

Abstract
Chemical modifications of messenger RNA (mRNA) may regulate many aspects of mRNA processing and protein synthesis. Recently, 2′-O-methylation of nucleotides was identified as a frequent modification in translated regions of human mRNA, showing enrichment in codons for certain amino acid. Here, using single-molecule, bulk kinetics and structural methods, we show that 2′-O-methylation within coding regions of mRNA disrupts key steps in codon reading during cognate transfer RNA (tRNA) selection. Our results suggest that 2′-O-methylation sterically perturbs interactions of ribosomal monitoring bases (G530, A1492 and A1493) with cognate codon-anticodon helices, thereby inhibiting downstream GTP-hydrolysis by elongation factor Tu (EF-Tu) and A-site tRNA accommodation, leading to excessive rejection of cognate aminoacylated-tRNAs in initial selection and proofreading. Our current and prior findings highlight how chemical modifications of mRNA tune the dynamics of protein synthesis at different steps of translation elongation.
Introduction
The number of known post-transcriptional modifications of messenger RNA (mRNA), regulating many aspects of protein synthesis1–3, is growing rapidly. In particular, modifications within the coding sequence of mRNA may affect translation by changing tRNA-mRNA or mRNA-ribosome interactions4, resulting in altered dynamics of translation elongation. Recently, extensive 2′-O-methylation sites were discovered within the human mRNA coding regions5. Although much of the mRNA modification mechanism and function are still unknown, 2′-O-methylation is common in ribosomal RNA (rRNA)6. These modifications are placed either by the stand-alone methyltransferases7 or the enzyme fibrillarin (FBL) guided by C/D-box small nucleolar RNA (snoRNA) for selected modification sites8–10. The function of 2′-O-methylation in rRNA has been suggested to be enhancement of translational fidelity6, possibly by thermodynamic and chemical stabilization of rRNA helices11,12. The presence of a 2′-O-methylated mRNA codon has been shown to decrease translation efficiency in vitro through an unknown mechanism13. 2′-O-methyl groups in a modified mRNA could potentially introduce steric clashes with the decoding machinery, by which universally-conserved nucleotides within the small ribosomal subunit (G530, A1492 and A1493 bases of the 16S rRNA) monitor formation of correct codon-anticodon RNA duplexes, in part through contacts with mRNA 2′-OH groups14,15.
Here, we used single-molecule and bulk kinetic methods to show that translation of 2′-O-methylated codons is severely impaired. By systematically testing the position and the context of modifications, we show that 2′-O-methylation impairs efficient decoding on the modified codon, and results in excessive rejection of cognate tRNA. The presence of aminoglycoside antibiotics such as paromomycin that hyper-activates the ribosomal monitoring bases alleviated the effect of 2′-O-methylation in both bulk and single-molecule assays. Using x-ray crystallography, we have determined structures of Thermus thermophilus ribosomal small subunit complexed with a short mRNA with the 2′-O-methylated codon and the cognate anticodon stem-loop (ASL) in the absence and presence of paromomycin. These structures collectively confirm that the presence of 2′-O-methylation severely disrupts canonical interaction between the codon-anticodon helix and the ribosomal monitoring bases in the absence of paromomycin. Combined with our previous findings4, our results suggest that post-transcriptional modification of mRNA coding regions are a potential regulator of translation-elongation dynamics.
Results
2′-O-methylation delays tRNA accommodation to the modified codon
We first measured the global effect of 2′-O-methylation on translation elongation using in vitro translation assays based on single-molecule Förster resonance energy transfer (smFRET). Single-molecule experiments were carried out on a zero-mode waveguide (ZMW)-based instrument16,17, which allows measurement at near physiological concentration of free labeled components. As previously reported4,18–20, we labeled two individually purified mutant Escherichia coli ribosomal subunits with a Cy3B and BHQ-2 donor-quencher pair (Cy3B-labeled 30S ribosomal subunit, or Cy3B-30S; BHQ-2-labeled 50S ribosomal subunit, or BHQ-2-50S). With this system, we monitor inter-subunit conformational changes that correspond to multiple rounds of the translation elongation cycle over many codons. The pre-initiation translation complex was formed using the Cy3B-30S and a biotinylated mRNA, where its coding region contains eight codons with one single-base 2′-O-methylated Lys codon (AAA, one of six over-represented 2′-O-methylated codons5) at three different base positions (AmAA, AAmA or AAAm, where a 2′-O-methylated base, A, is marked by letter m: Am). This complex was tethered onto the surface within the ZMW, and translational factors including the BHQ-2-50S and Lys-(Cy5)-tRNALys were delivered during data acquisition (Figure 1a). The use of Lys-(Cy5)-tRNALys allowed correlation of two independent fluorescence signals during translation elongation, as well as observing possible tRNA sampling dynamics on the modified Lys codon (Figure 1b).
Figure 1. Multiple cognate tRNAs are rejected during decoding of the 2′-O-methylated codon.

(a) Single-molecule experimental schematics. Fluorescently labeled pre-initiation complex is tethered to the surface of the ZMW well via biotinylated mRNA, and necessary translation factors are delivered in the beginning of the signal acquisition.
(b) Expected sequence of events between translocation from the previous codon and the peptidyl transfer reaction. Both translocation and peptidyl transfer events are detected from the ribosomal inter-subunit conformation change from the rotated state to the non-rotated state via change in the dye-quencher signal. Independently, tRNA binding events are detected from co-localized fluorescence from fluorescently-labeled tRNA.
(c) Representative experimental trace at 5 mM Mg2+ condition with the first-base modified codon (AmAA) shows a long stall between the translocation to the modified codon and its peptidyl transfer event.
(d) Quantification of stall on the different modified Lys codon at different Mg2+ condition. The stall duration measured as in (c) was normalized to the non-rotated state lifetimes for the unmodified codon, as specified in the Supplementary Note 1 (n = 147, 154, 77, 111, 94, 111, 120, and 108 from left to right; error bars were calculated from propagating s.e. from fitting the single-exponential distributions; * on AAmA codon at 5 mM Mg2+ condition indicates that out of all traces that showed translation leading to the modified codon, less than 5% showed a tRNA-binding/ribosome-rotation event on the modified codon, hindering calculation of the normalized stall duration).
The presence of 2′-O-methylation within the coding region caused a striking stall of elongation at modified codons (Figure 1c,d). Stalls in the order of hundreds of seconds occurred while the ribosome was in the non-rotated state, with the modified codon presented in the A site. No alterations to translocation kinetics, or a stall at any other nearby codons were caused by 2′-O-methylation (Supplementary Data Set 1), as observed previously with N6-methyladenosine (m6A) modification4. This allowed us to quantify the stall duration as a function of the position of 2′-O-methylation in a codon, normalized to non-rotated state lifetimes of unmodified Lys codons also present in the preceding coding region and statistically corrected for interruptions during observations due to photobleaching (Supplementary Note 1). The normalized stall duration at a 2′-O-methylated codon was the greatest at the second and the least at the third base-position modification (Figure 1d), partly in line with prior data using in vitro translation assay in a bacterial system13. The extremely long stall at the second position 2′-O-methylated codon in our near-physiological buffer condition (the polymix buffer21 with 5 mM total Mg2+ concentration) resulted in only 2% of total molecules translating the modified codon within the observation time defined by dye photobleaching17, hindering an accurate quantification of the stall at this condition (Supplementary Data Set 1).
