

Abstract
B6.SJL-PtprcaPepcb/Boy (CD45.1) mice have been used in hundreds of congenic competitive transplants, with the presumption that they differ from B6 mice only at the CD45 locus. In this study, we describe a point mutation in the Ncr1 locus fortuitously identified in the CD45.1 strain. This point mutation was mapped at the 40th nucleotide of the Ncr1 locus causing a single amino acid mutation from cysteine to arginine at position 14 from start codon, resulting in loss of NCR1 expression. We found that these mice were more resistant to cytomegalovirus due to a hyper innate IFNγ response in the absence of NCR1. In contrast, loss of NCR1 increased susceptibility to Influenza virus, a result which is consistent with the role of NCR1 in the recognition of influenza antigen, hemagglutinin. This work shed light on potential confounding experimental interpretation if this congenic strain is used as a tool for tracking lymphocyte development.
Introduction
The most commonly approach used for tracking immune cell development in vivo takes advantage of polymorphisms in the extracellular domain of the transmembrane receptor tyrosine phosphatase protein CD45 (Ptprc), a 220-kDa protein expressed on all subsets of leukocytes. Two isoforms have been identified in mice: the common form is CD45.2, which is expressed by the C57BL/6 (B6) strain and is encoded by the Ptprcb allele; and an additional allelic variant Ptprca which encodes the CD45.1 isoform, was identified in the SJL mouse strain. CD45.1 and CD45.2 alleles differ by only five amino acids within the extracellular domain, resulting in epitope changes that permit specific recognition by monoclonal antibodies. Backcrossing of mice expressing the CD45.1 allele into the B6 background has resulted in the development of the mouse strain B6.SJL-PtprcaPepcb/Boy (CD45.1). As CD45.1 mice have been backcrossed over many generations into B6 background, these mice have been termed congenic, with the presumption that they differ from the B6 strain only at the CD45 locus. Surprisingly, despite extensive backcrossing, genotypic analysis revealed that the congenic interval in which CD45.1 differs from CD45.2 mice is almost 43-mbp encoding 306 genes and at least 124 genetic polymorphisms (1).
In this study, we describe a point mutation in the Ncr1 locus of CD45.1 strain, resulting in loss of expression. A point of critical consequence of this mutation is the different susceptibility of this strain to viral infection. Thus, using this “congenic” strain, under the presumption that it differs from B6 strain only at the CD45 locus, probably has been, and most likely will be, conducive to confounding experimental interpretation.
Material and Methods
Mice
CD45.2 and CD45.1 mice were originally obtained from Jackson Laboratories and maintained in our animal facility at the University of Michigan. For BM mixed chimeras, CD45.2 recipient mice were lethally irradiated (1200 rad) and injected (i.v) with 10 million donor BM cells containing a mixture of CD45.2 and CD45.1 cells provided at 1:1 ratio. For the transfer of gut microbiota, gut content was harvested and provided to recipient mice by oral gavage. As indicated, mice were either infected with MCMV (3,500 PFU of Smith strain per gram body weight by i.p.), Influenza A virus (50 PFU of H1N1 per mouse by intranasal inhalation), or Citrobacter Rodentium (1010 CFU per mouse by oral gavage). All experiments were performed in accordance with University of Michigan Animal Care and Use Committee and approval to use mice was granted by the University of Michigan in accordance with the US National Institute of Health requirements for the care and use of animals.
Sequencing, Cloning and Transfection of Ncr1 gene
Ncr1 ORF region was amplified from cDNA using the following primers, and amplified products were sequenced by Sanger method (University of Michigan): 5′-GTTCAGCACTGGTCTGGCCACTGG-3′; 5′-GCCAAACTTGGTAACACTCCTACC-3′. For cloning and transfection of Ncr1 gene, Ncr1 ORF was amplified from cDNA using the following primers: 5′ GGAATTAGAGAGTTTCATGCTGCCAACACTCACTGC-3′; 5′-GAATTGTGGAAGTTTCTCACAAGGCCCCAGGAGTTG-3′. Amplified Ncr1 gene was cloned into pMSCV-neo vector, transfected into Bosc cells for viral packaging, infected into MEF cells, and then selected with 500 μg/ml of Geneticin for analysis.
