arm | Screen arm | Control arm | |
| Sample size | 72,891 | 89,352 | 38,343 | 38,350 |
| Age at randomization | 55-69 | 55-69 | 55-74 | 55-74 |
| Screen attendance | On average 82% MISCAN: By center and round FHCRC: By center |
Not applicable | By age and round on average 85% | Not applicable |
| Screen protocol | 2 years interval for Sweden, 4 years for other centers (7-years interval between rounds 1 and 2 for Belgium) Screening from age 55 to 69/71/74 depending on center MISCAN: PSA threshold of 3 ng/ml for all centers FHCRC: PSA threshold and DRE testing by center |
Not applicable | 1 year interval for 6 years, PSA threshold of 4 ng/ml Screening from age 55-74 FHCRC: also DRE testing |
Not applicable |
| Biopsy compliance | On average 86% MISCAN: By age, center and round FHCRC: By age, PSA, and center |
On average 86% MISCAN: 86% for all FHCRC: By age, PSA, and center |
By age and round, on average 35% FHCRC: also by PSA |
On average 35% |
| Biopsy sensitivity | 80% | 80% | Increasing from 70% in 1990 to 93% in 2000 | Increasing from 70% in 1990 to 93% in 2000 |
| Contamination | Pretrial screening: about 3-5% of participants had a PSA test No contamination during trial |
Pretrial screening: about 3-5% of participants had a PSA test During trial: about 17,000 tests |
Pretrial screening: about 50% of participants had a PSA test During trial: no contamination Post-trial screening: US population screening |
Pretrial screening: about 50% of participants had a PSA test During trial: about 2.7 test per participant 9 Post-trial screening: US population screening |
| Treatment of local-regional cases | By age, stage, and grade, on average 47% radical prostatectomy * 21% radiation therapy 32% conservative management or active surveillance |
By age, stage, and grade, on average 53% radical prostatectomy * 17% radiation therapy 30% conservative management or active surveillance |
By age, stage, and grade, on average 59% radical prostatectomy * 22% radiation therapy 19% conservative management or active surveillance |
By age, stage, and grade, on average 58% radical prostatectomy * 22% radiation therapy 20% conservative management or active surveillance |
| Life tables | MISCAN: by European country (Human Mortality Database) FHCRC: US life tables (Berkeley Mortality Database) |
MISCAN: by European country (Human Mortality Database) FHCRC: US life tables (Berkeley Mortality Database) |
US life tables (Berkeley Mortality Database) | US life tables (Berkeley Mortality Database) |
This category includes radical prostatectomy, radical prostatectomy with hormone therapy, and radiation therapy with hormone therapy, all assuming to have a hazard ratio of 0.62 on prostate cancer death.
Survival
Both models generated prostate cancer survival from clinical diagnosis in the absence of screening or localized treatment benefits. Prostate cancer survival was estimated using a common proportional hazards regression model with piecewise constant hazards24 fit to Surveillance, Epidemiology, and End Results (SEER) data for untreated cases diagnosed in 1983-1986, just prior to the advent of PSA screening. This baseline survival was improved for localized cases who received radical prostatectomy, or radiation therapy in combination with hormone therapy, using a hazard ratio of 0.62 and for non-metastatic cases who received radiation monotherapy using a hazard ratio of 0.7.25 Distributions of treatments depending on age, Gleason score, and stage were based on separate multinomial regression models fit to trial data (Supplementary Material 3). Other-cause survival was generated using US and European life tables.
Modeling screening benefit
The mortality benefit of PSA screening was modeled as a cure probability that depended on the lead time (years by which detection of the cancer is advanced by screening compared to the clinical situation) and was implemented only for screen-detected, non-metastatic, and non-overdiagnosed cases as cure probability = 1 – exp (-cure parameter × lead time). Thus the probability of cure increases with lead time, with diminishing incremental benefit for longer lead times. In the models, cured men were assigned to die at their independently generated date of other-cause death. Men who were not cured died at the same time they would have died if they had not been screened.