The stall duration induced by 2′-O-methylated codon depended both on the codon context and the modification position within the codon. We tested the second position 2′-O-methylated Phe codon (UUmC) in a similar assay, with Lys-(Cy5)-tRNALys and Phe-tRNAPhe substituted by Lys-tRNALys and Phe-(Cy5)-tRNAPhe (Figure 2a). As for modified lysine codons, we observed a long stall while decoding the phenylalanine codon containing the 2′-O-methylated uridine in its second base position (Figure 2b). The stall on the UUmC codon was substantially shorter than the stall observed on the AAmA codon, as nearly 80% of total molecules translated the UUmC codon within the observation time (Supplementary Data Set 2), compared to 2% for the AAmA codon. One possibility for this difference could be the presence of G-C pairing within the codon-anticodon helix. To explore the effect of the codon context further, we measured the stall duration on the second position 2′-O-methylated Leu codon (CUmC) and Pro codon (CCmC). Since fluorescently-labeled leucine or proline tRNAs were not available, we modified our single-molecule assay by substituting Lys-tRNALys with total tRNA charged without phenylalanine amino acid, but still using the ribosomal intersubunit FRET signal as well as Phe-(Cy5)-tRNAPhe binding and departure signals to measure elongation kinetics of each codons translated (Figure 2c,d). We observed a further reduction of the modification-dependent stall-time increase for decoding the CUmC codon (26 ± 8 fold-increase) and CCmC (19 ± 6 fold-increase) codon, compared to the UUmC codon (56 ± 15 fold-increase; Figure 2e). The observed codon-context dependent effect of mRNA 2′-O-methylation is of greater magnitude than that measured for mRNA m6A modification4.
Figure 2. Codon-context alters the magnitude of 2′-O-methylation-induced stall.

(a) mRNA sequence used to test the effect of 2′-O-methylation on the Phe codon, and the expected sequence of events between translocation from the previous codon and the peptidyl transfer reaction on Phe codon using Phe-(Cy5)-tRNAPhe.
(b) Representative experimental trace shows a long stall between the translocation to the modified codon and its peptidyl transfer event on the UUmC codon.
(c) mRNA sequence used to test the effect of 2′-O-methylation on the Leu and Pro codons, and the expected sequence of events between translocation from the previous codon and the peptidyl transfer reaction on the Leu or Pro codon using Phe-(Cy5)-tRNAPhe.
(d) Representative experimental trace shows a long stall between the translocation to the modified codon and its peptidyl transfer event on the CUmC and CCmC codon.
(e) Quantification of stall on the different modified codons at 5 mM Mg2+ condition (n = 131, 108 and 104 from left to right; error bars were calculated from propagating s.e. from fitting the single-exponential distributions).
2′-O-methylation impairs both the initial and proofreading selection of aa-tRNA
Fluorescently-labeled cognate tRNA repeatedly samples the ribosomal A site briefly without peptide-bond formation during the stall at all 3 modified AAA codons and UUmC codon. (Figure 1c; Figure 2b). With the AmAA codon, fluorescently-labeled cognate tRNAs sampled their ribosome-bound state about fifteen times in comparatively short but observable (lifetime of 0.22 ± 0.02 seconds) binding/dissociation events. Increasing the concentration of total Mg2+ to 10 mM or 15 mM incrementally decreased the duration of the stall at all codon positions, as previously observed for m6A-modified codon4 (Figure 1d), and also increased the sampling pulse durations (Figure 3). At high Mg2+ concentration (15 mM), the cognate tRNA remained bound to 2′-O-methylated codons on the ribosome for nearly one second prior to dissociation (Figure 3c).
Figure 3. 2′-O-methylation prolongs time between the initial tRNA binding and its full accommodation event.

(a) A representative trace from the 10 mM total Mg2+ experimental condition (AAmA codon) shows a time-lag between the initial tRNA binding and the ribosomal intersubunit rotation, which follows the peptidyl transfer reaction. tRNA samplings and rotation lags on the modified codon were observed in all 3 2′-O-methylated codons.
(b) Contour plots of the normalized Cy3B and Cy5 intensity trajectories, generated by post-synchronizing to the initial tRNA binding event (red; unbound to bound state) that leads to the ribosomal rotation (green; non-rotated to rotated state; n = 81).
(c) Lifetimes of futile tRNA samplings on the modified codon observed in AmAA, AAmA and AAAm codons. (n = 1008, 1188, 759, 1961, 3294, 1452, 731, 862 and 316 from left to right; error bars represent s.e. from fitting the single-exponential distributions)
(d) Average time between the initial tRNA binding and the ribosomal intersubunit rotational event on the modified codon observed in AmAA, AAmA and AAAm codons. (n = 65, 123, 42, 81, 64, 64, 124 and 92 from left to right; error bars represent s.e. from fitting the single-exponential distributions; * on AAmA codon at 5 mM Mg2+ condition indicates that out of all traces that showed translation leading to the modified codon, less than 5% showed a tRNA-binding/ribosome-rotation event on the modified codon, same as in Figure 1d).
(e) Average number of futile sampling events prior to the successful tRNA accommodation on the 2′-O-methylated codons (n = 147, 154, 77, 111, 94, 111, 120 and 108 from left to right).
While translating 2′-O-methylated codons, we observed a time-lag between the productive tRNA binding (marked by the fluorescently-labeled tRNA binding) and the ribosomal intersubunit rotation after the peptidyl transfer event (marked by the ribosomal intersubunit FRET efficiency change) (Figure 1c; Figure 2b; Figure 3a–c). Such behavior is not observed while translating unmodified codons, where the time between the cognate tRNA binding and the ribosomal conformational change is estimated as less than the temporal resolution of our experiment (100 milliseconds). Further, the time-lag depended on the concentration of Mg2+ (Figure 3d), similar to the sampling pulse durations.
Both the sampling behavior and time-lag between the initial tRNA binding and ribosomal intersubunit conformational change at the modified codon suggested that 2′-O-methylation hinders tRNA accommodation after the initial binding. To probe further into tRNA dynamics during interrupted decoding, we monitored smFRET between the P-site tRNA and the incoming tRNA to the A-site modified codon22. For this, we used a shorter mRNA construct with the AAA or AAmA codon next to the start codon to measure FRET efficiency between the P-site fMet-(Cy3)-tRNAfMet and the incoming A-site Lys-(Cy5)-tRNALys. In this experiment, we observed that the incoming tRNA binds to the A*/T site23 of the ribosome during this time-lag and sampling events, likely as a ternary complex (TC) with EF-Tu and GTP, before the successful accommodation to the A site (after the time-lag) or dissociation (after the sampling events) (Supplementary Figure 1).