Results and Discussion
CD45.1 and CD45.2 strains are not functionally equivalent
CD45.1 and CD45.2 mice were originally obtained from Jackson laboratories and then maintained by breeding in our animal facility. While performing experiments with CD45.2 and CD45.1 mice, we observed a pattern of responses that suggested inherent differences in their susceptibility to infection (Fig. 1). In response to MCMV infection, we found that CD45.1 mice were better equipped to fight lethal dose of MCMV. In response to Influenza virus infection, we observed opposites results with CD45.1 mice being less protected than CD45.2 mice. However, in response to Citrobacter rodentium infection, both CD45.1 and CD45.2 mouse strains were equally resistant to the enteric bacteria. Based on these results, we conclude that CD45.1 and CD45.2 mouse strains –that are housed in our animal facility– are not functionally equivalent.
Figure 1. CD45.2 and CD45.1 strains exhibit different susceptibility to infection.
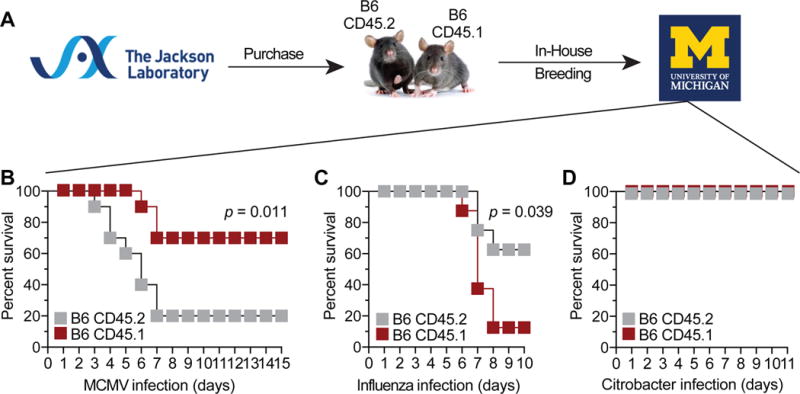
(A) History of in-house breeding of CD45.2 and CD45.1 mouse strains. Mouse survival after (B) MCMV, (C) influenza virus, or (D) Citrobacter rodentium infection. Data are from two independent experiments with n = 10 mice per group. Statistics were analyzed using Log-rank Mantel-Cox test.
Point mutation in the Ncr1 locus identified in CD45.1 strain
Detailed analysis of NK cell compartment landed one major difference between CD45.1 and CD45.2 strains: NCR1 expression was lost in CD45.1 strain (Fig. 2). Loss of NCR1 expression in CD45.1 strain was identified in NK cells from various organs (Fig. 2A-B) and ILCs from the intestine (Fig. 2C). We confirmed that the loss of NCR1 expression was not the result of intracellular sequestration, as shown by surface and intracellular staining and with monoclonal and polyclonal anti-NCR1 antibodies (Fig. 2D, I). Given the increasing evidence of associations between host gene expression and gut microbiota (2), we considered the possibility that the loss of NCR1 in CD45.1 strain is the result of a specific gut microbiota acquired during breeding in our animal facility. However, the transfer of gut content from CD45.2 mice failed to restore the expression of NCR1 in CD45.1 recipient NK cells (Fig. 2E). Finally, to demonstrate that the loss of NCR1 in CD45.1 strain is not environment-driven, we assessed the expression of NCR1 in mixed BM chimeras. Results showed no expression of NCR1 on CD45.1-derived NK cells, indicating a cell-autonomous defect (Fig. 2F).
Figure 2. CD45.1 strain bear a point mutation in the Ncr1 locus causing loss of NCR1 expression.