In a previous study modeling the PLCO trial, the models substantially over-projected observed prostate cancer mortality despite closely reproducing incidence and stage and grade patterns.19 Therefore, we included a baseline survival hazard ratio to improve the baseline survival, reasoning there have been improvements in disease management since the period 1983-1986 beyond screening or primary treatment. In this study, we jointly calibrated this hazard ratio with the cure parameter to the observed prostate cancer mortality data for both trials separately.
Model runs
Each model projected the mortality rate ratio for each trial by year of follow-up. Then, using the cure parameter calibrated to the ERSPC (because the published effect of screening was positive), the models systematically varied key characteristics of the trials. We first replaced observed characteristics (control arm contamination, attendance patterns, receipt of biopsies) in the ERSPC setting in a cumulative way with idealized versions of no control arm contamination, perfect attendance, perfect compliance with biopsy recommendations, then substituted the idealized ERSPC setting with the idealized PLCO setting, and finally inserted observed PLCO characteristics (Supplementary Material 4). In each run, the numbers of prostate cancer cases and prostate cancer deaths, and corresponding person-years of follow-up were projected, and the prostate mortality rate ratio was calculated. We quantified stochastic uncertainty around mortality rate ratio point estimates using ranges across 100 simulations and examined sensitivity to estimates of the cure parameter.
Results
Calibration results
Both calibrated models approximated the observed patterns of prostate cancer incidence, grade and stage distributions, and mortality in both arms of both trials (Figure 1 and Supplementary Material 5 and 6). The corresponding lead times are shown in Figure 2 for men aged 60-65 at screen detection and in Supplementary Material 7 for all age groups. The estimated cure parameter was 0.22 (Erasmus-MISCAN) and 0.18 (FHCRC) for the ERSPC. The corresponding cure probability by lead time is shown in Figure 3. Cancers detected early by screening were detected substantially earlier in both trials. For the PLCO trial, we estimated hazard ratios to improve baseline survival of 0.40 (Erasmus-MISCAN) and 0.31 (FHCRC) and for the ERSPC of 0.82 (Erasmus-MISCAN) and 0.77 (FHCRC), illustrating important differences in background risk for men enrolled in the two trials. Because there were more prostate cancer deaths in the screening arm than in the control arm of the PLCO, the estimated cure parameter for that trial was 0 for both models. Consequently, we examined sensitivity of the mortality reduction and PSA screening efficacy to trial population, protocols, and practice settings using the cure parameter estimated for the ERSPC.
Figure 1.
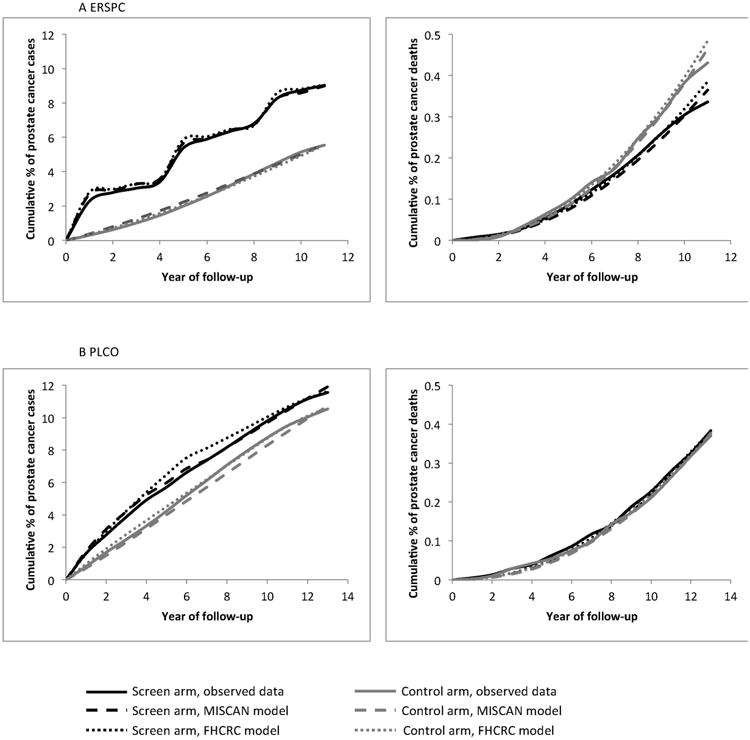
Observed and predicted cumulative percentage of prostate cancer incidence (left) and prostate cancer mortality (right) in the ERSPC (A) and PLCO (B) by year of follow-up.