Next, we used rapid-mixing bulk kinetics (quench-flow) to determine how a 2′-O-methylated codon affects the steps leading to tRNA accommodation and peptidyl transfer at near physiological Mg2+ concentrations (5 mM total). During initial selection, TC is either rejected or accepted for GTP hydrolysis by EF-Tu, followed by proofreading to reject near-cognate tRNA species24,25 before accommodation to the A site. By reacting [3H]GTP containing TC with ribosomes programmed with unmodified (AAA) or modified (AAmA) Lys codon in the A site (Supplementary Note 2), we observed a striking reduction in the kcat/Km of GTP-hydrolysis ((kcat/Km)GTP) by the 2′-O-methylation (300-fold reduction; Figure 4). The reduction in (kcat/Km)GTP means a reduced probability of GTP-hydrolysis per tRNA binding event, corresponding to the multiple cognate tRNA sampling events we observed in the smFRET assay (Figure 1c; Figure 2b). Further, by using f[3H]Met-tRNAfMet and following fMet-Lys dipeptide formation over time, we measured the proofreading factor, which quantifies the number of GTP-hydrolysis events per one successful dipeptide formation event. We observed an increase in the proofreading factor with the modified codon compared to that of unmodified codon (5-fold increase; Figure 5), which signifies increased tRNA rejection on the modified codon during proofreading. 2′-O-methylation did not induce substantial miscoding by other tRNA species (Supplementary Figure 2).
Figure 4. Bulk kinetics measurements of (kcat/Km)GTP with modified (AAmA) and unmodified (AAA) codons at 5 mM Mg2+.
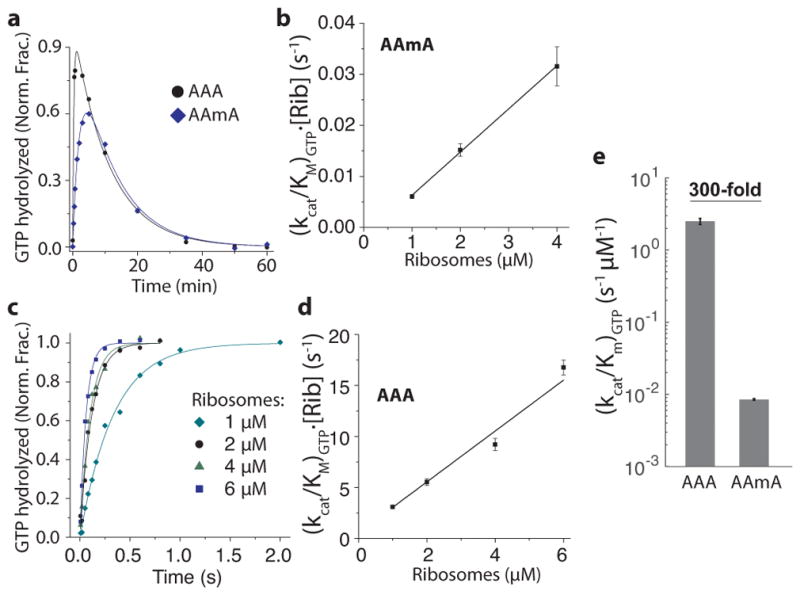
(a) A representative time course of GTP hydrolysis on AAmA codon. 0.7 μM Lys-tRNALys ternary complexes were reacted to 1 μM ribosomes initiated with AAA or AAmA codons in the A site. AAmA reaction was run in parallel with AAA reaction and the curves fitted with shared parameters to increase precision (see Online Methods). The decrease of the fraction of hydrolyzed [3H]GTP was due to its regeneration by pyruvate kinase.
(b) Ribosome titration to determine kcat/Km value of GTP hydrolysis on EF-Tu with AAmA codon in the A site (error bars represent s.d. from n = 3 technical replicates).
(c) Time courses of GTP hydrolysis on AAA codon at different ribosome concentrations. Lys-tRNALys ternary complexes (0.3 μM) were reacted to 70S programed with AAA codon in the A site.
(d) Ribosome titration to determine (kcat/Km)GTP with AAA codon in the A site (error bars represent s.d. from n = 3 technical replicates).
(e) Estimates of (kcat/Km)GTP from b and d (2.5 ± 0.2 and 0.0085 ± 0.0002 s-1 μM−1 for AAA and AAmA, respectively). Error bars represent s.d. from fitting procedure.
Figure 5. The effect of 2′-O-methylation on proofreading.
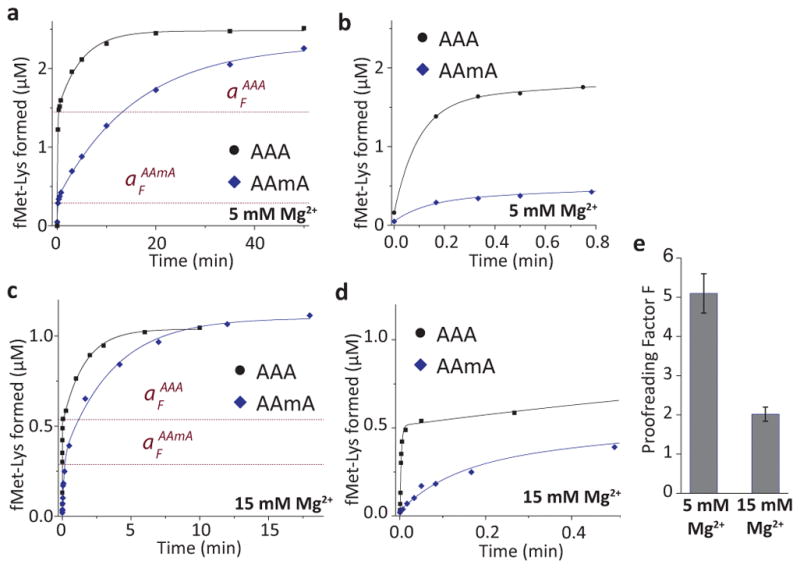
(a) Time courses of fMet-Lys dipeptide formation at 5 mM total Mg2+ (1.3 mM free). Parallel reactions with Lys-tRNALys ternary complexes (2 μM) were performed with ribosomes (4.2 μM) containing AAA or AAmA codon in the A site. The curves were fitted to a model with two exponentials (see Online Methods). The proofreading factor F is expressed as the ratio of the fast phase amplitudes (aF) on and . The aF values obtained from the fit are shown as red dotted lines. The lower aF for AAmA codon indicates the rejection of Lys-tRNALys after GTP hydrolysis in the presence of the modification.
(b) The first 0.8 min of the experiment in a expanded to highlight the fast phase of the reaction.
(c) The same reaction as in a, but at 15 mM total Mg2+ (7.5 mM free) with 1.5 μM ribosomes and 1 μM ternary complexes.
(d) The first 0.5 min of c, expanded to highlight the fast phase of the reaction.
(e) Proofreading factor measured at 5 mM Mg2+ condition (F = 5.1 ± 0.4), and at 15 mM Mg2+ condition (F = 2 ± 0.2; error bars represent s.d. from n = 3 technical replicates).
Our single-molecule and bulk kinetic data probe tRNA selection and incorporation from different perspectives and agree quantitatively (see Supplementary Note 3). From bulk kinetic parameters, we derived probabilities that a bound TC hydrolyzes GTP (pI from (kcat/Km)GTP) or proceeds to peptide formation (pA from (kcat/Km)pep). Both parameters are greatly decreased on 2′-O-methylated codons at the low Mg2+ condition (300-fold and 1500-fold decrease of pI and pA, respectively). Single-molecule experiments observe these probabilities directly as a great increase in number of futile tRNA sampling events before successful peptide bond formation, which is detected by the subsequent ribosomal intersubunit rotation. Lifetimes of these sampling events are Mg2+ dependent (Figure 3c), and reflect both forward and reverse rate constants for TC binding and forward progression during initial tRNA selection; higher Mg2+ concentrations favor slower TC dissociation from the ribosome, increasing TC occupancy times, and thus increasing pI. The prolonged lag-time prior to peptide bond formation observed during productive TC binding events is likely due to a competition between the slow dissociation rate (1–5 per second, depending on the Mg2+ concentration; Figure 3c), with the slower forward rate (0.1 per second at high and, by inference, also at low Mg2+ concentrations; Figure 3d), during the GTP-hydrolysis incompetent A*/T-site TC bound state23 (Supplementary Figure 1).
