

Abstract
Adipocytes possess remarkable adaptive capacity to respond to nutrient excess, fasting or cold exposure, and thus are an important cell type to maintain proper metabolic health. While the endoplasmic reticulum (ER) is a critical organelle for cellular homeostasis, the mechanisms that mediate adaptation of the ER in adipocytes to metabolic challenges are unclear. Here, we show that brown adipose tissue (BAT) thermogenic function requires an adaptive increase in proteasomal activity to secure cellular protein quality control, and identify the ER-localized transcription factor nuclear factor erythroid-2, like-1 (Nfe2l1, also known as Nrf1) as a critical driver of this process. We show that cold adaptation induced Nrf1 in BAT to increase proteasomal activity, and that this was crucial for maintaining ER homeostasis and cellular integrity, specifically when the cells are in a state of high thermogenic activity. In mice, under thermogenic conditions, brown adipocyte-specific deletion of Nrf1 resulted in ER stress, tissue inflammation, markedly diminished mitochondrial function and whitening of the BAT. In mouse models of both genetic and dietary obesity, stimulation of proteasomal activity by exogenously expressing Nrf1 or the proteasome activator PA28α in BAT resulted in improved insulin sensitivity. In conclusion, Nrf1 emerges as a novel guardian of brown adipocyte function, providing increased proteometabolic quality control for adapting to cold or to obesity.
Keywords: Endoplasmic reticulum, proteasome, proteostasis, brown adipose tissue, obesity, insulin resistance, inflammation
Introduction
BAT mediates non-shivering thermogenesis in mammals exposed to temperatures below thermoneutrality, an ambient environment in which basal metabolism is sufficient to maintain body temperature1. To sustain its exceptional oxidative metabolic activity, BAT consumes vast amounts of intracellular and circulating nutrients2, and in doing so, BAT activity exerts beneficial metabolic effects on obesity2, insulin resistance3, and atherosclerosis4,5. During cold acclimatization BAT undergoes an adaptive recruitment process and the tissue grows in size and in total metabolic capacity. This involves both new adipocyte differentiation as well as increased metabolic activity of existing adipocytes1,6-8. BAT is also activated by acute nutrient influx, in particular consumption of a high-fat diet (HFD)9,10, but the chronic exposure to excess nutrients that occurs in obesity results in whitening and reduced metabolic capacity of BAT in mice11 and humans12,13.
As the mechanisms underlying BAT deterioration in obesity remain insufficiently understood, identifying factors that protect thermogenic adipocytes from metabolic stress during the adaptation to cold or to obesity may hold great potential towards therapeutic approaches for metabolic diseases. The dramatic metabolic changes in BAT not only involve dissipation of energy-rich nutrients, but also the de novo synthesis of new proteins, lipids, and cellular organelles1. Thus, we hypothesized that cold adaptation might require adaptive mechanisms for increased quality control of these metabolic processes. In particular, enhanced protein production and folding as well as protection against aberrantly modified or oxidized proteins and other metabolites may require adipocyte-specific adaptive mechanisms to resist the rising cellular stress. The concept of specialized molecules and adaptive responses to achieve this challenging task is even more compelling in brown adipocytes, considering the unique cellular organization of high mitochondrial and lipid content and extremely high metabolic flux, yet relatively scarce levels of ER content.
As the site of protein and lipid synthesis, the ER is a critical organelle for the maintenance of metabolic and homeostatic balance. Perturbations of ER function are combated by the unfolded protein response (UPR), a molecular network that results in enhanced protein folding capacity and decreased protein translation, as well as by ER-associated protein degradation (ERAD) of damaged or dispensable proteins via the ubiquitin-proteasome system (UPS)14–18. Especially in metabolically active tissues such as the liver, obesity is associated with failure of the UPR or the ERAD pathway to secure ER function, resulting in activation of ER and inflammatory stress pathways, and ultimately leading to the disruption of systemic metabolic homeostasis19,20. However, the identity of the factors that support ER homeostasis and promote metabolic function in adipocytes, particularly in thermogenic adipocytes, remains unclear. In light of unique features of the ER in brown adipocytes, such as the apparently lower ER content as well as relatively limited ER surface area compared to for example hepatocytes21,22, ER adaptation in these cells may require alternative pathways other than chaperone-mediated protein folding and ER expansion. Indeed, some of the canonical UPR molecules, for example X-box binding protein 1 (Xbp1), seem to be dispensable in adipocytes23. Although the physiological significance of ERAD in brown adipocytes remains unknown, we considered proteasomal protein quality control as a possible alternative pathway by which these cells might secure ER homeostasis and their metabolic function. Here, we show that proteasomal protein quality control in brown adipocytes is essential for the thermogenic adaptation of BAT and identify Nrf1 (encoded by Nfe2l1), an ER membrane-embedded transcription factor24, as a fundamental regulator of the adaptation of BAT to increased metabolic stress in cold or in obesity through the function of the proteasome.
Results
Non-shivering thermogenesis requires proteasomal activity
The ER is a critical organelle for coordinating cellular homeostasis during states of high metabolism, but the specific pathways and ER-resident proteins that contribute to the adaptation of BAT to a cold environment remain elusive. In line with previous results23, we did not find differences in thermogenic energy expenditure stimulated by CL316,243 (CL), an adipocyte-specific β3-adrenergic agonist25, in mice with either adipocyte-specific Inositol-requiring enzme-1α (Ire1α) or Xbp1 deficiency (Supplementary Fig. 1a,b), indicating that this branch of the UPR is dispensable for BAT-mediated respiration.
As ERAD supports ER function by removing damaged or dispensable proteins14,15, we focused on protein degradation through the UPS as a potential alternate mechanism by which brown adipocytes secure ER homeostasis and ameliorate metabolic stress. To investigate whether cold adaptation is linked to proteasome function we first measured proteasomal activity in BAT from wild-type (WT) mice adapted to thermoneutrality (30 °C), room temperature (22 °C) or cold (4 °C) for 7 days. We found that cold adaptation either to 22 °C or 4 °C was associated with higher proteasomal activity in BAT compared to 30 °C (Fig. 1a). In contrast, in the liver proteasomal activity remained unchanged after cold adaptation (Supplementary Fig. 1c). To determine the physiological significance of this observation for non-shivering thermogenesis, we treated mice housed at 30 °C or 22 °C with bortezomib, a clinical proteasome inhibitor26,27. We observed that at 22 °C, a setting in which non-shivering thermogenesis in BAT is active, mice treated with bortezomib were less able to defend their core body temperature compared to controls treated with dimethyl sulfoxide (DMSO), while mice adapted to 30 °C and treated with bortezomib or DMSO displayed no difference in their ability to defend core body temperature (Fig. 1b). These data demonstrate that proteasome function is critically important for sustaining non-shivering thermogenesis. This effect was independent of changes in the mRNA expression of genes coding for uncoupling protein-1 (Ucp1) or peroxisome proliferator receptor-γ, co-activator 1-α (Pgc1α, encoded by Ppargc1a) (Fig. 1c) or the amount or histological appearance of BAT (Supplementary Fig. 1d).
Figure 1.
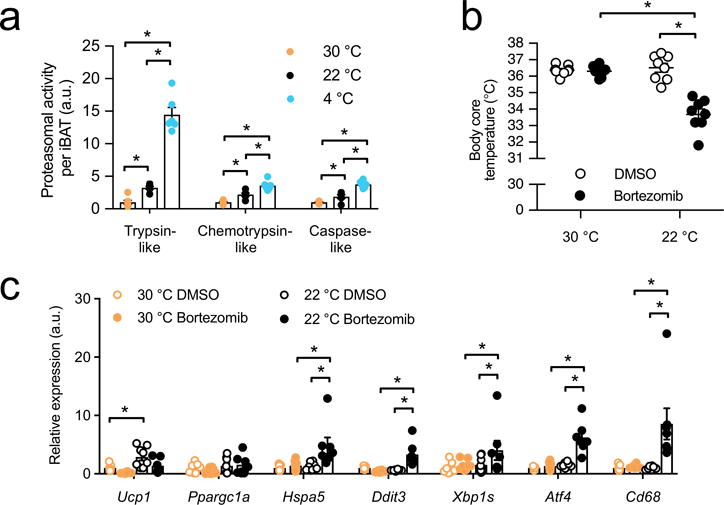
Proteasomal activity is induced during cold adaptation and is required for non-shivering thermogenesis in BAT. (a) Proteasome activity per total interscapular BAT (iBAT) from wild-type (WT) mice after adaptation to thermoneutrality (30 °C), room temperature (22 °C) or cold (4 °C) for 7 days (Biol. replicates n = 6, *P < 0.05 by 1-way ANOVA). (b) Body core temperature after 16 h bortezomib (2.5 mg/kg) or control dimethyl sulfoxide (DMSO) treatment in WT mice housed at 22 °C or 30 °C (biol. replicates n = 8, *P < 0.05 by 2-way ANOVA). (c) mRNA levels of brown fat activation and stress markers in BAT of WT mice housed at 22 °C or 30 °C 16 h after bortezomib (2.5 mg/kg) or control DMSO treatment in (Biol. replicates n = 7 for 30 °C DMSO and 22 °C bortezomib and n = 8 for 22°C DMSO and 30°C bortezomib, normalized by ΔCT, *P < 0.05 by 2-way ANOVA).
Proteasomal activity promotes ER homeostasis in BAT
In BAT of mice housed at 22 °C, bortezomib treatment led to higher mRNA expression of markers of stress pathway activation such as heat shock protein 5 (Hspa5, also known as GRP78 or BiP), DNA-damage inducible transcript 3 (Ddit3, also known as Chop), spliced Xbp1 (Xbp1s), activating transcription factor 4 (Atf4) and Cd68 antigen (Cd68) compared to DMSO-treated controls, whereas in BAT of mice housed at 30 °C this response was diminished (Fig. 1c), indicating that proteasome function is also required to limit cellular stress in BAT particularly in states of high metabolism. In contrast, housing at 30 °C did not abrogate the effect of proteasome inhibition on stress pathway activation in the livers of these mice (Supplementary Fig. 1e). As bortezomib may exhibit off-target protease inhibitor effects28, we additionally evaluated the effects of carfilzomib, a potent, structurally distinct and specific proteasome inhibitor in clinical use28. Treatment with carfilzomib produced similar findings compared to bortezomib as both proteasome inhibitors led to lower body temperature and higher ER stress compared to DMSO-treated controls only when mice were under thermal stress but not at 30 °C (Supplementary Fig. 1f,g).
In adipocytes expressing a reporter system for ER function29, we found that proteasome inhibition by bortezomib or epoxomicin resulted in a similar degree of ER functional impairment as treatment with thapsigargin, a known inducer of ER stress30 (Supplementary Fig. 2a), supporting a critical role for the proteasome in maintaining ER function in adipocytes. To explore whether proteasome function might be specifically required for brown adipocyte respiration, we directly tested the effect of proteasome inhibition on mitochondrial bioenergetics. Notably, ADP-stimulated (state 3), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP)-induced (maximal capacity) and GDP-sensitive (Ucp1-mediated) respiration were all lower in mitochondria isolated from BAT of bortezomib-treated mice compared to mitochondria isolated from BAT of DMSO-treated controls (Supplementary Fig. 2b,c). Also, in a brown adipocyte cell line, treatment with epoxomicin led to lower maximal mitochondrial respiration (Supplementary Fig. 2d-f), highlighting the importance of proteasomal activity for brown adipocyte oxidative capacity.
Nrf1 is highly expressed in BAT and induced by cold
As recent in vitro studies described that Nrf1 (encoded by Nfe2l1) is an adaptive stimulator of proteasomal activity by binding to the promoters of proteasome subunit genes and driving their expression27,31-33, we investigated the potential role of Nrf1 in cold-induced proteasomal activity in BAT. First, we examined the regulation of Nrf1 in human and mouse BAT and cultured brown adipocytes. In human primary differentiated brown adipocytes, NFE2L1 mRNA was highly expressed, at substantially higher levels than other members of the NFE2L family (Fig. 2a). In human adipose tissue biopsies from subcutaneous and carotid sheath areas34, we found that NFE2L1 mRNA levels strongly correlated with the brown fat character of the samples (Fig. 2b). We also analyzed a broad array of clonal cell lines derived from human adipose tissue34, in which we found that NFE2L1 gene expression was a predictor of thermogenic competency (Fig. 2c).