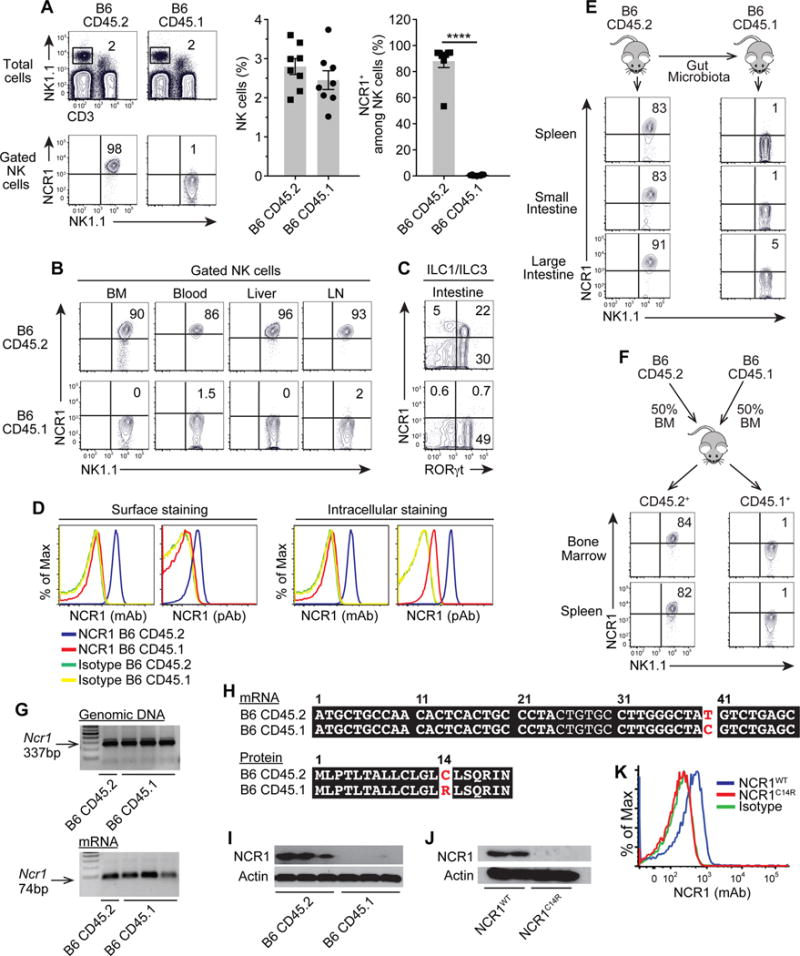
NCR1 expression on (A) spleen NK cells (CD3−NK1.1+), (B) NK cells from the indicated organs, and (C) intestinal ILCs. (D) Surface and intracellular expression of NCR1 using monoclonal (mAb) or polyclonal (pAb) antibodies. (E) NCR1 expression on NK cells from CD45.1 recipients 3 weeks after gut content transfer. (F) NCR1 expression on donor-derived NK cells from CD45.2+ or CD45.1+ cells. (G) Genomic DNA and mRNA transcripts of Ncr1 gene. (H) mRNA and protein sequence of Ncr1 gene. (I) Amounts of NCR1 proteins from sorted NK cells. (J, K) NCR1 expression in mutant (NCR1C14R) and wild-type (NCR1WT) transfected cells. Data are representative of three independent experiments with n = 4 per group. Statistics were analyzed using t-test, ****p < 0.0001.