2′-O-methylation disturbs interaction between the codon-anticodon helix and ribosomal monitoring bases
The intrinsic accuracy of tRNA initial selection and proofreading is amplified by the monitoring bases14,15,26 (A1942, A1943 and G530), which make structure-specific contacts with the ribose backbone of the codon-anticodon helix to activate GTP-hydrolysis in TC and promote subsequent accommodation step. Their accuracy enhancing effect may also be ascribed the creation of a water-free environment that enhances the specificity of codon-anticodon pairing27. The presence of aminoglycoside antibiotics such as paromomycin and neomycin that are known to induce missense errors in protein synthesis by hyper-activating the monitoring bases for binding to the codon-anticodon helix15, attenuated the effect of the 2′-O-methylated codon on translation elongation in both the single-molecule and bulk kinetics assays (Figure 6a–c, Supplementary Figure 3). Our results suggest that the 2′-O-methylation affects interactions between the monitoring bases and the codon-anticodon helix essential for tRNA selection.
Figure 6. The delay in GTP-hydrolysis by 2′-O-methylation is due to the inactivation of the monitoring bases.

(a) Kinetics of GTP hydrolysis at 15 mM Mg2+ condition. Lys-tRNALys ternary complexes (0.4 μM) were reacted to 70S ribosomes (1 μM) programed with AAA or AAmA codon in the A site. 10 μM neomycin (Neo) or paromomycin (Paro) were added where indicated.
(b) Estimates of (kcat)GTP from a. Error bars represent s.d. from n = 3 technical replicates.
(c) Non-rotated state lifetimes measured on either AAA or AAmA codon in the A site with or without 10 μM neomycin (Neo) or paromomycin (Paro) using single-molecule assay (n = 194, 172, 153, 201, 182 and 243 instances; error bars represent s.e. from fitting the single-exponential distribution)
(d–f) X-ray crystallography structures of the decoding center (UUU anticodon and A1492, A1493 and G530 monitoring bases) while decoding (d) the first base modified (AmAA), (e) the second base modified (AAmA), and (f) the third base modified Lys codon (AAAm), soaked in the 30S subunit crystals in the presence of paromomycin. Displayed distances indicate interatomic distance (dashed lines) for possible hydrogen bonds between the monitoring bases and 2′-O-moiety on the decoded codon (purple for the modified base and tan for the unmodified base). 1.3 Å shift in A1492 base position is indicated by black arrow in panel e.
To validate this, we determined seven Thermus thermophilus 30S ribosomal subunit structures in complex with a tRNA anticodon stem loop (ASL) of human tRNALys3 bound to unmodified or 2′-O-methylated codons in the A site with and without the aminoglycoside paromomycin using x-ray crystallography28–30. In the absence of paromomycin, we obtained well-defined electron density for only the unmodified mRNA and ASL pair. However, consistent with our studies above, we were not able to observe the electron densities that belongs to either of the three modified mRNA-ASL complexes due to possible steric clashes among the 2′-O-methyl group and the monitoring bases (Supplementary Figures 4, 5 and 6; Table 1). In contrast, when paromomycin is bound, we obtained well-defined electron densities for the unmodified (AAA) and AmAA or AAAm A-site codon mRNA-ASL complexes. In addition, modified (AAmA) codon and ASL pair resulted in less ordered but observable electron density map despite the presence of paromomycin. (Supplementary Figures 6 and 7; Table 1). The presence of paromomycin lead to stabilization of codon-anticodon pairing with 2′-O-methylation at base-positions 1 and 3 of Lys codon with minor distortions of the decoding site, likely to accommodate the bulkier 2′-O-methyl group. The largest decoding center perturbations and ASL disorder are observed when 2′-O-methylation is located at the base-position 2 in the presence of paromomycin. We observed a 1.3 Å shift and tilting of the monitoring residue A1492 to accommodate the 2′-O-methyl, extending and weakening the hydrogen bond with the N3 position of A1492 (overall coordinate error of this structure is 0.45 Å) (Figure 6e). These effects of 2′-O-methylation at all three positions observed in the presence of paromomycin are likely exacerbated in the absence of this drug, which stabilizes the monitoring bases in an activated conformation.
Table 1.
Data collection and refinement statistics (molecular replacement)
| Unmodified −Paro (5WNP) |
2′OMe +1 −Paro (5WNQ) |
2′OMe +2 −Paro (5WNR) |
2′OMe +3 −Paro (5WNS) |
2′OMe +1 +Paro (5WNT) |
2′OMe +2 +Paro (5WNU) |
2′OMe +3 +Paro (5WNV) |
|
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| Space group | P41212 | P41212 | P41212 | P41212 | P41212 | P41212 | P41212 |
| Cell dimensions | |||||||
| a, b, c (Å) | 400.1, 400.1, 173.4 | 400.5, 400.5, 173.2 | 400.1, 400.1, 173.3 | 401.3, 401.3, 173.1 | 400.4, 400.4, 173.4 | 400.4, 400.4, 173.1 | 400.3, 400.3, 173.6 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 30.0–3.30 (3.42–3.30) a | 30.0–3.50 (3.65–3.50) a | 30.0–3.50 (3.65–3.50) a | 30.0–3.50 (3.65–3.50) a | 30.0–3.30 (3.42–3.30) a | 30.0–3.40 (3.52–3.40) a | 30.0–3.30 (3.41–3.30) a |
| Rmerge | 0.41 (1.29) | 0.25 (1.35) | 0.47 (1.38) | 0.47 (1.34) | 0.37 (1.16) | 0.41 (1.39) | 0.40 (1.26) |
| I/σ(I) | 13.70 (1.03) | 24.10 (1.25) | 12.93 (1.24) | 17.25 (1.42) | 17.29 (1.56) | 14.08 (1.07) | 18.04 (1.77) |
| CC1/2 | 0.99 (0.39) | 1.00 (0.65) | 1.00(0.47) | 1.00(0.62) | 1.00 (0.68) | 0.99 (0.47) | 0.97 (0.68) |
| Completeness (%) | 99.3 (92.9) | 99.6 (99.2) | 99.8 (99.7) | 98.9 (98.4) | 99.9 (99.6) | 99.9 (99.9) | 100.0 (99.9) |
| Redundancy | 44.3 (40.9) | 38.6 (36.6) | 39.2 (39.3) | 35.1 (32.3) | 49.8 (43.3) | 41.9 (39.2) | 48.1 (44.9) |
| Refinement | |||||||
| Resolution (Å) | 30.0–3.30 (3.41–3.30) | 30.0–3.50 (3.65–3.50) | 30.0–3.50 (3.65–3.50) | 30.0–3.50 (3.65–3.50) | 30.0–3.30 (3.41–3.30) | 30.0–3.40 (3.53–3.40) | 30.0–3.30 (3.41–3.30) |
| No. reflections | 206225 (18875) | 171347 (18209) | 160112 (8126) | 162862 (10933) | 207232 (20401) | 184844 (16177) | 208300 (20468) |
| Rwork/Rfree | 0.191/0.224 (0.358/0.367) | 0.198/0.245 (0.373/0.352) | 0.214/0.255 (0.375/0.379) | 0.219/0.255 (0.393/0.403) | 0.173/0.211 (0.269/0.308) | 0.187/0.231 (0.355/0.382) | 0.179/0.202 (0.290/0.343) |
| No. atoms | |||||||
| Protein | 19121 | 19090 | 19090 | 19090 | 19121 | 19121 | 19121 |
| RNA | 32504 | 32644 | 32644 | 32644 | 32504 | 32504 | 32504 |
| Ligand | 508 | 493 | 495 | 493 | 543 | 543 | 542 |
| B factors | |||||||
| Protein | 129.96 | 128.20 | 136.06 | 125.36 | 132.17 | 125.28 | 131.16 |
| RNA | 117.44 | 116.83 | 121.07 | 118.30 | 124.20 | 120.85 | 128.82 |
| Ligand | 120.55 | 129.47 | 130.93 | 133.80 | 118.38 | 122.84 | 121.13 |
| R.m.s. deviations | |||||||
| Bond lengths (Å) | 0.002 | 0.002 | 0.002 | 0.002 | 0.002 | 0.002 | 0.002 |
| Bond angles (°) | 0.503 | 0.593 | 0.564 | 0.576 | 0.534 | 0.522 | 0.502 |
One crystal was used for each dataset.