Figure 2.
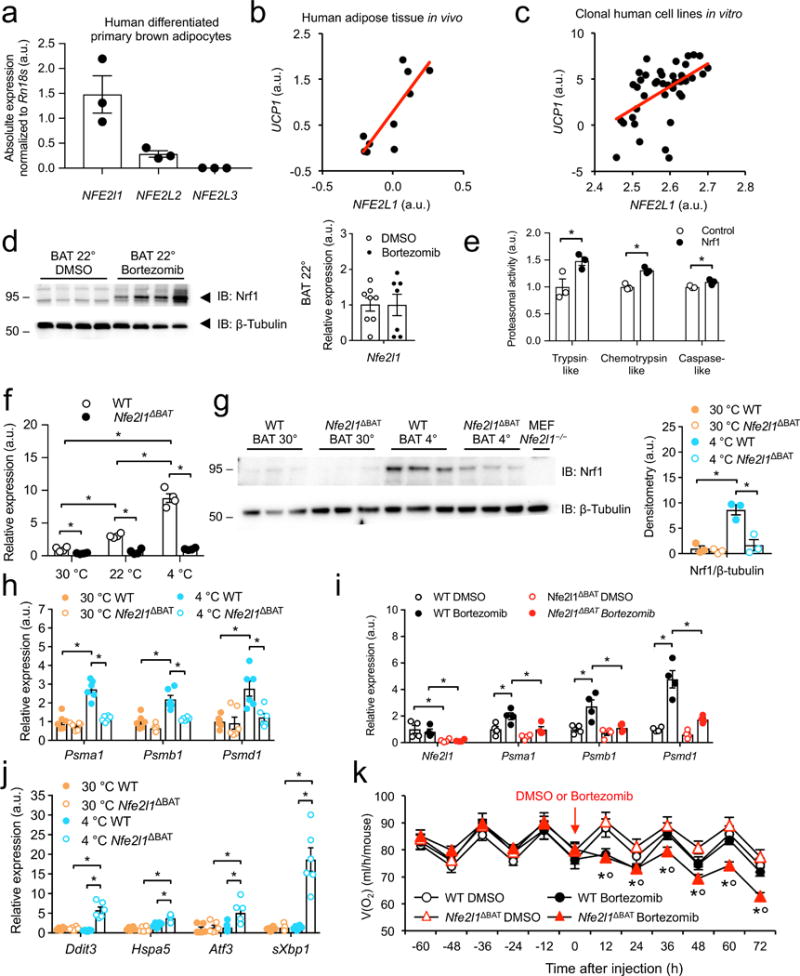
Nrf1 is a cold-inducible regulator of proteasome function in brown fat. (a) NFE2L1 gene expression in human differentiated primary brown adipocytes (Biol. replicates n = 3) (b) Human adipose tissue biopsy NFE2L1 gene expression correlated to UCP1 (Black circles represent individual subjects, biol. replicates n = 10; Spearman correlation: r = 0.08, P = 0.00469). (c) NFE2L1 gene expression in clonal cell lines derived from human adipose tissue correlated to UCP1 (Black circles represent individual clones, biol. replicates n = 42; Spearman correlation: r = 0.528, P = 0.00039). (d) Representative immunoblot of Nrf1 and Nfe2l1 mRNA levels in BAT after 16 h bortezomib (2.5 mg/kg) or DMSO treatment (Blot: biol. replicates n = 4, cropped images; mRNA: 22 °C, biol. replicates n = 8 for DMSO and n = 7 for bortezomib treatment, normalized by ΔCT). (e) Proteasomal activity in Nrf1-overexpressing and control brown adipocytes (Biol. replicates n = 3, *P < 0.05 by T-Test). (f,g) Nfe2l1 mRNA levels and representative immunoblot of Nrf1 after 7 days adaptation to 4 °C in BAT from WT mice or mice lacking Nrf1 in brown adipocytes (Nfe2l1ΔBAT; biol. replicates n = 4 in (f) and n = 3 in (g), normalized to Tbp, *P < 0.05 by 2-way ANOVA, cropped images). (h) mRNA levels of proteasome subunits expression in BAT of WT or Nfe2l1ΔBAT mice adapted to 4°C or 30 °C (Biol. replicates n = 6, normalized to Tbp, *P < 0.05 effect by 2-way ANOVA). (i) mRNA levels of proteasome subunits in BAT of WT or Nfe2l1ΔBAT mice after 16 h bortezomib (2.5 mg/kg) or DMSO treatment (22 °C; biol. replicates n = 4, normalized to Rn18s, *P < 0.05 by 2-way ANOVA). (j) mRNA levels of stress markers in BAT of WT or Nfe2l1ΔBAT mice adapted to 4 °C or 30 °C (Biol. replicates n=6, normalized to Tbp, *P < 0.05 by 2-way ANOVA). (k) Whole body oxygen consumption in WT or Nfe2l1ΔBAT mice treated with Bortezomib (1.25 mg/kg) or DMSO (22 °C, biol. replicates n = 8, *P < 0.05 by 2-way ANOVA).
In line with previous work in cultured cells27,31,32, we found that in BAT of mice treated with either bortezomib or carfilzomib Nrf1 was induced at the protein but not the mRNA level (Fig. 2d and Supplementary Fig. 2g), which demonstrates a functional relationship between Nrf1 and proteasomal activity in BAT. Furthermore, we generated a mouse brown adipocyte cell line, in which Nrf1 was overexpressed (Supplementary Fig. 2h), which led to higher proteasomal activity compared to control cells (Fig. 2e). Taken together, our results suggest that Nrf1 plays a key role in regulating adaptive proteasomal activity in brown adipocytes in vitro and in vivo. In mice, Nfe2l1 mRNA was enriched in BAT compared to white adipose tissue (WAT), and cold adaptation led to markedly higher Nfe2l1 expression in BAT compared to 30 °C and to a minor extent also in inguinal WAT (IngWAT) (Supplementary Fig. 2i), an adipose depot prone to beige adipocyte formation and browning35-37.
Nrf1 drives cold-inducible proteasomal activity in BAT
To evaluate the significance of cold-induced Nrf1 for proteasomal activity and thermogenic function of BAT we generated a mouse model in which the Nfe2l1 gene was disrupted specifically in all Ucp1-expressing adipocytes, for example in BAT and IngWAT (Nfe2l1ΔBAT) (Supplementary Fig. 3a). In this model, after cold adaptation Nfe2l1 mRNA and Nrf1 protein in BAT were markedly lower compared to WT controls (Fig. 2f,g), showing that the induction of Nrf1 in BAT by cold takes place in brown adipocytes. Of note, also in WT mice at 30 °C Nrf1 expression in BAT was very low compared to 30 °C (Fig. 2f,g), suggesting that thermoneutrality is a state of natural Nrf1 deficiency. Notably, we observed that compared to 30 °C, cold adaptation led to higher levels of both the ER-localized form of Nrf1 protein as well as the cleaved, transcriptionally active form in BAT (Supplementary Fig. 3b,c).
In support of our hypothesis that Nrf1 is an important physiological regulator of proteasomal activity in BAT, cold adaptation led to higher mRNA expression of proteasome subunits (encoded by Psm genes) compared to 30 °C in WT mice but not Nfe2l1ΔBAT mice (Fig. 2h). In addition, treatment with bortezomib led to higher proteasome subunit mRNA expression in BAT of WT controls but this induction was blunted in Nfe2l1ΔBAT mice (Fig. 2i) whereas in the livers of these mice the Nrf1-proteasome pathway was still functional (Supplementary Fig. 3d). Similar to chemical inhibition of the proteasome, we found that cold adaptation led to higher mRNA expression of stress pathways in BAT of Nfe2l1ΔBAT mice, while the BAT of wild-type littermates exhibited no stress response to cold (Fig. 2j). Acclimating mice from 22 °C to 30 °C alleviated the activation of stress pathways in the Nfe2l1ΔBAT mice, showing the reversibility and adaptive nature of this response (Supplementary Fig. 3e). In addition, we observed perturbations in ER morphology and increased levels of heat shock proteins and autophagy in BAT from Nfe2l1ΔBAT mice compared to WT controls (Supplementary Fig. 4). To evaluate the role of Nrf1-mediated proteasome function in BAT for whole-body respiration, we used Nfe2l1ΔBAT mice and control littermates that were born at thermoneutrality, placed at 22 °C and then treated with low-dose bortezomib. In these experiments, Nfe2l1ΔBAT mice were uniquely sensitive to proteasome inhibition, as only Nfe2l1ΔBAT mice displayed an unrecoverable bortezomib-induced drop in respiration whereas WT mice fully recovered (Fig. 2k).
BAT activity requires Nrf1 for metabolic adaptation
Although the BAT in Nfe2l1ΔBAT mice appeared grossly normal at birth, when raised at 22 °C the tissue became progressively discolored and lipid-laden, appearing white in adult mice compared to the BAT of WT littermates (Fig. 3a,b and Supplementary Fig. 5a). In line with the concept that Nrf1 and proteasome function are dispensable at thermoneutrality, BAT of Nfe2l1ΔBAT mice and WT littermates born and raised at 30 °C was indistinguishable (Supplementary Fig. 5b-d). Importantly, neither changes in BAT Ucp1 protein levels (Supplementary Fig. 3b,c) nor changes in BAT mRNA levels of critical brown adipocyte regulators such as Ucp1, Ppargc1a, Ppargc1b, Ppara or Pparg were detected between genotypes, either at 30 °C or at 22 °C (Supplementary Fig. 5e). However, in line with results above (Fig. 2j), ER stress measured by Ddit3 expression was increased at 22 °C but not at 30 °C in Nfe2l1ΔBAT mice (Supplementary Fig. 5e). Notably, BAT-mediated whole-body respiration was lower in Nfe2l1ΔBAT mice raised at 22 °C compared to WT control mice at 22 °C, but not at 30 °C, (Supplementary Fig. 5f).
Figure 3.