Figure 2.
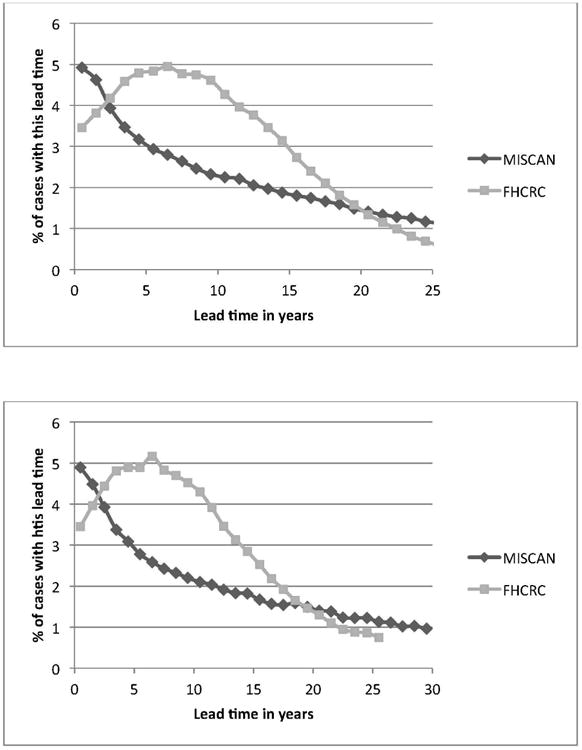
Lead time distribution of screen-detected cases in the models, using the base ERSPC model (A) or the PLCO model (B), for men aged 60-65 at prostate cancer diagnosis. This is defined as the time from detection (screen and interval) until clinical detection before age 100 in the absence of death from other causes. In the Erasmus-MISCAN and FHCRC models, 31% and 20%, respectively, of cases were clinically detected and therefore had a lead time of 0 (and corresponding cure probability of 0). Results for other ages at diagnosis were similar.
Figure 3.
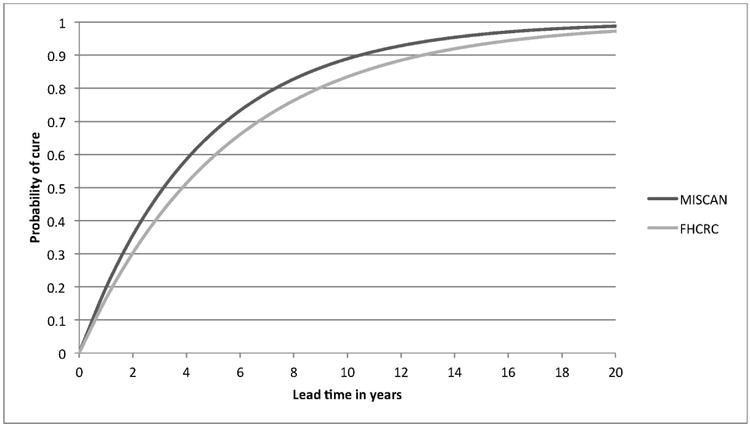
The cure probability for screen-detected cases by lead time in the ERSPC trial as estimated by the two models. In the models, cured men were assigned to die at their independently generated date of other-cause death. Men who were not cured died at the same time they would have died if they had not been screened. So, for example, 60% (FHCRC) to 70% (Erasmus-MISCAN) of men with a lead time of 5 years will not die from prostate cancer and the remaining 30% to 40% will die at the same time and from the same cause as if they had not been screened.