Values in parentheses are for highest-resolution shell.
Discussion
Synthesizing our results from distinct methods, we propose a mechanistic model of how 2′-O-methylated codons stall translation elongation. 2′-O-methylation does not disrupt the early stage of tRNA binding to the T site23 (Figure 7), and may allow formation of a stable A-form codon-anticodon duplex. This contrasts with modification such as m1A or m6A that destabilize Watson-Crick base pairing31,32; in fact 2′-O-methylation stabilizes RNA helical structures11,12. The monitoring bases are only fully engaged with a codon-anticodon complex in the A/T site, as shown recently by cryoEM23. Steric clash caused by the 2′-O-methyl group perturbs the correct stereochemistry of the monitoring bases, which delays EF-Tu GTPase activation by the ribosome (reduced kcat-value for GTP hydrolysis at high and, by inference, low Mg2+ concentrations; Figure 6a,b). Thus 2′-O-methylation decreases the probability of bound TC proceeding to successful GTP hydrolysis and subsequent tRNA accommodation. Mg2+ decreases the dissociation rates of TC on 2′-O-methylated codons, leading to prolonged ribosomal sampling time of TC in the GTPase-inactivated A*/T site, and reduced modification-dependent stalling time before the eventual GTP hydrolysis. In both these regards, 2′-O-methylation makes the kinetics of a cognate TC resemble that of a near-cognate TC to an unmodified codon. Our crystal structures above represent the stereochemistry in the final accommodated state. After GTP-hydrolysis, the incorrect stereochemistry of monitoring bases may result in the hyper-accurate proofreading, where the kinetic loss due to slow GTP-hydrolysis is amplified by tRNA dissociation during the proofreading.
Figure 7.

Mechanism of epitranscriptomic-induced translation elongation stall. (Top) The schematic of translation decoding, starting from aa-tRNA TC binding event to codon recognition event, monitoring base activation, GTP-hydrolysis, two-step proofreading and full accommodation/peptidyl-transfer reaction (left to right). While 2′-O-methylation does not interrupt codon-anticodon RNA duplex formation (the faded-out step), it sterically hinders dynamics of monitoring bases, delaying GTPase activation and causing rejections of multiple tRNA molecules during proofreading. (Bottom) m6A modification disrupts codon-anticodon RNA duplex stability during initial selection and for proofreading.
Thus, our results with 2′-O-methylation illuminate the general process of initial tRNA selection and mechanism of recently-proposed two-step proofreading33. In this model, two independent proofreading mechanisms exist after GTP-hydrolysis to increase accuracy of decoding prior to peptidyl transfer reaction. While these two proofreading steps may be affected differently by the Mg2+ concentrations, the relative weights between the codon-anticodon interaction and the monitoring bases used in each step are unknown. 2′-O-methylation increases a proofreading factor at the modified codon, whose magnitude largely depends on the free Mg2+ concentration (Figure 5). Such Mg2+ concentration dependence is the hallmark of the first proofreading step, where the accommodation pathway of the EF-Tu·GDP dissociation kinetically competes with the rejection pathway of aa-tRNA·EF-Tu·GDP dissociation33 (Figure 7). Engagement of the monitoring bases with the codon-anticodon helix may play an essential role in this proofreading step. (Supplementary Figure 8).
Our findings here, combined with our previous report4 on m6A, reveals the epitranscriptome as a possible regulator of translation-elongation dynamics. While stalls induced by 2′-O-methylation of mRNA during translation elongation resemble those caused by m6A modification, the magnitude and the mechanism of the stall differ. The distinct effects of m6A and 2′-O-methylation on translational decoding arise from their separate influences on codon-anticodon stability and monitoring base interactions (Figure 7), which respond differently to base-positions within a codon as well as its codon-context4 (Figure 1,2). Fine-tuning the translation-elongation rate at a specific mRNA region may lead to different folding pathways of a nascent protein or to the formation of polyribosomes, which may play an important role in developmental biology34. Future discovery of additional chemical modifications within the coding region may broaden our understanding of dynamic regulatory elements involved during translation elongation.
Online Methods
Reagents and buffers for single-molecule experiments
Reagents and buffers were prepared as previously reported4,19,22. Briefly, each small and large subunit were mutated to include a weakly forming RNA hairpin at helix 44 and helix 101, which was used to attach Cy3B/BHQ-2 labeled DNA oligonucleotides via RNA/DNA hybridization (DNA sequences for short oligonucleotides are: 5′-GAGGCCGAGAAGTG-(BHQ-2)-(BHQ-2)-3′ and 5′-GGGAGATCAGGATA-(Cy3B)-3′; both purchased from Trilink). Individual tRNA species used were purchased from Chemical Block Ltd. tRNALys was labeled at acp3U47 position with Cy5 using NHS chemistry as previously described18,35, with Cy5-NHS-ester purchased from GE Healthcare. tRNAfMet was labeled at s4U8 position using maleimide chemistry as previously described18,35 (Cy3-Maleimide was purchased from GE Healthcare). Synthetic biotinylated RNA oligonucleotide including 2′-O-methylated base were synthesized from Stanford PAN facility, with a 5′-UTR adapted from gene32 of the T4 phage (5′-CAACCUAAAA CUUACACACG CCCCGGUAAG GAAAUAAAA-3′), followed by the coding region specified in the text and 3′-UTR (UAAUUU-3′). Translational factors, ribosomal S1 protein, and aminoacylated tRNAs were prepared as previously reported4,19,22.