Since levels of the Ncr1 transcripts were comparable in both mouse strains (Fig. 2G), we sequenced the mRNA and identified a point mutation, TGT to CGT, in the 40th nucleotide from the start codon leading to a single amino acid mutation from cysteine to arginine at the 14th amino acid (C14R), which is localized in the region of the signal peptide of NCR1 (Fig. 2H). To evaluate the effects of this signal peptide mutation, we constructed expression vectors with mutant (NCR1C14R) or wild-type (NCR1WT) forms and assessed outcomes on protein expression. Unlike wild-type control, no protein was detected from the mutated construct, indicating that this signal peptide mutation was sufficient to abrogate NCR1 expression (Fig. 2J-K). Signal peptides are responsible for numerous functions, including recognition and binding of the protein to the cytosolic signal recognition particle, its insertion into the ER-protein-conducting channels, and protein maturation processes such as quality control and addition of N-linked glycans (3). Examples from signal peptide mutations showed that changes in the signal peptide hydrophobicity interfere with the binding of the mutant preproteisn to the signal recognition and translocation into the ER (4). Even for the fraction of the mutant preproteins that enter the ER, their cleavage by the signal peptidase is inefficient; and as a result, the uncleaved preproteins could not reach the glycosylation machinery and therefore were retained in the ER (4). Alternatively, the mutant preproteins are removed from the ER by a process of active degradation via the proteasome pathway (5). Although further studies are required to identify the exact mechanism, our results imply a similar mechanism for the C14R signal peptide mutation resulting in degradation and loss of NCR1. Accordingly, we termed this mutant strain, Ncr1C14R mice.
Mutant Ncr1C14R confers hyper IFNγ response without altering the lytic activity
NCR1 contains two extracellular Ig domains, a type I trans-membrane domain, and a short cytoplasmic tail lacking immune-receptor tyrosine-based activating motifs (ITAMs). Given the lack of inherent signaling motifs, NCR1 relies on adaptor molecules FCεRIγ and CD3ζ to impart positive signals through their ITAMs (6). Based on this structure, NCR1 has been assumed to act solely as an activating receptor delivering positive signals to NK cells (7-9). However, such a role has been challenged by opposite results arguing instead for a regulatory role of NCR1 in NK cell activities (10). Given the controversial data reported from Ncr1gfp/gfp and Ncr1Noe/Noe mice (11, 12), we sought to re-examine the role of NCR1 using the Ncr1C14R mutant mice.
While the efficacy of lytic activity was not altered in the absence of NCR1 (Fig. 3A-C), IFNγ response was higher in Ncr1C14R compared to wild-type NK cells (Fig. 3D-F). To examine how this gain of function translates in vivo, mutant and wild-type mice were infected with a lethal dose of MCMV (Fig. 3G-M). We found that Ncr1C14R mice were more resistant to MCMV infection than wild-type mice, as indicated by higher survival rates (Fig. 1A) and reduced viral loads in the liver and spleen (Fig. 3G). Analysis of NK cells on 1.5 and 5 days post-infection showed higher production of IFNγ both in the liver and spleen of Ncr1C14R compared to wild-type mice, and both in Ly49H+ and Ly49H− NK cell subsets lacking NCR1 expression (Fig. 3H-M). Notably, neither proliferation nor production of GzmB was altered in the absence of NCR1 (Supplementary Figure 1A), indicating a selective role of NCR1 in the regulation of innate IFNγ. One could raise the concern that, in these experiments, the control mice are not the mutant’s littermates. To address this issue, we repeated the above experiments using mutant (F2 Ncr1C14R) and wild-type (F2 Ncr1WT) littermates derived from F2 progeny (Fig. 3N-R; Supplementary Figure 1B-E), and confirmed the gain of NK cell IFNγ response in the absence of NCR1. Interestingly, expression of NCR1 in F1 heterozygotes was intermediate indicating that normal expression requires both functional alleles. Consistent with our data, stimulation of human NK cells in the presence of anti-NCR1 blocking antibody resulted in copious amounts of IFNγ (13). Of note, this gain of function was specific to NCR1 since blocking NCR2 or NCR3 showed no effects on IFNγ (13).
Figure 3. Ncr1C14R mutant mice mount a stronger innate IFNγ response.