Prostate cancer mortality reduction adjusted for different trial characteristics
Starting with the observed prostate cancer mortality reduction in the ERSPC trial of 21% (95%CI 9%-32%) after 11 years of follow-up (run 0, Erasmus-MISCAN: 21%, FHCRC: 19%), the projected mortality reduction increased as the settings became more idealized (Figure 4). The largest screening effect in ERSPC was predicted under no contamination, 100% attendance, 100% receipt of biopsy for positive screens, and annual screening, with mortality reductions of 43% (Erasmus-MISCAN; uncertainty range 34%-52%) and 37% (FHCRC; uncertainty range 16%-59%) after 11 years of follow-up (run 5). Sensitivity analyses using the 95%CI of the point estimate of the ERSPC for fitting the cure parameter, indicated a 20%-64% prostate cancer mortality reduction in run 5 (Supplementary Material 8). Sensitivity analyses of uncertainty in the joint estimation of the cure parameter and improvement in baseline prostate cancer survival indicated a 16%-65% prostate cancer mortality reduction in run 5 (Supplementary Material 9).
Figure 4.
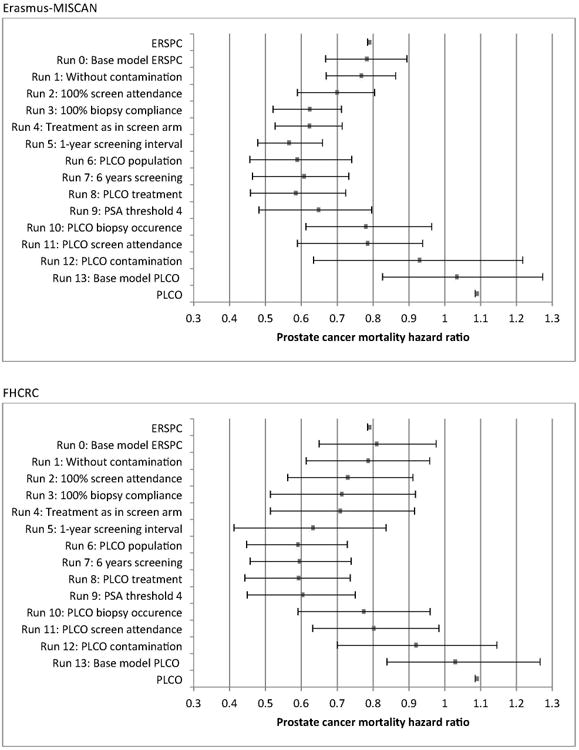
Step-by-step prostate cancer mortality rate ratios and simulation-based uncertainty ranges for the Erasmus-MISCAN and FHCRC models. The changes in the models are cumulative. In run 13 a cure parameter of 0 is used, in all other runs, the ERSPC-based cure parameter is used (0.22 for Erasmus-MISCAN, 0.18 for FHCRC). Supplementary Material 9 provides intervals that incorporate variability in the estimated cure rate parameter (FHCRC model). For each run 0-13, 100 simulations of a single ERSPC or PLCO trial population were performed to generate sample mortality rate ratios; the bracketed line (uncertainty range) and dot represent, respectively, the range and mean of the sample mortality rate ratios observed over the 100 simulations.
In run 0-5 a follow-up of 11 years is used, in run 6-13 the follow-up is 13 years. In each step the listed implementation change is added to the previous step.