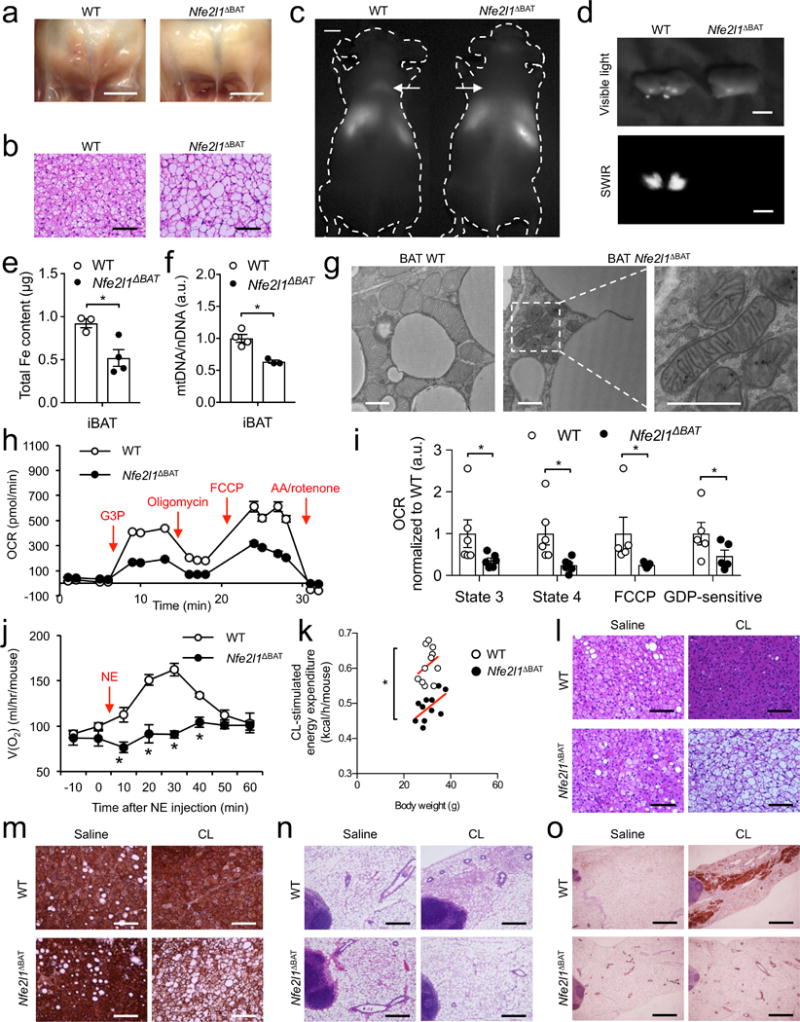
Nrf1 is a fundamental regulator of BAT adaptation. (a) Photographs of iBAT (Scale bars, 5 mm) and (b) Hematoxylin & Eosin (H&E)-stained histology (Scale bars, 100 μm) of WT and Nfe2l1ΔBAT mice at 22 °C. (c) Short-wave infrared (SWIR) imaging of WT mice and Nfe2l1ΔBAT. Arrows indicate interscapular BAT (d) Ex vivo SWIR and visible light imaging of iBAT from (c) (Scale bars in c,d, 5 mm). (e) Iron and (f) mitochondrial DNA content in iBAT of WT and Nfe2l1ΔBAT mice at 22 °C (Biol. replicates (e) n = 3 for WT and n = 4 for Nfe2l1ΔBAT and (f) n = 4 for WT and n = 3 for Nfe2l1ΔBAT, *P < 0.05 by Student’s t-Test). (g) Representative electron microscopy of iBAT of WT and Nfe2l1ΔBAT mice at 22 °C (biol. replicates n = 3, Scale bars, 1 μm) (h) Oxygen consumption rate (OCR) in mitochondria isolated from WT and Nfe2l1ΔBAT mice at 22 °C (G3P: Glycerol-3-phosphate, FCCP: Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone, AA: Antimycin A; representative traces) (i) State 3, state 4, FCCP-induced and GDP-sensitive OCR in mitochondria isolated from WT and Nfe2l1ΔBAT mice at 22 °C (Biol. replicates n = 6 for state 3 and state 4, n = 5 for FCCP and GDP, *P < 0.05 by Mann-Whitney-U rank test). (j) Norepinephrine (NE)-stimulated whole-body oxygen consumption in WT and Nfe2l1ΔBAT mice at 22 °C (Biol. replicates n = 4, *P < 0.05 by Student’s t-Test). (k) CL316,243 (CL)-stimulated whole-body energy expenditure in WT and Nfe2l1ΔBAT mice at 22 °C (Biol. replicates n = 8, *P < 0.05 by T-Test). (l-o) Representative H&E histology (l) and Ucp1 immunohistochemistry (IHC) (m) of iBAT as well as H&E histology (n) and Ucp1 IHC (o) of inguinal white adipose tissue from WT and Nfe2l1ΔBAT mice at 30 °C treated with CL or saline (Biol. replicates n = 4. Scale bars in l,m, 100 μm; in n, 0.4 mm; in o, 1 mm).
To characterize the metabolic activity of Nrf1-deficient BAT, we investigated the ability of Nfe2l1ΔBAT mice at 22 °C to metabolize triglyceride-rich lipoproteins, a major substrate for BAT activity2. Using a novel imaging platform with shortwave infrared (SWIR)-emitting quantum dots38, we observed that while there was a pronounced uptake of lipoproteins in cold-activated BAT of WT mice, Nfe2l1ΔBAT mice did not display any detectable uptake in BAT (Fig. 3c,d and Supplementary Movies 1,2). In line with the discoloration, the abnormal BAT in Nfe2l1ΔBAT mice at 22 °C was characterized by lower iron and mitochondrial content (Fig. 3e,f). Using transmission electron microscopy, we observed that the mitochondria in brown adipocytes of Nfe2l1ΔBAT mice displayed abnormal shape, irregular cristae organization and signs of mitochondria-associated membrane (MAM) formation compared to WT tissue (Fig. 3g). Furthermore, similar to chemical inhibition of the proteasome, isolated mitochondria from BAT of Nfe2l1ΔBAT mice were characterized by lower state 3, oligomycin-resistant (State 4), maximal respiratory capacity, as well as GDP-sensitive Ucp1-dependent respiration compared to mitochondria isolated from WT controls (Fig. 3h,i and Supplementary Fig. 5g). However, neither chemical inhibition of the proteasome nor Nrf1 deficiency was associated with reduced protein levels of components of the respiratory chain in isolated mitochondria per se (Supplementary Fig. 6a).
To evaluate the impact of impaired BAT function in Nfe2l1ΔBAT mice on systemic metabolism, we determined the effect of well-established BAT activators25 on metabolic parameters in Nfe2l1ΔBAT mice and controls. Stimulation of whole-body oxygen consumption by norepinephrine (NE), a major physiologic BAT activator, was markedly diminished in Nfe2l1ΔBAT mice (Fig. 3j). In line with this, CL-stimulated energy expenditure was lower in Nfe2l1ΔBAT mice compared to WT controls (Fig. 3k). However, lipolysis in BAT and WAT explants remained unchanged by Nfe2l1 deletion (Supplementary Fig. 6b,c). While these results were obtained in mice adapted to 22 °C, we also tested whether in mice at 30 °C Nrf1 was required for the adaptation to chronic pharmacological BAT activation with CL. BAT from saline-treated Nfe2l1ΔBAT mice and WT littermates was histologically indistinguishable at 30 °C (Fig. 3l,m). BAT from WT mice showed the expected CL-induced lipid depletion compared to WT controls, indicating BAT activation4, whereas BAT from Nfe2l1ΔBAT mice show a diminished, if not an opposite phenotype compared to WT when treated with CL (Fig. 3l,m). In addition, CL-induced browning of IngWAT in WT controls was diminished in Nfe2l1ΔBAT mice (Fig. 3n,o), supporting the concept that thermogenic adipocyte activation itself rather than cold is required and sufficient for the Nfe2l1ΔBAT phenotype to emerge.
While our results consistently indicate BAT dysfunction in Nfe2l1ΔBAT mice, we did not observe general cold intolerance in Nfe2l1ΔBAT mice, likely due to thermoregulatory compensation by lower peripheral heat loss (Supplementary Fig. 6d). To further validate the inducible and adaptive nature of the Nrf1-proteasome pathway as well as to rule out potential developmental defects in the Nfe2l1ΔBAT model, we utilized a second mouse model, in which the Nfe2l1 gene was inactivated with tamoxifen-inducible Cre in adult mice housed at 22 °C. Similar to the constitutive Nfe2l1ΔBAT model, Nrf1 was required to sustain BAT mRNA levels of proteasome subunits (Supplementary Fig. 6e), as well as to limit activation of cellular stress pathways (Supplementary Fig. 6f). Within 1 week, deletion of Nfe2l1 in adult mice led to abnormal macroscopic and histological appearance of BAT (Supplementary Fig. 6g) as well as diminished BAT-mediated whole body respiration (Supplementary Fig. 6h).
The BAT ubiquitome
We next sought to identify the cellular pathways that are linked to Nrf1 and proteasome function in BAT. Proteasomal degradation of proteins requires their posttranslational modification with ubiquitin39, particularly formation of K48-linked ubiquitin chains40 and conversely, insufficient proteasomal activity is linked to accumulation of K48-modified proteins41,42. As Nrf1 was critical for cold-induced proteasomal activity, we tested whether Nrf1 modulates the levels of ubiquitinated proteins in BAT. The quantity of ubiquitinated proteins in BAT was equivalent in WT and Nfe2l1ΔBAT littermates at 30 °C, however, at 22 °C we observed higher levels of ubiquitinated proteins in BAT of WT controls compared to 30 °C, which was even higher in BAT from Nfe2l1ΔBAT mice (Fig. 4a). In order to identify ubiquitinated proteins differentially modulated by cold adaptation in the setting of Nrf1 deficiency, we performed a systematic and quantitative analysis of ubiquitinated proteins using multiplexed quantitative mass spectrometry (MS)41 in BAT of WT and Nfe2l1ΔBAT mice. In line with results above (Fig. 4a), this MS quantification confirmed the higher abundance of ubiquitinated proteins in BAT from Nfe2l1ΔBAT mice compared to WT controls (Fig. 4b). Ubiquitin linkages are formed by one of seven lysine residues in ubiquitin40. K48 modification was the most abundant ubiquitin linkage identified in the BAT samples, accounting for ~63 % of all modifications, followed by K6 (~15 %) and K63 (~10 %) (Supplementary Fig. 7a and Supplementary Table 1). K48, K11 and K708 ubiquitin linkages, which have been previously linked to ERAD-selective proteasomal degradation40, were all higher in Nfe2l1ΔBAT mice compared to WT controls (Fig. 4b). In contrast, putative non-degradative, regulatory linkages such as K27, K29 and K6340 were unchanged in Nfe2l1ΔBAT mice. As insufficient proteasomal activity might alter the abundance of specific proteins or we detected higher ubiquitination because the respective proteins were more abundant, we normalized the ubiquitination for each identified lysine residue to the amount of the respective protein determined by conventional proteomics. This allows determination of the ubiquitination status of each identified protein independent of the actual protein amount. In the analysis of this differential ubiquitome, we found that Nrf1 deficiency led to higher ubiquitination of proteins (Fig. 4c and Supplementary Table 2), indicating a hyperubiquitinated state in BAT of Nfe2l1ΔBAT mice. Remarkably, ~34 % (418/1229) of all hyperubiquitinated sites in BAT of Nfe2l1ΔBAT mice were listed in the Mitocarta (Fig. 4c), a proteomic inventory of mitochondria43, including components of the respiratory chain as well as Ucp1 (Fig. 4c), which has been earlier described to be regulated by proteasomal degradation44.
Figure 4.
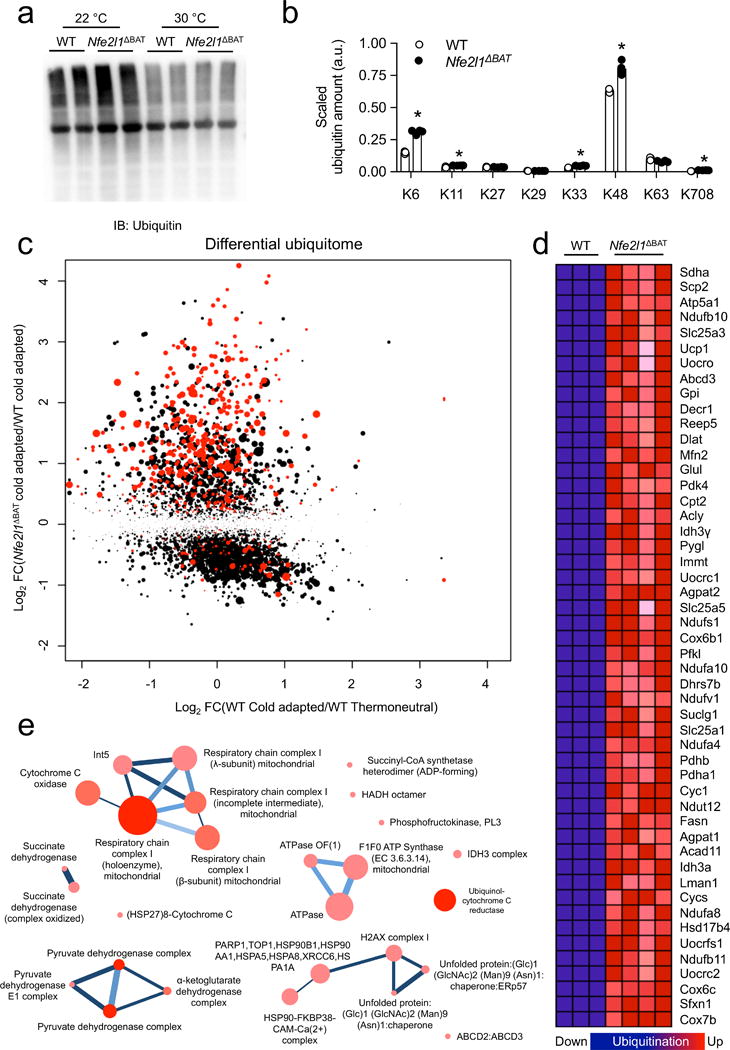
The brown fat ubiquitome. (a) Representative immunoblot of ubiquitin in BAT from WT and Nfe2l1ΔBAT mice born and raised at 22 °C or 30 °C (Biol. replicates n=2, cropped image). (b) Proteomic quantification of ubiquitin lysine (K)-linkages in BAT from cold-adapted (4 °C for 7 days) WT and Nfe2l1ΔBAT mice (Biol. replicates WT n = 3, Nfe2l1ΔBAT n = 4, *P < 0.05 by Student’s t-Test). (c) Scatter plot of differentially ubiquitinated protein lysine site (FC: fold change) in BAT from thermoneutral or cold-adapted WT as well as cold-adapted Nfe2l1ΔBAT mice (Biol. replicates. n = 3 for WT 30 °C, n = 3 for WT 4 °C and n = 4 for 4 °C Nfe2l1ΔBAT mice; Significance cold-adapted WT vs Nfe2l1ΔBAT mice is indicated, higher significance indicated by larger dot size, red colour indicates mitocarta protein) (d) List of top 50 hyperubiquitinated proteins in BAT from cold-adapted Nfe2l1ΔBAT mice compared to cold-adapted WT. (e) Functional protein complex prediction of hyperubiquitinated proteins in Nfe2l1ΔBAT mice.