All experiments were conducted in a Tris-based polymix buffer consisting of 50 mM Tris-acetate (pH 7.5), 100 mM potassium chloride, 5 mM ammonium acetate, 0.5 mM calcium acetate, 5 mM magnesium acetate, 0.5 mM EDTA, 5 mM putrescine-HCl, and 1 mM spermidine, with additional 4 mM GTP. The free Mg2+ concentrations were calculated as 1.9 mM, taking account of chelation by GTP and Mg2+ present in buffers containing translation factors. Additional 5 and 10 mM magnesium acetate was added to buffer where specified, resulting in 7 and 12 mM free Mg2+ concentrations.
Single-molecule experiments on ZMW instrument
Detailed development and specification of the Pacific Bioscience RSII instrument as a platform for single-molecule fluorescence microscope has been published previously17. Immediately before each experiment, small 30S and large 50S ribosomal subunits were mixed with respective fluorescently labeled DNA oligonucleotide at 1:1.2 stoichiometric ratio with 1 uM final concentration of the subunit in the polymix buffer and incubated in the 37°C water bath for 10 minutes, followed by incubation in the 30 °C water bath for 20 minutes. Afterward, the tube containing the large 50S subunit was kept on ice, while the small 30S subunit was incubated with S1 ribosomal protein at 1:1 stoichiometric ratio with 0.5 μM final concentration in the 37°C water bath for 5 minutes, as S1 ribosomal proteins may not have been purified with the small subunit.
Using the labeled small 30S ribosomal subunit, the pre-initiation complex was formed by mixing it with biotinylated-mRNA, initiation factor 2, amino-acylated formyl-methionine tRNA at 1:2:13:4 with 150 nM final concentration of the small subunit in the polymix buffer, supplemented with 4 mM GTP, and incubating in the 37°C water bath for 5 minutes. Formed complex was diluted to 10 nM in the polymix buffer supplemented with 4 mM GTP and the imaging mix (2.5 mM of PCA (protocatechuic acid), 2.5 mM of TSY, and 2× PCD (protocatechuate-3,4-dioxygenase), purchased from Pacific Bioscience; PCD added last), and incubated in the zero-mode waveguide chip treated with Neutravidin at room temperature for 3 minutes, which bound to the Biotin-PEG (Polyethylene glycol) on the chip. After immobilizing the pre-initiation complex, the chip was washed three-times using the same buffer without the complex to remove unbound complexes, and loaded onto the instrument (RSII, purchased from Pacific Bioscience).
At the same time, the delivery solution, a polymix buffer supplemented with 4 mM GTP, the imaging mix, 200 nM of tRNA ternary complexes, 80 nM of EF-G, and 200 nM of the BHQ-2 labeled large ribosomal subunit was prepared, and loaded onto the instrument. tRNA ternary complex was formed by incubating tRNA with 100 μM EF-Tu·GTP within the polymix buffer without the spermidine and the putrescine, supplemented with fresh 1 mM GTP, and incubated in the 37°C water bath for 1 minutes.
At the start of the experiment, the instrument delivered the delivery solution to the chip, and recorded 8- to 12-minute movie with frame rate 10 frame per second, illuminated by 60 mW per mm2 of 532 nm laser and 10 mW per mm2 of 642 nm laser for dual illumination experiments, or by 0.32 mW per mm2 of 532 nm laser for smFRET experiments, both with chip temperature clamped to 20 °C. Resulting movies were analyzed using in-house-written MATLAB (MathWorks) scripts, as previously described17,36. Briefly, traces from each zero-mode waveguide wells were filtered based on the presence of both fluorophores at different time points (signal from immobile fluorophores on the ribosome was expected to be present at the beginning of the movie, while signal from fluorophores attached to tRNA was expected not to be), lifetimes, and a single photobleaching step for each fluorophores. Both rotated and non-rotated state lifetimes were calculated by fitting a single-exponential distribution to the measured state lifetimes using maximum-likelihood estimation in MATLAB.
For generating post-synchronized plots in dual illumination experiments, each traces were preprocessed by subtracting background fluorescence first and normalized across the specified time window around the synchronized event in each channel, which accentuated a single state transition. For tRNA-tRNA smFRET experiments, normalization for different fluorescence channels for gamma-value correction was applied across the entire trace, to preserve actual FRET values calculated after the background correction.
Bulk kinetics experiments
The 5 mM Mg2+ buffer was polymix21 buffer, containing 95 mM KCl, 5 mM NH4Cl, 0.5 mM CaCl2, 8 mM putrescine, 1 mM spermidine, 5 mM potassium phosphate, 1 mM DTE and 5 mM Mg(OAc)2 and supplemented with energy regeneration components: 1 mM ATP and 1 mM GTP for the ribosome mixture or 2 mM ATP for the ternary complex mixture, 10 mM phosphoenolpyruvate (PEP), 50 μg/ml pyruvate kinase and 2 μg/ml myokinase. For observing GTP hydrolysis, 0.5 μM [3H]GTP (Perkin-Elmer) was added to the ternary complex mixture. The 15 mM Mg2+ buffer contained 10 mM additional Mg(OAc)2. Assuming that 1 ATP or GTP molecule chelates 1 Mg2+ and PEP chelates Mg2+ with a Kd of 6 mM37, the free Mg2+ concentrations were 1.3 mM in 5 mM Mg2+ buffer and 7.5 mM in 15 mM Mg2+ buffer.
Fast kinetic measurements were performed at 20°C in a temperature controlled quench-flow instrument (RQF-3; Kintek Corp.). Ribosome and ternary complex mixtures were pre-incubated 15 min at 37 °C, cooled down to 20 °C prior to the reaction, then rapidly mixed and the reaction stopped at different time points by quenching with 17% (final) formic acid. The mixtures were prepared essentially as previously described4. Ribosome mixture contained E. coli 70S ribosomes (variable concentrations as indicated), [3H]fMet-tRNAfMet (1.5 × ribosomes), mRNA (encoding AUG-AAA-Stop or AUG-AAmA-Stop; 1.5 × ribosomes), IF1 (1.5 × ribosomes), IF2 (0.5 × ribosomes) and IF3 (1.5 × ribosomes). Ternary complex mixture contained EF-Tu (variable concentrations as indicated), E. coli bulk tRNA (Roche) charged with 0.2 mM lysine and 0.5 μM Lys-tRNA synthetase. The amount of charged Lys-tRNALys in bulk tRNA was determined by forming [3H]fMet-Lys dipeptide and was in excess over EF-Tu. The amount of [3H]GDP and [3H]fMet-Lys formed was quantified by HPLC equipped with a β-RAM Model 4 radioactivity detector (IN/US Systems) as described38.
To determine (kcat/Km)GTP, ternary complexes were titrated with ribosomes in excess so that the rate was limited by ribosome concentration. For AAmA codon, 0.7 μM and 3 μM Lys-tRNALys were used and each reaction performed in parallel with AAA reaction using the same ternary complex mixture and both curves jointly fitted with shared parameters to increase precision as described39. For AAA codon, 0.3 μM Tu and 3 μM Lys-tRNALys were used and data fitted to a single exponential model. For GTP hydrolysis experiments at 15 mM total [Mg2+], 0.6 μM EF-Tu and 3 μM Lys-tRNALys were used. 10 μM (final) neomycin or paromomycin were added where indicated.