The projected reduction diminished substantially as the idealized PLCO setting was systematically replaced with observed characteristics, to 8% (Erasmus-MISCAN) and 6% (FHCRC) under observed settings for all characteristics after 13 years of follow-up (run 12). These projections approach the published ratio in PLCO (9% increase; 95%CI 13% reduction to 36% increase). When a cure parameter of 0 was used, an increase in prostate cancer mortality was found (run 13, Erasmus-MISCAN 3% and FHCRC 5%). Both models found that infrequent receipt of biopsies (runs 9 vs 10) and high contamination (runs 11 vs 12) increased the prostate cancer mortality rate ratio considerably. Although the models generally agreed, different effects were predicted for some trial characteristics, especially for 100% receipt of biopsy in the ERSPC and for the PSA threshold of 4 ng/ml in the PLCO.
Discussion
Efficacy is the extent to which a specific intervention produces a beneficial result under ideal conditions. In practice, true efficacy is rarely estimated as such. Randomized controlled trials, the gold standard for assessing screening interventions, can only assess efficacy limited by the circumstances of the implementation. Our results show that, by explicitly accounting for differences in implementation and settings between ERSPC and PLCO, it is possible to partially reconcile their seemingly different results. In particular, the infrequent receipt of biopsies after a positive test and the high contamination rate in the control arm of the PLCO are the main factors explaining why, even in the presence of a screening benefit such as that observed in the ERSPC, the PLCO could have yielded a negative result.
In addition to allowing us to examine differences between the trials, the models also afford insights into the mortality benefit that might potentially result from an ideal screening regimen. If all men in the ERSPC were screened annually (ignoring selection effects), received a biopsy after a positive test, and there was no contamination, the models predict that the prostate cancer mortality reduction due to screening would have been about 40% after 11 years. Extrapolating this to the European population setting suggests that 1 screen at age 55 could lead to 6,657 (5%) fewer prostate cancer deaths annually and biennial screening for ages 55-69 to 62,529 (44%) fewer deaths annually (Supplementary Material 10).
Earlier studies investigated explanations for the apparently different results of the trials.7,26-28 We previously found that contamination in the PLCO substantially lowered its power.19 Questions have been raised about possible differences in treatment men received in the screening and control arm of the ERSPC.29 However, after correcting for age and tumor stage, no significant differences in treatment were found.30 Our analysis shows that, if all patients in the control arm received treatment according to the frequencies (by age, tumor stage and grade) observed in the screening arm, the prostate cancer mortality reduction would remain unchanged. A similar result holds in the PLCO.
The level of contamination in the ERSPC has not been systematically reported and therefore had to be estimated from earlier published studies, which showed contamination ranging from 7-40% per year across centers.21-23,31 The only study investigating the level of screening before the start of the ERSPC is a study of the Finnish center.22 In this study, 10% of the men in the intervention arm had been screened before. However, both pre-trial and contamination estimates include PSA tests conducted because of symptoms, which could have accounted for up to half of the PSA tests performed.21,23 Also, not all PSA tests were followed by a biopsy. For example, in the Rotterdam control arm, only 8% of positive opportunistic PSA tests were followed by biopsy.23 We did not assess the influence of other less important characteristics separately, for example, population size, age distribution and enrollment patterns, other cause mortality, DRE testing, or biopsy sensitivity. However, we believe we have accounted for the characteristics most likely to be influential.
Using the cure parameter estimated for the ERSPC in the PLCO setting, we obtained a prostate cancer mortality reduction of 6-8%. This means that if PSA testing in the PLCO had been as efficacious as in the ERSPC, the circumstances of its implementation (e.g., infrequent receipt of biopsies, high contamination, healthy screenee effect) would likely have resulted in a modest reduction in prostate cancer mortality. This result is consistent with our prior study, in which we showed that contamination increased the mortality rate ratio and decreased the power of the trial to detect a mortality difference from 40–70% to 9–25%.19
Initially, we planned to consider a symmetric approach, by also starting from the PLCO cure parameter and working towards more ideal situations, and back to the ERSPC. However, the best fit cure parameter for the PLCO was 0, and when there is no benefit, it is impossible to examine how benefit depends on the circumstances of implementation. A limitation is that this result depends on how much of the lower-than-expected mortality is attributed to changes in baseline survival relative to the pre-PSA era (e.g., due to improvements in care) rather than screening benefit in both arms. We feel our approach and prediction is valid, in the situation that one trial has shown an effect of earlier treatment of screen-detected lesions, and that the other trial has been underpowered.