Next, we investigated whether in addition to mitochondrial proteins, other specific cellular processes were overrepresented in the hyperubiquitinated state of Nrf1 deficiency, and performed unbiased computational gene set enrichment analysis45,46 of the major hyperubiquitinated proteins. We found that 27 processes were represented in 228 hyperubiquitinated proteins (Fig 4d), most notably related to mitochondria and organelle homeostasis, in particular ER proteins and energy metabolism (Supplementary Fig. 7b and Supplementary Table 3). We expanded our analysis to identify functional clusters in these processes using the ConsensusPathDB (CPDB) interaction database47 and found that most notably respiration and lipid metabolism were represented in the hyperubiquitinated state (Supplementary Fig. 7c). Using CPDB we also explored whether the major hyperubiquitinated proteins belong to functional complexes and found among others the respiratory chain, pyruvate dehydrogenase and the ER folding machinery (Fig. 4e). Taken together, these data demonstrate that Nrf1 is a critical regulator of brown adipocyte proteome health, enabling the proper turnover of ubiquitinated proteins. In addition, these results indicate that the thermogenic machineries for energy metabolism and respiration are particularly sensitive to imbalances in proteasomal activity, and that cold adaptation requires efficient turnover of mitochondrial proteins.
BAT proteasomal activity is impaired in obesity
BAT thermogenic activation exerts beneficial effects on systemic metabolism2–4 but chronic obesity results in reduced metabolic capacity of BAT in mice11. As it is not known whether proteasomal activity is altered and linked to the metabolic response to HFD-induced obesity (DIO), we investigated the relevance of Nrf1-mediated proteasomal activity in thermogenic adipocytes for obesity and associated metabolic dysfunction. First, we measured proteasomal activity in BAT of lean or HFD-fed WT and Nfe2l1ΔBAT mice at 22 °C. We found that in BAT of WT mice, chemotrypsin-like and caspase-like proteasomal activities were specifically lower in DIO, and this effect was amplified in BAT of Nfe2l1ΔBAT mice in both lean and obese conditions (Fig. 5a). In lean mice housed at 22 °C and fed a regular chow diet, we did not find differences in body weight or glucose tolerance between Nfe2l1ΔBAT mice and WT controls (Supplementary Fig. 8a,b). However, Nfe2l1ΔBAT mice fed HFD gained more weight and displayed higher adiposity compared to WT littermates fed HFD (Fig. 5b). Under these conditions, Nfe2l1ΔBAT mice displayed impaired glucose tolerance and insulin resistance (Fig. 5c,d), which was also reflected in higher plasma insulin and leptin concentrations compared to WT controls (Supplementary Table 4). Plasma adiponectin, triglycerides, cholesterol as well as transaminase concentrations were unchanged between genotypes (Supplementary Table 4).
Figure 5.
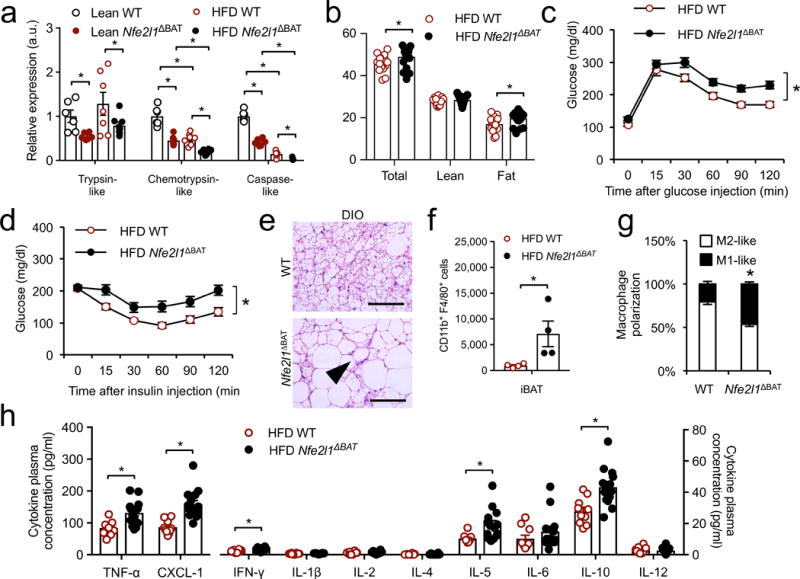
Nrf1-mediated proteasomal activity is linked to obesity-associated disorders. (a) Proteasomal activity in lean and high-fat diet (HFD) WT and Nfe2l1ΔBAT mice. (Biol. replicates n = 8, *P < 0.05 by 2-way ANOVA). (b) Body weight and composition of Nfe2l1ΔBAT mice and WT controls after HFD feeding (Biol. replicates WT n = 15, Nfe2l1ΔBAT n = 21, *P < 0.05 by Student’s t-Test). (c) Glucose tolerance test (1 g/kg) in HFD-fed WT and Nfe2l1ΔBAT mice (Biol. replicates WT n = 9, Nfe2l1ΔBAT n = 14, *P < 0.05 area under the curve different by Student’s t-Test). (d) Insulin tolerance test (1 U/kg) in HFD-fed WT and Nfe2l1ΔBAT mice (Biol. replicates WT n = 11, Nfe2l1ΔBAT n = 16, *P < 0.05 area under the curve different by Student’s t-Test). (e) Representative H&E histology of BAT from HFD-fed WT and Nfe2l1ΔBAT mice (Biol. replicates n = 15; Scale bars, 100 μm, arrow indicating crown-like structure). (f,g) Fluorescent flow cytometery quantification of (f) total macrophage content and (g) macrophage polarization in BAT from HFD-fed WT and Nfe2l1ΔBAT mice (Biol. replicates n = 4, *P < 0.05 by Student’s t-Test). (h) Cytokine plasma concentrations from HFD-fed WT and Nfe2l1ΔBAT mice. (Biol. replicates n = 10 for WT, n = 14 for Nfe2l1ΔBAT, *P < 0.05 by Student’s t-Test).
In addition to higher lipid accumulation compared to WT controls on HFD, we observed adipose tissue inflammation as indicated by abundant crown-like structures in BAT from Nfe2l1ΔBAT mice (Fig. 5e), resulting from inflammatory immune cell infiltration, similar to what has previously been described exclusively in WAT from obese animals48,49. We confirmed the higher macrophage infiltration in BAT of Nfe2l1ΔBAT mice compared to WT controls by flow cytometry analysis (Fig. 5f and Supplementary Fig. 8c), which also pointed to a more proinflammatory polarization50 in BAT of Nfe2l1ΔBAT mice compared to WT controls (Fig. 5g and Supplementary Fig. 8c). Consistent with this observation, short-term inhibition of the proteasome in cultured brown adipocytes led to higher mRNA expression of several chemokines and pro-inflammatory cytokines compared to vehicle (Supplementary Fig. 8d), which was heightened in the absence of Nrf1 in vivo (Supplementary Fig. 8e). This indicates that adipocyte stress in the setting of Nrf1 deficiency or proteasome inhibition induces the expression of chemokines, explaining the strong immune cell infiltration observed in vivo. Compared to WT controls, Nfe2l1ΔBAT mice also exhibited higher plasma concentrations of several proinflammatory cytokines including TNF-α, an important mediator linking adipose tissue inflammation to insulin resistance51 (Fig. 5h and Supplementary Table 4).
Enhancing proteostasis in BAT alleviates insulin resistance
Finally, to address the reversibility of the impact of genetic Nrf1-deficiency, as well as to examine the potential metabolic benefits of enhancing Nrf1 function in adult mice, we used an intra-tissue adenovirus (AV) injection strategy to drive Nfe2l1 overexpression in BAT. First, AV-Nrf1 injection into BAT of lean Nfe2l1ΔBAT mice led to higher BAT protein and mRNA expression of Nfe2l1 as well as Psm genes compared to the contralateral side of the interscapular BAT depot of the same mice, which was injected with AV-LacZ control (Fig. 6a,b and Supplementary Fig. 9a). Re-expression of Nrf1 in BAT of Nfe2l1ΔBAT mice partially restored proteasomal activity compared to control-injected WT mice (Fig. 6c). WT mice injected with AV-Nrf1 also showed moderately higher chemotrypsin-like activity compared to control-injected contralateral BAT (Fig. 6c). Nrf1 re-expression in BAT of Nfe2l1ΔBAT induced a more brown-like phenotype based on the appearance (Fig. 6d), histology (Fig. 6e) and Ucp1 immunohistochemistry (IHC) compared to the contralateral side (Fig. 6f). Ucp1 analysis by immunoblot, however, indicated similar Ucp1 total levels (Supplementary Fig. 9a). To explore whether these changes in BAT of Nfe2l1ΔBAT mice treated with AV-Nrf1 translate into changes in BAT metabolic activity, we performed a combined oral glucose and fat tolerance test with radiolabeled glucose and fatty acid tracers2. Re-expression of Nrf1 in BAT of Nfe2l1ΔBAT mice partially restored fatty acid uptake and completely rescued glucose uptake compared to control-injected WT controls (Fig. 6g).
Figure 6.