To measure proofreading during peptide bond formation, two ribosome mixtures, one containing AAA and the other AAmA codon in the A site, were reacted to the same ternary complex mixture in parallel. At 5 mM total [Mg2+] 4.2 μM ribosomes, 2 μM EF-Tu, 3 μM Lys-tRNALys were used, while at 15 mM total [Mg2+] 1.5 μM ribosomes, 1 μM Tu and 1.2 μM Lys-tRNALys were used. Kinetics data of [3H]fMet-Lys dipeptide formation over time dip(t) were fitted to a model dip(t) = aF · (1 − e−kF·t) + aS · (1 − e−kS·t) where aF was the amplitude of the fast phase, which corresponded to [3H]fMet-Lys formation with preformed Lys-tRNALys-Tu-GTP complex. Lower aF indicated rejection of Lys-tRNALys after GTP hydrolysis. Proofreading factor F was calculated as . The slow phase (S) was rate-limited by slow GDP exchange to GTP39 on EF-Tu due to absence of EF-Ts.
The [3H]fMet consumption over time was estimated to determine if 2′-O-methylation induced miscoding. The experiment was performed in polymix buffer containing 15 mM total Mg2+. 100 μM total E. coli tRNA was charged either with all 19 amino acids except lysine or only lysine. 100 μM each amino acid, 0.002 v/v corresponding aminoacyl-tRNA synthetase and 2 μM EF-Tu were used in all cases. The charged tRNA was reacted to 0.2 μM ribosomes initiated with [3H]fMet-tRNAfMet and containing AAA or AAmA codon in the A site. The decrease of the [3H]fMet-tRNAfMet fraction over time revealed a corresponding increase in peptide bond formation.
Purification and crystallization of 30S ribosomal subunits
We purified and crystallized 30S ribosomal subunits from T. thermophilus HB8 strain essentially as described29,30. The 2′-O-methylated mRNA fragments (with codon sequences underlined), 5′AmAAUUU3′, 5′AAmAUUU3′, 5′AAAmUUU3′ and native mRNA 5′AAAUUU3′ were purchased from Dharmacon. The presence of 2′-O-methylation was confirmed by mass spectrometry. The , (with anticodon sequence underlined GCAGACU(mcm5s2U)UU(ms2t6A)AΨCUGC ), is a generous gift of Paul Agris (University of Albany). 30S crystals were sequentially transferred to the final buffer with 26% v/v 2-methyl-2,4-pentanediol (MPD) for cryoprotection and soaks were performed in the final buffer supplemented with 200 μM of each mRNA oligos, and in the presence or absence of 80 μM paromomycin for 48 hours, as indicated. Crystals were flash-frozen for data collection by plunging them directly into liquid nitrogen.
Data collection and refinement of structure obtained by x-ray crystallography
X-ray diffraction data were collected from a single crystal for each conditions. All datasets were collected with a Pilatus 6M detector at beamline BL12-2 at the Stanford Synchrotron Radiation Lightsource (SSRL) in Menlo Park, CA. From each crystal four 360 degrees very low-dose datasets collected at 10% transmission. Diffraction data sets were processed and scaled with the XDS40. Low-dose and high redundancy data collection resulted in higher R merge values (Table 1). Coordinates of the 30S subunit structure excluding mRNA and ASL (PDB accession code 3T1H28 with additional 30S rRNA and protein modifications were used for initial rigid body refinement with Phenix41 for all data sets. After simulated-annealing refinement, individual coordinates, three group B factors per residue, and TLS parameters were refined. Potential positions of magnesium or potassium ions were compared with those in a high-resolution (2.5 Å) 30S subunit structure (PDB accession code 2VQE42) in program COOT43, and positions with strong difference density were retained. All magnesium atoms were replaced with magnesium hexahydrate. Water molecules located outside of significant electron density were manually removed. A similar refinement protocol was used for all datasets. Structure alignments were performed using the alignment algorithm of PyMOL with the default 2σ rejection criterion and five iterative alignment cycles. All X-ray crystal structure figures were produced with PyMOL44.
Statistics and Reproducibility
Measurements from single-molecule fluorescence assay were resulting from specified number (n) of molecules from a single experiment. Measurements from bulk-kinetics assay were resulting from three technical replicates.
Data Availability statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes [5WNP, 5WNQ, 5WNR and 5WNS] for data sets unmodified mRNA (AAAUUU), mRNA-1 (AmAAUUU), mRNA-2 (AAmAUUU) and mRNA-3 (AAAmUUU) in the absence of paromomycin and [5WNT, 5WNU and 5WNV] for data sets mRNA-1 (AmAAUUU), mRNA-2 (AAmAUUU) and mRNA-3 (AAAmUUU) in the presence of paromomycin, respectively. Source data for figures are available as accompanying Source Data for each figures. All the other experimental data are available upon request.
Supplementary Material
Acknowledgments
This work was supported by US National Institutes of Health (NIH) grants GM51266 and GM099687 to J.D.P.; by grants from the Knut and Alice Wallenberg Foundation (RiboCORE), the Swedish Research Council and the Human Frontier Science Program to M.E.; by grants from the Kahn Family Foundation to D.D. and G.R.; by grants from the Ernest and Bonnie Beutler Research Program, Flight Attendant Medical Research Institute (FAMRI) and the Israeli Centers of Excellence (I-CORE) Program (ISF grants no. 41/11 and no. 1796/12) to G.R.; by a Human Frontier Science Program long-term fellowship to D.D.; by the NSF Science and Technology Centers grant NSF-1231306 (Biology with X-ray Lasers, BioXFEL) to H.D.; by a Stanford Bio-X fellowship to J.C. and A. Prabhakar; by a Knut and Alice Wallenberg Foundation postdoc fellowship to J.W. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility operated by Stanford University on behalf of the US Department of Energy, US Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the US Department of Energy, Office of Biological and Environmental Research, NIH, US National Center for Research Resources, Biomedical Technology Program, and the US National Institute of General Medical Sciences. C.H. is an investigator of the Howard Hughes Medical Institute (HHMI). G.R. is a member of the Sagol Neuroscience Network and holds the Djerassi Chair for Oncology at the Sackler Faculty of Medicine, Tel-Aviv University, Israel. We thank P. Agris (University of Albany) for a human ASL reagent and members of Puglisi laboratory for discussion.
Footnotes
Author Contributions
J.C., G.I., H.D. and K.-W.I. performed all the experiments and the data analysis; J.C. performed single-molecule experiments, with the help of J.W., A. Prabhakar and A. Petrov in material preparation; G.I., and K.-W.I. performed bulk kinetics experiments; H.D. performed x-ray crystallography. D.D., G.R., and C.H. provided reagents and conceived the project with J.C., H.D., K.-W.I., M.E. and J.D.P. J.C., G.I., H.D., J.W., M.E., and J.D.P. wrote manuscript with input from all authors.
Competing Financial Interests Statement
The authors declare no competing financial interests.