In assessing the efficacy of any screening test, it is important to recognize that results will depend on how the test is implemented. If we start with a cure parameter estimated for the ERSPC, then under idealized circumstances (no control arm contamination, perfect attendance, perfect compliance with biopsy recommendations, run 5), the models predicted about a 40% mortality reduction after 11 years, which is greater than the 21% observed. However, under real-world circumstances of control arm contamination, and less-than-perfect attendance and biopsy compliance as in the PLCO trial, the models predicted a much reduced mortality reduction, of the order of 6-8%. Thus, the trials are likely less inconsistent than their results suggest. Further, the benefit of PSA screening under idealized circumstances is likely more than the trial results suggest. It could be as high as 40% which has previously been reported to imply a net benefit and a reasonably favorable tradeoff when accounting for the main harms of PSA screening.16,32 However, specialized methods will be required to extract an estimate of what this idealized benefit might be based on the data from both trials.
Supplementary Material
Acknowledgments
The authors thank the National Cancer Institute for access to the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial database. The interpretation and reporting of these data are solely the responsibility of the authors.
Funding: This work was supported by the National Cancer Institute [Grant Number U01CA157224] as part of the Cancer Intervention and Surveillance Modeling Network (CISNET). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute.
Dr. de Koning reports grants from CanCon, during the conduct of the study; grants from Beckman Coulter, outside the submitted work; Dr. Auvinen reports grants from Hybritech, during the conduct of the study; personal fees from Epid Research Inc, personal fees from MSD, outside the submitted work;Dr. Andriole reports grants from National Cancer Institute, during the conduct of the study; grants from Medivation (Clinical Research), grants from Progenics (Clinical Research), grants from Blue Earth Diagnostics (Clinical Research), grants from NIH-NIDDK, grants from Prostate Cancer Foundation, grants from Peter Michael Foundation, grants from St. Louis Men's Group Against Cancer, outside the submitted work; Dr. Berg reports personal fees from Medial Early Sign, LLC, personal fees from GRAIL, Inc, outside the submitted work; Dr. Kwiatkowski reports personal fees from Myriad, personal fees from Astellas, personal fees from Janssen, outside the submitted work; Dr. LUJAN reports grants from “Fondo de Investigación Sanitaria” (FIS): 93/0903, 96/0248, 96/1800, 99/0245, 02/0732, andÐ6/0831., grants from “Fundación para la Investigación en Urología” (FIU)., during the conduct of the study; Dr. Heijnsdijk report grants from Beckman Coulter, outside the submitted work;
Footnotes
Authors' contributions Conceptualization: HdK, RG, PP, AT, AB, EH and RE; Data curation and funding acquisition: SM, JH, CB, AA, GA, MR, EC, VN, MK, MZ, ML, AV; Formal analysis: RG, SM, AT, TdC, EH; Investigation: RG, TdC, EH; Methodology: HdK, RG, SM, PP, TdC, EF, AT, AB, EH and RE; Writing, original draft: HdK, RG, EH, RE ; Writing, review and editing: all authors
Disclosures: The other authors have nothing to disclosure.