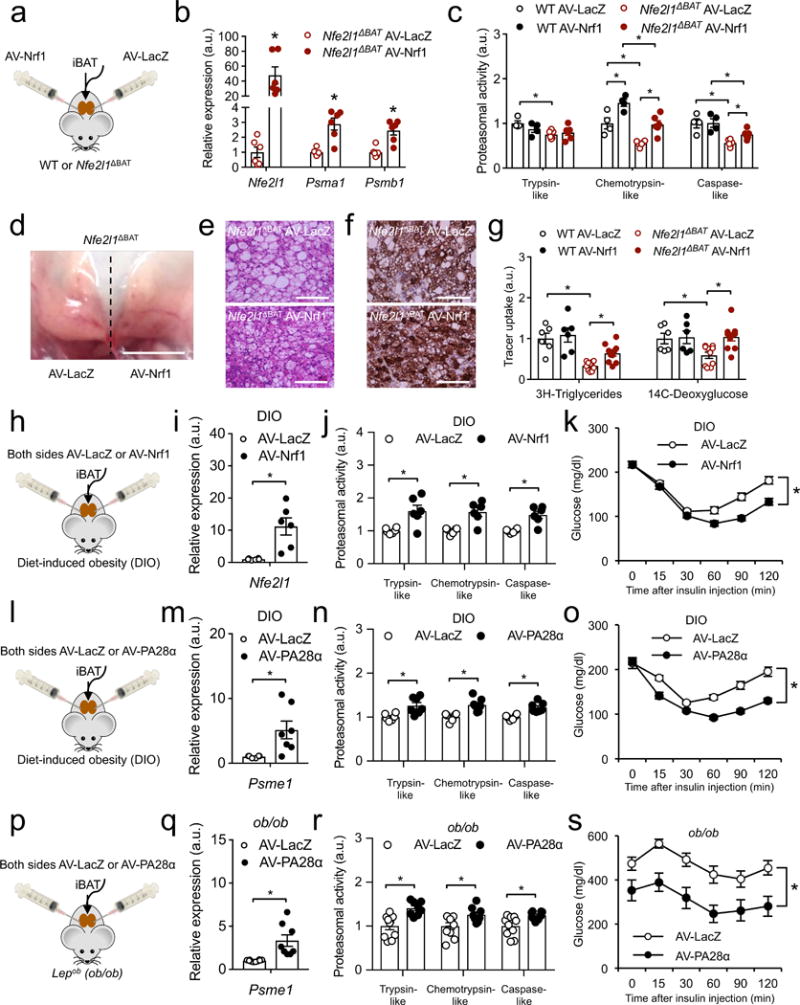
Enhancing proteostasis in BAT alleviates insulin resistance in DIO and ob/ob mice. (a) Strategy for contralateral intraBAT injection of adenovirus carrying LacZ (AV-LacZ) and Nrf1 (AV-Nrf1) into WT and Nfe2l1ΔBAT mice at 22 °C. (b) mRNA levels of Nfe2l1 and proteasome subunits in BAT of Nfe2l1ΔBAT mice BAT-injected with AV-LacZ and AV-Nrf1 (Biol. replicates n = 6, normalized to 18s, *P < 0.05 by Student’s t-Test). (c) Proteasome activity in WT and Nfe2l1ΔBAT mice BAT-injected with AV-LacZ and AV-Nrf1 (Biol. replicates n = 4 for WT and n = 6 for Nfe2l1ΔBAT, *P < 0.05 by 2-way ANOVA). (d) Photograph of iBAT of a Nfe2l1ΔBAT mouse injected with AV-LacZ (left) and AV-Nrf1 (right) contralaterally (Scale bar, 5 mm). (e) Representative H&E histology and (f) Ucp1 IHC of Nfe2l1ΔBAT mice injected with AV-LacZ and AV-Nrf1 (Biol. replicates n = 3; Scale bars, 100 μm). (g) BAT uptake of radiolabelled tracers after a combined oral fat and glucose tolerance test in WT and Nfe2l1ΔBAT mice injected with AV-LacZ and AV-Nrf1 (Biol. replicates n = 6 for WT and n = 10 for Nfe2l1ΔBAT, *P < 0.05 by 2-way ANOVA. (h) Strategy for intraBAT injection of AV-LacZ or AV-Nrf1 into diet-induced obese (DIO) mice. (i) mRNA levels of Nfe2l1 in BAT of DIO mice BAT-injected with AV-LacZ or AV-Nrf1 (Biol. replicates n = 6, normalized to Tbp, *P < 0.05 by Student’s t-Test). (j) Proteasome activity in DIO mice BAT-injected with AV-LacZ or AV-Nrf1 (biol. replicates n = 6, *P < 0.05 by Student’s t-Test). (k) Insulin tolerance test in DIO mice BAT-injected with AV-LacZ or AV-Nrf1 (Biol. replicates n = 10, *P < 0.05 by Student’s t-Test on AUC). (l) Strategy for intraBAT injection of AV-LacZ or AV-PA28α into diet-induced obese (DIO) mice. (m) mRNA levels of PA28α (encoded by Psme1) in BAT of DIO mice BAT-injected with AV-LacZ or AV-PA28α (Biol. replicates n = 6 for AV-LacZ and n = 7 for AV-PA28α, normalized to Tbp, *P < 0.05 by Student’s t-Test). (n) Proteasome activity in DIO mice BAT-injected with AV-LacZ or AV-PA28α (Biol. replicates n = 6 for AV-LacZ and n = 7 for AV-PA28α, *P < 0.05 by Student’s t-Test). (o) Insulin tolerance test in DIO mice BAT-injected with AV-LacZ or AV-PA28α (Biol. replicates n = 5, *P < 0.05 by Student’s t-Test on AUC). (p) Strategy for intraBAT injection of AV-LacZ or AV-PA28α into ob/ob mice. (q) mRNA levels of PA28α (encoded by Psme1) in BAT of ob/ob mice BAT-injected with AV-LacZ or AV-PA28α (Biol. replicates n = 10 for AV-LacZ and n = 8 for AV-PA28α, normalized to Tbp, *P < 0.05 by Student’s t-Test). (r) Proteasome activity in ob/ob mice BAT-injected with AV-LacZ or AV-PA28α (Biol. replicates n = 10 for AV-LacZ and n = 8 for AV-PA28α, *P < 0.05 by Student’s t-Test). (s) Insulin tolerance test in DIO mice BAT-injected with AV-LacZ or AV-PA28α (Biol. replicates n = 10 for AV-LacZ and n = 8 for AV-PA28α, *P < 0.05 by Student’s t-Test on AUC).
To address the translational relevance of Nrf1 targeting, we next tested whether increasing Nrf1 activity in BAT of mice with obesity-associated insulin resistance might enhance proteasome function, and thus confer beneficial systemic metabolic effects. Therefore, we evaluated the effects of AV-Nrf1 delivery into BAT of WT mice with HFD-induced obesity (Fig. 6h). As expected, Nfe2l1 mRNA and proteasomal activity were higher after treatment with AV-Nrf1 in DIO mice compared to control-injected animals (Fig. 6i,j), which led to a body weight-independent improvement of insulin tolerance just 9 days after injection compared to control-injected animals (Fig. 6k and Supplementary Table 5). We further validated the beneficial effect of AV-Nrf1 injection into BAT in an independent model of obesity, insulin resistance, and associated disorders, the leptin-deficient ob/ob mouse52. We found that AV-Nrf1 treatment improved insulin tolerance independently of body weight or Ucp1 levels in BAT compared to control-injected ob/ob animals (Supplementary Fig. 9b-d) and Supplementary Table 5).
In order to prove that proteostasis per se is a critical aspect of BAT adaptation and has therapeutic relevance we designed an Nrf1-independent strategy targeting proteasome function. To this end, we used a similar intraBAT adenovirus injection strategy to deliver PA28α (encoded by Psme1), a regulatory component of the proteasome enhancing its activity53, into BAT of DIO mice (Fig. 6l). We found that injection with AV-PA28α led to higher mRNA expression of Psme1 in BAT (Fig. 6m) and higher BAT proteasomal activity compared to control-injected animals (Fig. 6n). Similar to the effects of AV-Nrf1 in this model (Fig. 6h-k), PA28α expression improved insulin tolerance compared to control-injected animals, independently of body weight (Fig. 6o and Supplementary Table 5). Finally, we independently validated the effects of AV-PA28α delivery into BAT of ob/ob mice (Fig. 6p), which led to higher BAT expression of Psme1, and higher proteasomal activity (Fig. 6q,r) and also improved insulin tolerance compared to control-injected ob/ob animals (Fig. 6s and Supplementary Table 5).
Discussion
ER homeostasis is a critical component of metabolic health, especially in the context of obesity18. However, the mechanisms that protect the ER from metabolic stress in adipocytes remain unclear. Moreover, while thermogenic fat cells possess exceptional metabolic properties, accounting for roughly 65 % of total oxidative metabolism in mice when fully recruited1, it is unclear whether brown adipocytes require specific mechanisms to mitigate cellular stress in this state of high metabolism. Here we find that cold adaptation of brown fat requires proteasomal activity, which is transcriptionally driven by cold-inducible Nrf1. In this way, Nrf1 in thermogenic fat cells is a metabolic guardian, preventing tissue stress and inflammation independently of BAT differentiation, mass or expandability.
One could argue that the phenotypes associated with Nrf1 deficiency in BAT are simply a reflection of a defect in a fundamental process, which compromises the general integrity of the tissue. However, several lines of observations make this possibility very unlikely. First, genetic deletion of Nrf1 in BAT does not result in complete proteasome deficiency; it only limits the inducible component of proteasomal activity required for thermogenic adaptation. Second, the effects of proteasome inhibitors on non-shivering thermogenesis in mice are not associated with changes in Ucp1, BAT mass, or histological appearance. Third, we do not find any effect of Nrf1 deficiency on key brown fat regulators such as Ucp1, Pgc1α/β, or Ppargα/γ. Fourth, mice lacking Nrf1 in brown adipocytes are born normal but alterations in the BAT of Nfe2l1ΔBAT develop as a function of cold and age. At thermoneutrality, where BAT activity is minimal, Nrf1 levels in BAT are very low and in this environment neither proteasome inhibition nor genetic deficiency of Nrf1 are deleterious for the tissue, indicating the Nrf1-mediated program is not vital in BAT but rather is required for adapting to rising thermogenic activity. This is further corroborated by the phenotype of tamoxifen-induced deletion of Nrf1 in adult mice. Indeed, BAT thermogenic activation following cold adaptation or treatment with the β3-agonist CL at thermoneutrality requires an Nrf1-mediated increase in proteasomal activity in order to fulfil its metabolic potential.
In many tissues, metabolic or proteotoxic stress is combatted by the activation of classical UPR pathways. Our finding that proteasomal protein degradation contributes more to the metabolic function of brown adipocytes than chaperone-assisted protein folding (exemplified here by the Ire1-Xbp1 pathway), might be partially related to a limited expandability and folding capacity of the ER in brown adipocytes. Indeed, in Nfe2l1ΔBAT mice we observe a compensatory response to the failure of the UPS to keep pace with proteometabolic demand during cold adaptation, which altogether was unable to restore ER homeostasis in the absence of sufficient proteasomal activity. Of note, this process is partially reversible, as placing Nfe2l1ΔBAT mice from colder to warmer temperatures alleviates stress responses in BAT, further supporting that the regulation of proteasomal activity by Nrf1 serves as a physiological adaptation to high metabolism associated with thermogenic BAT activity.
Based on evidence from cultured cells, it is well accepted that proteasomal protein degradation is important for clearing damaged or dispensable proteins16, but little is known about the plasticity and physiological relevance of this process. Indeed, the proteasome is often considered a static protein complex, and most mechanistic investigations have been performed with chemical inhibitors27,31,32. In this light, our finding that cold adaptation naturally engages the UPS through Nrf1 is a remarkable example of physiologically stimulated proteasomal protein degradation. We also find that in the general absence of phenotypic abnormalities in Nfe2l1ΔBAT mice born at thermoneutrality, Nrf1 in BAT is still required to cope with proteotoxic stress induced by proteasome inhibitors once the tissue is activated by cold. This further emphasizes the adaptive role of and mechanistic link between Nrf1, the proteasome, and non-shivering thermogenesis. Interestingly, even though Nrf1 is ubiquitously expressed in cells and tissues, regulation of its expression and activation as well as down-stream mechanisms engaged by Nrf1 are poorly understood. The marked cold-induced increase in Nrf1 is specific for brown and beige adipocytes, possibly pointing to a cell-type specific regulation of the Nfe2l1 gene. While in the liver Nrf1 and proteasomal function have been linked to feeding and steatosis20,54,55, there was no induction of proteasomal activity by cold in this tissue. Our study globally identifies proteins and cellular pathways modulated by the Nrf1-proteasome pathway, suggesting that proteasomal protein turnover is a prerequisite for cold-induced metabolic activity and remodelling of BAT. Beyond the role of the Nrf1-proteasome pathway in maintaining ER homeostasis, we find that the hyperubiquitinated proteins in BAT of Nfe2l1ΔBAT mice represented a broad array of brown fat metabolic processes, ranging from carbohydrate to lipid metabolism and, surprisingly, included a large number of mitochondrial proteins. This is directly linked to mitochondrial respiratory capacity in terms of oxidative substrate turnover, leak respiration and Ucp1 function. In addition to enhanced ubiquitination of general mitochondrial components we discovered here ubiquitination of numerous known regulators of non-shivering thermogenesis as well as other proteins with unknown function in BAT, altogether emphasizing the fundamental role of Nrf1-mediated proteasomal activity for proteostasis and metabolic quality control in BAT. In conjunction with earlier reports on Ucp1 ubiquitination44, our study identifying numerous cold-regulated ubiquitination sites provides the biochemical basis for understanding the turnover of BAT proteins in response to cold adaptation.