References
- 1.Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2016;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu N, et al. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15:293–306. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- 4.Choi J, et al. N6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat Struct Mol Biol. 2016;23:110–115. doi: 10.1038/nsmb.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai Q, et al. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat Methods. 2017 doi: 10.1038/nmeth.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decatur WA, Fournier MJ. rRNA modifications and ribosome function. Trends Biochem Sci. 2002;27:344–351. doi: 10.1016/s0968-0004(02)02109-6. [DOI] [PubMed] [Google Scholar]
- 7.Somme J, et al. Characterization of two homologous 2′-O-methyltransferases showing different specificities for their tRNA substrates. Rna. 2014;20:1257–1271. doi: 10.1261/rna.044503.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavaillé J, Nicoloso M, Bachellerie JP. Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature. 1996;383:732–735. doi: 10.1038/383732a0. [DOI] [PubMed] [Google Scholar]
- 9.Kiss-László Z, Henry Y, Bachellerie JP, Caizergues-Ferrer M, Kiss T. Site-specific ribose methylation of preribosomal RNA: A novel function for small nucleolar RNAs. Cell. 1996;85:1077–1088. doi: 10.1016/s0092-8674(00)81308-2. [DOI] [PubMed] [Google Scholar]
- 10.Shubina MY, Musinova YR, Sheval EV. Nucleolar Methyltransferase Fibrillarin: Evolution of Structure and Functions N terminal domain. Biochemistry. 2016;81:941–950. doi: 10.1134/S0006297916090030. [DOI] [PubMed] [Google Scholar]
- 11.Cummins LL, et al. Characterization of fully 2 V. Nucleolar Methyltransferase Fibrillarin -modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res. 1995;23:2019–2024. doi: 10.1093/nar/23.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majlessi M, Nelson NC, Becker MM. Advantages of 2 V. Nucleolar Methyltransferase Fibrillarin -O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998;26:2224–2229. doi: 10.1093/nar/26.9.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoernes TP, et al. Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code. Nucleic Acids Res. 2016;44:852–862. doi: 10.1093/nar/gkv1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshizawa S, Fourmy D, Puglisi JD. Recognition of the Codon-Anticodon Helix by Ribosomal RNA. Science (80- ) 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]
- 15.Ogle JM, et al. Recognition of Cognate Transfer RNA by the 30S Ribosomal Subunit. Science (80- ) 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 16.Uemura S, et al. Real-time tRNA transit on single translating ribosomes at codon resolution. Nature. 2010;464:1012–7. doi: 10.1038/nature08925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, et al. High-throughput platform for real-time monitoring of biological processes by multicolor single-molecule fluorescence. Proc Natl Acad Sci U S A. 2014;111:664–9. doi: 10.1073/pnas.1315735111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorywalska M, et al. Site-specific labeling of the ribosome for single-molecule spectroscopy. Nucleic Acids Res. 2005;33:182–189. doi: 10.1093/nar/gki151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aitken CE, Puglisi JD. Following the intersubunit conformation of the ribosome during translation in real time. Nat Struct Mol Biol. 2010;17:793–800. doi: 10.1038/nsmb.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Tsai A, Petrov A, Puglisi JD. Nonfluorescent quenchers to correlate single-molecule conformational and compositional dynamics. J Am Chem Soc. 2012;134:5734–7. doi: 10.1021/ja2119964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurland C, Ehrenberg M. Optimization of translation accuracy. Prog Nucleic Acid Res Mol Biol. 1984;31:191–219. doi: 10.1016/s0079-6603(08)60378-5. [DOI] [PubMed] [Google Scholar]
- 22.Blanchard SC, Jr, RLG, Kim HD, Chu S, Puglisi JD. tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol. 2004;11:1008–1014. doi: 10.1038/nsmb831. [DOI] [PubMed] [Google Scholar]
- 23.Loveland AB, Demo G, Grigorieff N, Korostelev AA. Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature. 2017;546:113–117. doi: 10.1038/nature22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ninio J. Kinetic amplification of enzyme discrimination. Biochimie. 1975;57:587–595. doi: 10.1016/s0300-9084(75)80139-8. [DOI] [PubMed] [Google Scholar]
- 25.Hopfield JJ. Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc Natl Acad Sci. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–32. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- 27.Satpati P, Sund J, Åqvist J. Structure-based energetics of mRNA decoding on the ribosome. Biochemistry. 2014;53:1714–1722. doi: 10.1021/bi5000355. [DOI] [PubMed] [Google Scholar]
- 28.Vendeix FaP, et al. Human tRNA(Lys3)(UUU) is pre-structured by natural modifications for cognate and wobble codon binding through keto-enol tautomerism. J Mol Biol. 2012;416:467–85. doi: 10.1016/j.jmb.2011.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demirci H, et al. Modification of 16S ribosomal RNA by the KsgA methyltransferase restructures the 30S subunit to optimize ribosome function. RNA. 2010;16:2319–2324. doi: 10.1261/rna.2357210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wimberly BT, et al. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, et al. m1A and m1G disrupt A-RNA structure through the intrinsic instability of Hoogsteen base pairs. Nat Struct Mol Biol. 2016;23:803–810. doi: 10.1038/nsmb.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roost C, et al. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc. 2015;137:2107–15. doi: 10.1021/ja513080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ieong K-W, Uzun Ü, Selmer M, Ehrenberg M. Two proofreading steps amplify the accuracy of genetic code translation. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1610917113. 201610917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richter JD, Coller J. Pausing on Polyribosomes: Make Way for Elongation in Translational Control. Cell. 2015;163:292–300. doi: 10.1016/j.cell.2015.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marshall RA, Dorywalska M, Puglisi JD. Irreversible chemical steps control intersubunit dynamics during translation. Proc Natl Acad Sci U S A. 2008;105:15364–15369. doi: 10.1073/pnas.0805299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, Petrov A, Tsai A, O’Leary SE, Puglisi JD. Coordinated conformational and compositional dynamics drive ribosome translocation. Nat Struct Mol Biol. 2013;20:718–27. doi: 10.1038/nsmb.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wold F, Ballou CE. Studies on the enzyme enolase. I. Equilibrium studies. J Biol Chem. 1957;227:301–312. [PubMed] [Google Scholar]
- 38.Johansson M, Zhang J, Ehrenberg M. Genetic code translation displays a linear trade-off between efficiency and accuracy of tRNA selection. Proc Natl Acad Sci U S A. 2012;109:131–6. doi: 10.1073/pnas.1116480109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Ieong KW, Johansson M, Ehrenberg M. Accuracy of initial codon selection by aminoacyl-tRNAs on the mRNA-programmed bacterial ribosome. Proc Natl Acad Sci U S A. 2015;112:9602–7. doi: 10.1073/pnas.1506823112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabsch W. Xds. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurata S, et al. Modified uridines with C5-methylene substituents at the first position of the tRNA anticodon stabilize U.G wobble pairing during decoding. J Biol Chem. 2008;283:18801–18811. doi: 10.1074/jbc.M800233200. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schrödinger. The PyMOL Molecular Graphics System, Version 1.7.4. Schrödinger, LLC; 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes [5WNP, 5WNQ, 5WNR and 5WNS] for data sets unmodified mRNA (AAAUUU), mRNA-1 (AmAAUUU), mRNA-2 (AAmAUUU) and mRNA-3 (AAAmUUU) in the absence of paromomycin and [5WNT, 5WNU and 5WNV] for data sets mRNA-1 (AmAAUUU), mRNA-2 (AAmAUUU) and mRNA-3 (AAAmUUU) in the presence of paromomycin, respectively. Source data for figures are available as accompanying Source Data for each figures. All the other experimental data are available upon request.