References
- 1.Schröder FH, Hugosson J, Roobol MJ, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320–8. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- 2.Schröder FH, Hugosson J, Roobol MJ, et al. Prostate-cancer mortality at 11 years of follow-up. N Engl J Med. 2012;366:981–90. doi: 10.1056/NEJMoa1113135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schröder FH, Hugosson J, Roobol MJ, et al. Screening and prostate cancer mortality: results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet. 2014;384:2027–35. doi: 10.1016/S0140-6736(14)60525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andriole GL, Crawford ED, Grubb RL, 3rd, et al. Prostate cancer screening in the randomized Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial: mortality results after 13 years of follow-up. J Natl Cancer Inst. 2012;104:125–32. doi: 10.1093/jnci/djr500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barry MJ. Screening for prostate cancer--the controversy that refuses to die. N Engl J Med. 2009;360:1351–4. doi: 10.1056/NEJMe0901166. [DOI] [PubMed] [Google Scholar]
- 6.La Rochelle J, Amling CL. Prostate cancer screening: what we have learned from the PLCO and ERSPC trials. Curr Urol Rep. 2010;11:198–201. doi: 10.1007/s11934-010-0109-5. [DOI] [PubMed] [Google Scholar]
- 7.Schröder FH, Roobol MJ. ERSPC and PLCO prostate cancer screening studies: what are the differences? Eur Urol. 2010;58:46–52. doi: 10.1016/j.eururo.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 8.Pinsky PF, Black A, Parnes HL, et al. Prostate cancer specific survival in the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial. Cancer epidemiology. 2012;36:e401–6. doi: 10.1016/j.canep.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinsky PF, Blacka A, Kramer BS, Miller A, Prorok PC, Berg C. Assessing contamination and compliance in the prostate component of the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial. Clin Trials. 2010;7:303–11. doi: 10.1177/1740774510374091. [DOI] [PubMed] [Google Scholar]
- 10.Grubb RL, Pinsky PF, Greenlee RT, et al. Prostate cancer screening in the Prostate, Lung, Colorectal and Ovarian cancer screening trial: update on findings from the initial four rounds of screening in a randomized trial. BJU Int. 2008;102:1524–30. doi: 10.1111/j.1464-410X.2008.08214.x. [DOI] [PubMed] [Google Scholar]
- 11.Chou R, Croswell JM, Dana T, et al. Screening for prostate cancer: a review of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155:762–71. doi: 10.7326/0003-4819-155-11-201112060-00375. [DOI] [PubMed] [Google Scholar]
- 12.Djulbegovic M, Beyth RJ, Neuberger MM, et al. Screening for prostate cancer: systematic review and meta-analysis of randomised controlled trials. BMJ (Clinical research ed. 2010;341:c4543. doi: 10.1136/bmj.c4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ilic D, O'Connor D, Green S, Wilt TJ. Screening for prostate cancer: an updated Cochrane systematic review. BJU Int. 2011;107:882–91. doi: 10.1111/j.1464-410X.2010.10032.x. [DOI] [PubMed] [Google Scholar]
- 14.Gulati R, Gore JL, Etzioni R. Comparative effectiveness of alternative PSA-based prostate cancer screening strategies. Ann Intern Med. 2013;158:145–53. doi: 10.7326/0003-4819-158-3-201302050-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulati R, Inoue L, Katcher J, Hazelton W, Etzioni R. Calibrating disease progression models using population data: a critical precursor to policy development in cancer control. Biostatistics. 2010;11:707–19. doi: 10.1093/biostatistics/kxq036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heijnsdijk EA, de Carvalho TM, Auvinen A, et al. Cost-effectiveness of prostate cancer screening: a simulation study based on ERSPC data. J Natl Cancer Inst. 2015;107:366. doi: 10.1093/jnci/dju366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wever EM, Draisma G, Heijnsdijk EA, et al. Prostate-specific antigen screening in the United States vs in the European Randomized Study of Screening for Prostate Cancer-Rotterdam. J Natl Cancer Inst. 2010;102:352–5. doi: 10.1093/jnci/djp533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Draisma G, Postma R, Schröder FH, van der Kwast TH, de Koning HJ. Gleason score, age and screening: modeling dedifferentiation in prostate cancer. Int J Cancer. 2006;119:2366–71. doi: 10.1002/ijc.22158. [DOI] [PubMed] [Google Scholar]