An important question emerging from our study is how the Nrf1-proteasome pathway might relate to obesity and associated diseases in humans. A recent epigenome-wide association study linked altered methylation of the human NFE2L1 locus to BMI and adverse outcomes of obesity56, although future mechanistic studies are needed to understand how this epigenetic variant might impact Nrf1 and proteasome activity. Additionally, we demonstrate here that there are high levels of NFE2L1 expression in human BAT, which correlates with brown functionality of human clonal adipocyte cell lines or tissue samples. Moreover, in mice we find that proteasomal activity in BAT is compromised in DIO, and that Nrf1-mediated proteasomal activity is required to maintain ER homeostasis and limit the activation of cellular stress pathways, tissue inflammation, and obesity-induced metabolic dysfunction (Supplementary Fig. 10). In exploring the translational potential of the Nrf1-proteasome pathway, we find that increasing proteasomal activity by adenoviral delivery of Nrf1 or the proteasome activator PA28α into BAT of mice with dietary or genetic obesity results in improved systemic insulin sensitivity and glucose homeostasis. In addition, these experiments suggest that other proteolytic, ubiquitin-independent cellular machineries might also be part of global proteostatic regulation in BAT, which opens possible future avenues for further exploration of their role in metabolic regulation.
An important consideration for future studies will be developing Nrf1-targeted therapies. We find that cold induces Nrf1 expression at both the mRNA and protein levels, and while the cleavage ratio is unchanged, this overall increase produces higher levels of the active form. Hence, identification of this physiological signal mechanism governing Nrf1 gene expression or identifying factors that lead to more active Nrf1 protein will be interesting future avenues. Additionally, Nrf1 exhibits several features that to our knowledge do not exist in most other transcriptional regulators, which may offer multiple options for therapeutic targeting. For example, in addition to raising Nrf1 abundance to generate therapeutic efficacy as shown in our study here, Nrf1 has a multitude of posttranslational regulatory stages that are involved in mobilizing Nrf1 from the ER into the nucleus, which includes proteolysis, glycosylation, and potentially phosphorylation and ubiquitination27,31,32,57. In addition, we have recently shown that cholesterol in the ER membrane of hepatocytes influences the transcriptional activity of Nrf158. This multitude of regulatory stages will likely be accessible to a variety of modern day therapeutic modalities. In fact, others have identified natural compounds that can also modulate Nrf159,60 but their specificity and in vivo efficacy for obesity and related metabolic disorders remains unexplored.
Taken together our study demonstrates that proteostasis and brown adipocyte health are critical to secure systemic metabolic integrity during states of high activity. This represents an important conceptual advance, as currently there is scarce understanding of the pathways that sustain the high metabolic demands imposed on BAT during thermogenesis relative to those involved in BAT activation or development. Furthermore, our study introduces the concept that Nrf1 is a fundamental adaptive regulator of brown adipocyte thermogenic function and may act as a general cellular guardian of metabolic health in obesity and beyond.
Methods
Mice and treatments
This section is also summarized in the Life Sciences Reporting Summary. All experimental procedures involving animals were approved by the Harvard T.H. Chan School of Public Health (HSPH) Institutional Animal Care and Use Committee or in accordance with approved institutional protocols of the Massachusetts Institute of Technology, respectively. We purchased WT mice C57BL/6J (Stock no. 000664), C57BL/6J HFD-DIO mice (Stock no. 380050) and Lepob (ob/ob) mice (Stock no. 000632) from Jackson lab (Jax, jax.org). Adipocyte-specific (Adipoq-Cre) Xbp1-deficient mice were described previously23. Floxed Ire1α mice were provided by T. Iwawaki (Gunma University, Japan) and crossed with Adipoq-Cre mice (Jax stock no. 024671) to yield adipocyte-specific Ire1α deficient mice. We generated mice carrying floxed alleles of Nfe2l1 from Nfe2l1tm1a(EUCOMM)Hmgu ES cells (International Mouse Phenotyping Consortium, http://www.mousephenotype.org/). In cooperation with the Harvard Stem Cell Institute, C57BL/6N-A/a ES cells were injected into blastocysts, giving rise to chimeric mice, of which males were continuously bred until germ line transmission was successful in producing founders carrying the Nfe2l1tm1a allele. This allele caused a whole body null gene with Flp recombinase sites that could be utilized to re-insert the wild type allele containing Cre recombinase sites surrounding exon 4 and 5. This locus includes the coding region for the DNA-binding domain of Nrf1. We bred mice with C57BL/6J Flp recombinase transgenic mice (Jax), after which the Flp transgene was bred out, leaving floxed Nfe2l1 alleles only mice. These were crossed with hemizygous transgenic mice expressing Cre under the control of the Ucp1 promoter (Ucp1-Cre61) or with hemizygous mice expressing tamoxifen-inducible Cre under control of the chicken beta actin promoter/enhancer coupled with the cytomegalovirus (CMV) immediate-early enhancer (CAGGCre-ER™, Jax stock no. 004682), allowing for conditional Nrf1 deletion. After backcrossing with C57BL/6J mice we used homozygous floxed Nfe2l1 alleles with Ucp1-Cre (Nfe2l1ΔBAT mice) and WT control littermates carrying floxed Nfe2l1 alleles but no Cre or CAGGCre-ER™ (Ind.Nfe2l1−/−) and WT littermates, respectively. We performed genotyping by PCR or Transnetyx (Supplementary Table 6). If not indicated otherwise mice were bred and housed in the HSPH animal facility at 22 °C with ad libitum access to standard laboratory chow diet (PicoLab Mouse Diet 20 #5058, LabDiet) and a 12 h light/dark cycle. We induced obesity by feeding mice HFD ad libitum beginning at 6 weeks of age for 16 weeks before assessment of metabolic disease (D12492i, Research Diets) in 3 independent cohorts of mice. We bred and/or housed (7 days) thermoneutral mice in a Memmert HPP750 LIFE Chamber at 30 °C with ad libitum access to standard laboratory chow diet (PicoLab Mouse Diet 20 #5053, LabDiet) and a 12 h light/dark cycle. For cold adaptation (4 °C for 7 days) we housed mice in single cages with ad libitum access to standard laboratory chow diet (PicoLab Mouse Diet 20 #5058, LabDiet) and a 12 h light/dark cycle. We used 12 week-old male mice if not indicated otherwise. No blinding was performed. Sample sized was estimated by pilot experiments that showed trends of effects and their sizes. In most cases, an n=6 was the minimal amount used, only where effect size was large, an n=4 yielded significant results. Mice were excluded for poor body condition (e.g. after surgery or during feeding studies). We randomly used age-matched littermates for the study of adult mice. We performed glucose tolerance tests after 16 h of fasting by intraperitoneal injection of 1 g/kg glucose in PBS. We performed insulin tolerance tests after 6 h of fasting by intraperitoneal injection of 0.75 U/kg (DIO mice) or 1.25 U/kg (ob/ob mice) insulin (Eli Lilly Humulin) in 0.2 % w/v BSA in PBS. We combined oral glucose and fat tolerance tests by gavage of a mixture of liposomes containing 6.17 KBq [9,10-3H(N)]-triolein /g body weight and glucose (1 g/kg body weight) traced with 0.12 KBq 2-desoxy-D-[14C]-glucose/g body weight in H2O as described previously2. We measured body composition in anesthetized (100 mg/kg ketamine and 10 mg/kg xylazine) mice with dual-energy x-ray absorptiometry (DEXA Lunar PIXImus, GE). We measured energy expenditure, respiration and activity using a Columbus Instruments’ Oxymax-Comprehensive Lab Animal Monitoring System (CLAMS) system according to guidelines for measuring energy metabolism in mice62. We measured BAT-mediated energy expenditure by subcutaneous injection of NE (Sigma, 1 mg/kg in 0.9 % w/v NaCl) or CL316,243 (CL, Tocris, 0.5 mg/kg in 0.9 % w/v in NaCl) according to guidelines for measuring BAT activity25 around 1.30 p.m. We performed chronic CL treatment by subcutaneous injection of CL (0.5 mg/kg) for 7 days in 8 week-old female mice. We injected bortezomib (Selleckchem, 2.5 mg/kg in 10% v/v DMSO in PBS) or 10 % v/v DMSO in PBS as vehicle intraperitoneally at 5 pm and body core temperature, mitochondrial isolation and necropsy of WT B6 mice were performed 16 h after. To test the effect of bortezomib in WT and Nfe2l1ΔBAT littermates, mice born and raised at 30 °C were transferred to 22 °C to the CLAMS system at 8 weeks of age and after 72 h, bortezomib (Selleckchem, 1.25 mg/kg in 10 % v/v DMSO in PBS) or 10 % v/v DMSO in PBS as vehicle were injected intraperitoneally. We injected carfilzomib (Selleckchem, 4 mg/kg in 33% v/v DMSO in H2O) or 33 % v/v DMSO in H2O as vehicle into the tail vein of single-caged WT B6 mice at 10 am and performed body core temperature measurement and necropsy 4 h and 6 h later, respectively. We measured rectal body core temperature using a Physitemp Thermalert TH-5 probe and BAT and skin temperature using a FLIR Infrared camera (we took the average of two sequential images to calculate BAT and skin temperature per individual mouse). We performed standardized necropsies after 4 h fasting around noon. Mice were anesthetized with 300 mg/kg ketamine and 30 mg/kg xylazine in PBS, blood was withdrawn by cardiac puncture and mice were perfused with either PBS containing 50 U/ml heparin, formalin (Fisher Scientific) or electron microscopy grade fixative (Electron Microscopy Science). Organs were harvested and immediately preserved in TRIzol (Invitrogen), formalin, electron microscopy grade fixative or snap-frozen in liquid nitrogen and stored at −80 °C. All experiments were reproduced in independent cohorts at least twice.
Proteasomal activity
We used the Proteasome Activity Fluorometric Assay (UBP Bio) according to the manufacturer’s instructions for chemotrypsin-, trypsin- and caspase-like activity. In each assay, for each sample we used MG132 (Sigma) controls to determine the specific proteasomal activity Δ(slope) over time.
Gene recombination and expression analysis
We detected Cre-mediated recombination of floxed Nfe2l1 alleles in genomic DNA by PCR (Supplementary Table 6). We disrupted cells or tissues in TRIzol (Invitrogen) using TissueLyser (Qiagen). We isolated total RNA using NucleoSpin RNA II kit (Macherey & Nagel). We synthesized Complementary DNA using iScript RT Supermix kit (Biorad). We performed quantitative real-time PCR in triplicates on a ViiA7 system (Applied Biosystems) using SYBR green and custom-made primer sets designed by the Roche Universal ProbeLibrary Assay Design Center and verified by In-silico PCR (https://genome.ucsc.edu/cgi-bin/hgPcr) (Supplementary Table 6). We normalized gene of interest cycle thresholds (Cts) to TATA-box binding protein (Tbp) or 18S ribosomal RNA (Rn18s) house keeper levels by the ΔΔCt method and displayed the results as absolute or relative copies per Tbp or Rn18s or as relative expression normalized to experimental control groups.