- 19.Gulati R, Tsodikov A, Wever EM, et al. The impact of PLCO control arm contamination on perceived PSA screening efficacy. Cancer Causes Control. 2012;23:827–35. doi: 10.1007/s10552-012-9951-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mariotto AB, Etzioni R, Krapcho M, Feuer EJ. Reconstructing PSA testing patterns between black and white men in the US from Medicare claims and the National Health Interview Survey. Cancer. 2007;109:1877–86. doi: 10.1002/cncr.22607. [DOI] [PubMed] [Google Scholar]
- 21.Bokhorst LP, Bangma CH, van Leenders GJ, et al. Prostate-specific antigen-based prostate cancer screening: reduction of prostate cancer mortality after correction for nonattendance and contamination in the Rotterdam section of the European Randomized Study of Screening for Prostate Cancer. Eur Urol. 2014;65:329–36. doi: 10.1016/j.eururo.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Ciatto S, Zappa M, Villers A, Paez A, Otto SJ, Auvinen A. Contamination by opportunistic screening in the European Randomized Study of Prostate Cancer Screening. BJU Int. 2003;92(2):97–100. doi: 10.1111/j.1464-410x.2003.04407.x. [DOI] [PubMed] [Google Scholar]
- 23.Otto SJ, van der Cruijsen IW, Liem MK, et al. Effective PSA contamination in the Rotterdam section of the European Randomized Study of Screening for Prostate Cancer. Int J Cancer. 2003;105:394–9. doi: 10.1002/ijc.11074. [DOI] [PubMed] [Google Scholar]
- 24.Friedman M. Piecewise Exponential Models for Survival Data with Covariates. Ann Statist. 1982;10:101–13. [Google Scholar]
- 25.Etzioni R, Gulati R, Tsodikov A, et al. The prostate cancer conundrum revisited : Treatment changes and prostate cancer mortality declines. Cancer. 2012;118:5955–63. doi: 10.1002/cncr.27594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croswell JM, Kramer BS, Crawford ED. Screening for prostate cancer with PSA testing: current status and future directions. Oncology (Williston Park) 2011;25:452–60. 63. [PubMed] [Google Scholar]
- 27.Studer UE, Collette L. What can be concluded from the ERSPC and PLCO trial data? Urologic oncology. 2010;28:668–9. doi: 10.1016/j.urolonc.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 28.Palma A, Lounsbury DW, Schlecht NF, Agalliu I. A System Dynamics Model of Serum Prostate-Specific Antigen Screening for Prostate Cancer. American journal of epidemiology. 2016;183:227–36. doi: 10.1093/aje/kwv262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haines IE, Gabor Miklos GL. Prostate-specific antigen screening trials and prostate cancer deaths: the androgen deprivation connection. J Natl Cancer Inst. 2013;105:1534–9. doi: 10.1093/jnci/djt248. [DOI] [PubMed] [Google Scholar]
- 30.Wolters T, Roobol MJ, Steyerberg EW, et al. The effect of study arm on prostate cancer treatment in the large screening trial ERSPC. Int J Cancer. 2010;126:2387–93. doi: 10.1002/ijc.24870. [DOI] [PubMed] [Google Scholar]
- 31.Lujan M, Paez A, Pascual C, Angulo J, Miravalles E, Berenguer A. Extent of prostate-specific antigen contamination in the Spanish section of the European Randomized Study of Screening for Prostate Cancer (ERSPC) Eur Urol. 2006;50:1234–40. doi: 10.1016/j.eururo.2006.04.015. discussion 9-40. [DOI] [PubMed] [Google Scholar]
- 32.Heijnsdijk EA, Wever EM, Auvinen A, et al. Quality-of-life effects of prostate-specific antigen screening. N Engl J Med. 2012;367:595–605. doi: 10.1056/NEJMoa1201637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.