Human data
This study followed the institutional guidelines of and was approved by the Human Studies Institutional Review Boards of Beth Israel Deaconess Medical Center and Joslin Diabetes Center. Details on procedures of human subject collection were described previously63. All subjects gave written informed consent before taking part in the study. Neck adipose tissue (carotid sheath) biopsies from 10 different subjects were studied; 4 samples were used for primary cell culture and immortalization as described below, all 10 were used for μ-array analysis as described below. Isolation of primary stromal vascular fraction (SVF) from human neck adipose tissue was described previously63. Owing to the limited amount of human BAT tissue collected from surgeries, we pooled adipose depots from the same subject to obtain sufficient numbers of SVF cells for primary culture as well as immortalization as described previously34. We performed μ-array analysis of gene expression using GeneChip PrimeView (Affymetrix, Santa Clara, CA) on paired white and brown adipose tissue biopsy samples from 10 individuals as well as 42 highly adipogenic clones. RNA was isolated from tissue using Direct-zol RNA miniPrep kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. We evaluated the quality of total RNA by A260/A280 ratio, which was within the value of 1.9 to 2.0 (defined as high-quality total RNA). Biotin-labeled cRNA was synthesized, purified and fragmented using GeneChip 3IVT Express Kit (Affymetrix, Santa Clara, CA). We assessed integrity and fragmented cRNA by running aliquots on the Agilent 2100 Bioanalyzer before proceeding further. The high-quality cRNA met the following criteria: the A260/A 280 fell within the values of 1.9 – 2.0; the 28S/18S rRNA bands (from the gel) were crisp and the intensity of the 28S band was roughly twice the intensity of the 18S band.
Protein isolation and immunoblot analysis
We lysed tissues in RIPA buffer containing 150 mM NaCl, 50 mM Tris-HCl pH 7.5, 0.1 % w/v SDS, 0.5 % w/v Na-Deoxycholate, 1 % v/v Nonidet p40, 1 mM EDTA, 1 mM EGTA, 2.5 mM Na-pyrophosphate, 1 mM NaVO4 and 10 mM NaF using the Tissue Lyser II (Qiagen). For each isolation process, we added fresh protease inhibitors (Sigma) in a 1:100 ratio. For isolation of proteins from adipose tissue, the supernatant was centrifuged until the supernatant was cleared from fat contaminations at 4 °C for 20 min at 13,000 g. Protein lysates were stored at −20 °C until further use. Protein concentrations were determined by using the Pierce BCA kit (Life Technologies) according to instructions. For Western Blot we used lysates in a final concentration of 1 μg/μL in 50 % v/v Laemmli buffer. 4-12 % Bis-Tris gels (Life Technologies) with MES buffer were used for SDS-PAGE. After separation, proteins were transferred onto a nitrocellulose membrane (Bio-Rad) using the wet blot technique (Bio-Rad), and blocked in 5 % w/v milk and primary antibodies (Supplementary Table 6) applied in 0.5 % w/v milk over night at 4 °C. After washing 4 × for 15 min with TBS-T, secondary antibodies (Santa Cruz) were applied for 1 h in 5 % w/v milk at RT. Then membranes were washed again 4 × times for 15 min in TBS-T and developed using Super Signal™ West Femto Maximum Sensitivity Substrate (Life Technologies) and a FluorChem M imager (proteinsimple). We analysed immunoblot digital images with the software of the device. Uncropped images can be found in Supplementary Fig. 11-13.
Phenotypic ER reporter assay
We previously described the asialoglycoprotein receptor 1 (ASGR1)-Cypridina noctiluca luciferase (Cluc) fusion protein system to monitor ER protein folding that also expresses Gaussia luciferase (Gluc) as an internal control29. For this study we used 3T3-L1 adipocytes stably expressing the reporter system and performed the experiment as previously described29. We measured luciferase secretion after 4 h of incubation with the indicated compounds.
Primary cell preparation, genetic modification and culture
All cell culture experiments were reproduced at least twice. Representative data are shown for Seahorse and Immunoblot analysis. We isolated primary preadipocytes from interscapular BAT of 6 week-old mice by the method of Rodbell64, which were cultured for 48 h and infected with lentivirus carrying human telomerase (hTERT). To generate a lentiviral construct expressing hTERT, we amplified cDNA of hTERT (isoform 1 from pBABE-hTERT (Addgene plasmid 1771) with NEB Q5 Taq polymerase and primers to insert XbaI (5′) and SalI (3′) restriction sites) by PCR. We cloned hTERT cDNA into the XbaI and SalI restriction sites of pLenti CMV GFP Hygro (Addgene plasmid 17446). We acquired mouse Nfe2l1 cDNA from Open Biosystems, as previously described50. We generated mammalian cell expression plasmids encoding hemagglutinin (HA)-tagged Nrf1 by performing an LR recombination reaction between pENTR1A and destination vector pLenti/CMV/Puro-DEST using Gateway LR Clonase Enzyme Mix (Life Technologies, 11791-019). To generate lentivirus particles, we transfected 293T Lenti-X cells (Clonetech) with second-generation packaging and envelope (psPAX2 and pMD2.G) and pLenti/CMV/Hygro/hTERT or pLenti/CMV/Puro/Nrf1 plasmids, respectively. Lentiviral supernatant was collected at 48 h and 72 h after the initial transfection and filtered through 0.45 μm syringe filters. The viral supernatant was diluted 1:2 with fresh DMEM and supplemented with 2 μg/ml polybrene prior to performing 2 rounds of infection on pre-adipocytes. At 48 h after the final infection, pre-adipocyte media was changed to fresh DMEM containing 10 % v/v FBS and penicillin/streptomycin. Selection with Hygromycin B (Invitrogen) was performed at 125 μg/ml for 2 weeks. For differentiation of preadipocytes into brown adipocytes, cells were induced for two days and differentiated for 4 days following one day of cultivation in standard maintenance media, before conducting experiments. The composition of the differentiation media was established by titrating the various compounds over time (data not shown). The induction medium was composed of DMEM high glucose, 10 % CCS, 1 % penicillin/streptomycin, 20 nM insulin, 2 nM T3, 150 μM ascorbate, 1 μM rosiglitazone, 500 μM IMBX and 1 μM Dexamethasone. Differentiation medium was made up of DMEM high glucose, 10 % CCS, 1 % penicillin-streptomycin, 20 nM insulin, 2 nM T3, 150 μM ascorbate and 1 μM rosiglitazone. To ensure proper growth and adequate supply with nutrients, we changed the medium every two days.
Seahorse analysis of respiration in isolated mitochondria and cells
We adhered to guidelines for assessing mitochondrial function65,66. Mitochondria were purified as described previously66 with modifications: All steps were carried out at 4˚C. BAT from 3-4 WT littermate controls and from 5-6 Nfe2l1ΔBAT 6 week-old animals or from 4 DMSO and 4 Bortezomib-treated WT mice were minced in a glass petri dish containing 2 mL Sucrose-Tris-EGTA (STE) buffer (250 mM sucrose, 5 mM Tris-HCl, 2 mM EGTA, pH 7.4) and then transferred to a 10 mL glass homogenizer with Teflon pestle containing 5 mL STE supplemented with 1 % w/v FA-free BSA. These isolations and subsequent analysis were reproduced at least 4 times and the data pooled. Tissues were homogenized manually (up to six strokes) the volume was brought to 25 mL and centrifuged at 8500 g for 10 min. The top fat layer was removed and the tube walls were wiped to remove excess fat. The pellet was resuspended in 1 mL BSA-free STE and transferred to a clean tube; the volume was brought to 25 mL and centrifuged at 800 g for 5 min. The supernatant was collected and centrifuged at 8500 g for 10 min. The pellet was washed and resuspended in 150 μL STE. Protein concentration was determined by BCA (Pierce). Respiration with 20 mM glycerol-3-phosphate as substrate67 was measured in 50 mM KCl, 10 mM TES, 1 mM EGTA medium containing 0.4 % (w/v) fatty acid-free bovine serum albumin, 1 mM KH2PO4, 2 mM MgCl2 and 0.46 mM CaCl2. The oxygen consumption rates were monitored in a Seahorse XF24 instrument at 2.5 μg mitochondrial protein per well. Baseline measurements (state 2) were performed with 1 mM ADP and 3 mM GDP, followed by injection of 20 mM glycerol-3-phosphate on port A and then 1 μg/ml oligomycin on port B. FCCP was titrated to 9 μM and injected on port C. Mitochondrial oxygen consumption was inhibited by the injection of 4 μM rotenone and 2 μM antimycin A on port D. GDP-sensitive respiration (UCP 1 activity) was calculated by subtracting basal uncoupled respiration in the presence of 20 mM glycerol 3-phosphate by the rates in the presence of 3 mM GDP plus 2 % FA-free BSA. For seahorse measurements in brown adipocytes, cells were seeded at 40,000 cells per well in a 24-well seahorse V7 plate in 2 independent experiments. In order to ensure even distribution, cells were seeded in 100 μL of media and kept for one hour at room temperature, following two hours at 37 °C. Then 150 μL media were added and cells were allowed to fully attach overnight. Epoxomicin (100 nM) was applied for 16 h with 150 μL media and cells were incubated overnight. The corresponding amount of DMSO was added as control treatment. On the day before the experiment, seahorse cartridges with the sensors were equilibrated in XF calibrant solution at 37 °C in a non-CO2 chamber overnight. Seahorse media was always prepared fresh on the day of the experiment and was composed of XF base media, 2 % BSA, 25 mM glucose, 2 mM pyruvate, and 2 mM L-glutamine. The pH was adjusted to 7.4 using 1 M NaOH and the media was filter sterilized before applying on cells. Cells were washed twice with 1 mL seahorse media, before incubating in 500 μL of seahorse media for 1 hour at 37 °C without CO2, prior to the experiment. Concentrations of oligomycin and FCCP were determined by thorough titration (data not shown). Final concentrations were 2.5 μM oligomycin, 22 μM FCCP and 0.5 μM rotenone/antimycin A. The standard protocol was 3 min mix, 2 min wait and 3 min measure. For the normalization of respiration to live cell count after the measurement, cells were washed gently using 1 × PBS, following fixation using 150 μL methanol for each well. After 20 min incubation at RT, cells were washed with 1× PBS. In order to stain DNA of cells, 25 μL of a 0.1 % w/v crystal violet (Sigma) solution was applied on fixed cells and incubated for 30 min at RT. Afterwards cells were washed 3 × with H2O and the plate was allowed to air dry. For lysis, 25 μL a 1 % v/v Triton X-100 in PBS solution were applied. The plate was placed on a shaker for 15 min at RT. The lysate was diluted 1:5 with PBS and the absorbance was measured 595 nm. Cell count was determined by using a standard curve of absorbance of defined cell numbers.
Mitochondrial iron and DNA measurement
For quantitative iron analysis, we homogenized BAT samples from 1 cohort of mice in acid solution and iron content compared to a standard solution was determined by inductively coupled plasma mass spectrometry (ICP-MS) on an Elan DRC-II machine (Perkin-Elmer). For mitochondrial DNA content analysis, we isolated total DNA from BAT by phenol-chlorofom extraction and amplified mitochondrial as well as nuclear DNA by quantitative real-time PCR with SYBR green (Roche) as described previously22.
Lipolysis assay ex vivo
As described previsously68, we removed BAT and WAT depots from 1 representative cohort of 6-week old littermates, minced them in DMEM high glucose and washed them 5 × with DMEM high glucose supplemented with 2 % w/v fatty acid-free bovine serum albumin (FA-free BSA DMEM, Sigma). We placed 5-6 pieces of adipose tissue into 12-well plates in 0.5 ml 2 % w/v FA-free BSA DMEM and incubated for 1 h at 37 °C before stimulation with 10 μM isoproterenol (Sigma). Samples were taken at indicated time points and 15 μl supernatant were assayed for non-esterified fatty acid (NEFA) concentration. After the assay, we isolated DNA from the explants by phenol-chloroform extraction and the fatty acid release was normalized to DNA content.
Serum and plasma parameters
We measured glucose using the Bayer Breeze2 platform. We measured insulin with Mouse Ultrasensitive Insulin ELISA (ALPCO), adiponectin (Mouse Adiponectin/Acrp30 Quantikine ELISA Kit) and leptin (Mouse/Rat Leptin Quantikine ELISA Kit) were measured with R&D ELISAs. Liver transaminases, triglycerides and cholesterol were determined using the Lipid Panel Plus Piccolo Xpress clinical chemistry platform (Abaxis). Cytokines were determined using the V-PLEX Proinflammatory Panel 1 (mouse) Kit (Mesoscale) according to instructions with undiluted plasma. We calculated HOMA-IR using the formula (Insulin[ng/ml]/0.0347)×Glucose[mg/dl]/405.
Adenoviral manipulation of BAT gene expression
We constructed the AV vector using the Adeasy system69 with the full-length mouse 741 AS ORF of Nrf1 (Open Biosystems). Virus production was carried out by Vector Biolabs, AV carrying LacZ served as control particles (AV-LacZ). AV infection was carried out by surgically accessing BAT under anesthesia. The injections were reproduced in independent cohorts. For the infection of 8-week-old WT or Nfe2l1ΔBAT mice, we locally injected 5×109 phage forming units of AV-Nrf1 into the right side of iBAT whereas the same amount of AV-LacZ particles were injected into the contralateral left side of the same mouse. For the infection of 24 week-old DIO mice, we injected 5×109 phage forming units of AV-Nrf1 into each of the iBAT sides whereas control mice received AV-LacZ into both iBAT sides. For the infection of 8 week-old ob/ob mice, we injected 5×109 phage forming units of AV-Nrf1 into each of the iBAT sides whereas control mice received AV-LacZ into both iBAT sides. We performed insulin tolerance tests 9 days, necropsies 14 days after surgery. AV carrying PA28α was a kind gift from Xuejun Wang and was produced and used as described above for the AV-Nrf1; AV-LacZ served as control.
Statistics
Data were expressed as mean ± s.e.m. Sample sized was estimated by pilot experiments that showed trends of effects and their sizes. In most cases for in vivo experiments in mice, an n = 6 was the minimal amount used, only where effect size was large, an n = 4 yielded significant results. Two-tailed, unpaired Student’s t-Test was used for pair-wise comparison of genotypes or treatments and corrected for multiple testing by the method of Holm-Sidak where appropriate as indicated in the figure legends. For non-parametric comparison Mann-Whitney U-Test was used. 1-way ANOVA and 2-ANOVA were used when comparing three or more groups as indicated in the figure legends and otherwise. Analysis was performed using Microsoft Excel and/or GraphPad Prism. P < 0.05 was considered significant as indicated by asterisks in the figures legends. Computational GESA and CPDB analysis, which were done on the differential ubiquitome are described on the respective homepages (GSEA: www.broadinstitute.org/gsea; CPDB: http://cpdb.molgen.mpg.de/MCPDB). Mice were excluded for poor body condition (e.g. after surgery or during feeding studies, total n = 9 animals during HFD studies, n = 7 after surgery).
Supplementary Material
Acknowledgments
We are grateful to Kathryn Claiborn for her expert editorial input. We thank Martin McGrath for his support of the project. We thank Mustafa Yilmaz, Yankun Lee and Vanessa Byles for assistance with experiments. We acknowledge the prosperous scientific environment in the Hotamisligil lab and thank all lab members for their critical input and discussions. We thank Klaus Seedorf, Philippe Delerive and Servier for their continuous support of the project. We thank Roderick Bronson and the Harvard Medical School Rodent Histopathology Core. We thank Maria Ericson and the Harvard Medical School Electron Microscopy Facility, Nicola Lupoli and the Harvard T.H. Chan School of Public Health Trace Metals Laboratory. We thank Ryan Kunz and the Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School and the Harvard T.H. Chan School of Public Health Animal Facility team for their support of the project. We are grateful to Xuejun Wang for generously sharing the PA28α adenovirus. We thank Evan Rosen for providing us with the Ucp1-Cre as well as the Adipoq-Cre mouse model. We thank T. Iwawaki for the conditional Ire1α-deletion mouse model. A.B. was supported by a Deutsche Forschungsgemeinschaft Research Fellowship (BA 4925/1-1). S.B.W. was supported by a Canadian Institutes of Health Research fellowship. C.S. was supported by a University Medical Center Hamburg-Eppendorf MD/PhD program fellowship. R.L.S.G. was supported by the Barth Syndrome Foundation. This work has received support in part from the National Institutes of Health through the Laser Biomedical Research Center, grant number P41-EB015871 (M.G.B.), 5-U54-CA151884 (M.G.B.), the National Science Foundation ECCS-1449291 (D.F., M.G.B.) and the Massachusetts Institute of Technology through the Institute for Soldier Nanotechnologies, grant number W911NF-13-D-0001 (M.G.B.). D.F. was supported by a fellowship of the Boehringer Ingelheim Fonds. O.T.B. was supported by a European Molecular Biology Organization Long-term Fellowship. Y.-H.T. and L.O.L. were supported in part by the US National Institutes of Health grants R01DK077097, R01DK102898, and P30DK036836. M.D.L. was supported by the US National Institutes of Health grants T32DK007260, F32DK102320 and K01DK111714. A.P.A. was supported by the Pew Foundation. We apologize to colleagues whose work we could not cite due to space limitations.
Footnotes
Competing Interests
The authors declare no competing interest. The study was supported by an industry-sponsored research agreement between Harvard University and Servier.
Data availability
Uncropped immunoblot images are presented in Supplementary Figures 11-13. A Life Sciences Reporting Summary is available. Source data for Figure 4 is available in Supplementary Tables 1-3.
Authors’ Contributions
A.B. and S.B.W. designed the study, were involved in all aspects of the experiments, analyzed all data and wrote the manuscript. C.S. and K.J. performed in vitro experiments. R.L.S.G. performed mitochondrial isolations and Seahorse experiments. K.E performed flow cytometer experiments. A.W.F. performed immunohistochemistry. G.P., N.A.S. and T.B.N. were involved in animal experiments. O.T.B., D.F. and M.G.B. developed quantum dots and performed SWIR imaging. L.O.L. performed thermal imaging. M.D.L. and Y.-H.T. performed bioinformatics. A.P.A. and K.E.I. helped with design of the study and animal experiments. G.S.H. supervised the design and execution of the study, interpreted the results and wrote the manuscript. All authors discussed the results and commented on the manuscript.
References
- 1.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 2.Bartelt A, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 3.Stanford KI, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. 2013;123:215–223. doi: 10.1172/JCI62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berbee JF, et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun. 2015;6:6356. doi: 10.1038/ncomms7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartelt A, et al. Thermogenic adipocytes promote HDL turnover and reverse cholesterol transport. Nat Commun. 2017;8:15010. doi: 10.1038/ncomms15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19:1252–1263. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- 7.Kajimura S, Spiegelman BM, Seale P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015;22:546–559. doi: 10.1016/j.cmet.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gnad T, et al. Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature. 2014;516:395–399. doi: 10.1038/nature13816. [DOI] [PubMed] [Google Scholar]
- 9.Rothwell NJ, Stock MJ. A role for brown adipose tissue in diet-induced thermogenesis. Nature. 1979;281:31–35. doi: 10.1038/281031a0. [DOI] [PubMed] [Google Scholar]
- 10.Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu I, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest. 2014;124:2099–2112. doi: 10.1172/JCI71643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saito M, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Travers KJ, et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 15.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 17.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 18.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, et al. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. 2015;349:500–506. doi: 10.1126/science.aaa0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otoda T, et al. Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes. 2013;62:811–824. doi: 10.2337/db11-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wikstrom JD, et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014;33:418–436. doi: 10.1002/embj.201385014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arruda AP, et al. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20:1427–1435. doi: 10.1038/nm.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregor MF, et al. The role of adipocyte XBP1 in metabolic regulation during lactation. Cell Rep. 2013;3:1430–1439. doi: 10.1016/j.celrep.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, Chan JY. Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain. Inhibition of nuclear translocation and transacting function. J Biol Chem. 2006;281:19676–19687. doi: 10.1074/jbc.M602802200. [DOI] [PubMed] [Google Scholar]
- 25.Cannon B, Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. J Exp Biol. 2011;214:242–253. doi: 10.1242/jeb.050989. [DOI] [PubMed] [Google Scholar]
- 26.Adams J, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- 27.Sha Z, Goldberg AL. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol. 2014;24:1573–1583. doi: 10.1016/j.cub.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arastu-Kapur S, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res. 2011;17:2734–2743. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- 29.Fu S, et al. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci Transl Med. 2015;7:292ra298. doi: 10.1126/scitranslmed.aaa9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 31.Steffen J, Seeger M, Koch A, Kruger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell. 2010;40:147–158. doi: 10.1016/j.molcel.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Radhakrishnan SK, et al. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010;38:17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kahn NW, Rea SL, Moyle S, Kell A, Johnson TE. Proteasomal dysfunction activates the transcription factor SKN-1 and produces a selective oxidative-stress response in Caenorhabditis elegans. Biochem J. 2008;409:205–213. doi: 10.1042/BJ20070521. [DOI] [PubMed] [Google Scholar]
- 34.Xue R, et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat Med. 2015;21:760–768. doi: 10.1038/nm.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol. 2014;10:24–36. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- 36.Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature. 2014;510:76–83. doi: 10.1038/nature13477. [DOI] [PubMed] [Google Scholar]
- 37.Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruns OT, et al. Next-generation in vivo optical imaging with short-wave infrared quantum dots. Nat Biomed Eng. 2017;1 doi: 10.1038/s41551-017-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- 40.Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- 41.Kim W, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagner SA, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.013284. M111 013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mootha VK, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 44.Clarke KJ, et al. A role for ubiquitinylation and the cytosolic proteasome in turnover of mitochondrial uncoupling protein 1 (UCP1) Biochim Biophys Acta. 2012;1817:1759–1767. doi: 10.1016/j.bbabio.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 45.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 46.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamburov A, et al. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 52.Kim JY, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li J, et al. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. 2011;121:3689–3700. doi: 10.1172/JCI45709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, et al. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513:440–443. doi: 10.1038/nature13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee CS, Ho DV, Chan JY. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J. 2013;280:3609–3620. doi: 10.1111/febs.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wahl S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86. doi: 10.1038/nature20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, Xiang Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem J. 2016;473:961–1000. doi: 10.1042/BJ20151182. [DOI] [PubMed] [Google Scholar]
- 58.Widenmaier SB, et al. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell. 2017;171:1094–1109 e1015. doi: 10.1016/j.cell.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 59.Tsujita T, et al. Discovery of an NRF1-specific inducer from a large-scale chemical library using a direct NRF1-protein monitoring system. Genes Cells. 2015;20:563–577. doi: 10.1111/gtc.12248. [DOI] [PubMed] [Google Scholar]
- 60.Li F, Gao B, Dong H, Shi J, Fang D. Icariin induces synoviolin expression through NFE2L1 to protect neurons from ER stress-induced apoptosis. PLoS One. 2015;10:e0119955. doi: 10.1371/journal.pone.0119955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Online methods references
- 61.Kong X, et al. IRF4 is a key thermogenic transcriptional partner of PGC-1alpha. Cell. 2014;158:69–83. doi: 10.1016/j.cell.2014.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tschop MH, et al. A guide to analysis of mouse energy metabolism. Nat Methods. 2012;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cypess AM, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. 2013;19:635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodbell M. Metabolism of Isolated Fat Cells. I. Effects of Hormones on Glucose Metabolism and Lipolysis. J Biol Chem. 1964;239:375–380. [PubMed] [Google Scholar]
- 65.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cannon B, Nedergaard J. Studies of thermogenesis and mitochondrial function in adipose tissues. Methods Mol Biol. 2008;456:109–121. doi: 10.1007/978-1-59745-245-8_8. [DOI] [PubMed] [Google Scholar]
- 67.Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J Biol Chem. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haemmerle G, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 69.Luo J, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2:1236–1247. doi: 10.1038/nprot.2007.135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.