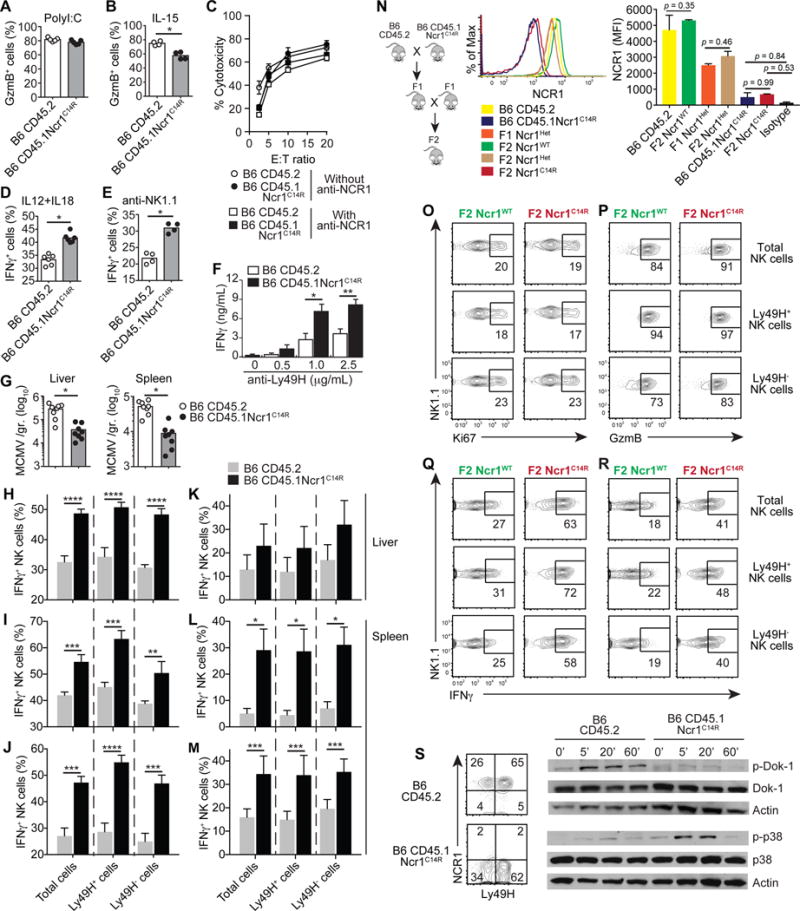
Frequency of GzmB+ cells among NK cells (A) from PolyI:C-treated mice, or (B) after 24 hr. simulation with IL-15 (20ng/ml). (C) Percent of NK specific lysis of YAC-1 cells in the absence or absence of 10 μg/ml of anti-NCR1 antibody. Frequency of IFNγ+ cells among NK cells stimulated with (D) IL-12 (1ng/ml) plus IL-18 (2ng/ml), or (E) plate-bound anti-NK1.1 (1μg/ml) antibody for 5 hrs. (F) Amounts of IFNγ secreted by NK cells stimulated with anti-Ly49H antibody for 6 hrs. (G) MCMV titers on day 3. Frequency of IFNγ+ cells among NK cells on days 1.5 (H, I, J) and 5 (K, L, M) post-infection. Results are from in vitro re-stimulation with 0.5ng/ml (L) or 1ng/ml (H, I, K, M) of IL-12 supplemented with 2ng/ml of IL-18, or PMA (20ng/ml) plus Ionomycin (1 μg/ml) (J). (N) NCR1 expression in F1 and F2 progeny and parental CD45.1Ncr1C14R and CD45.2 mice. Mutant and wild-type mice from F2 progeny were infected with MCMV and spleen NK cells were analyzed on d1.5 post-infection for proliferation (O) and GzmB expression (P). IFNγ expression is shown after 5 hr. stimulation with (Q) IL-12 (1ng/ml) plus 2ng/ml of IL-18 (2ng/ml) or (R) PMA/Ionomycin. (S) Dok-1 and p38 phosphorylation from NK cells stimulated with anti-Ly49H antibody for the indicated time points (min). Data are representative of three to four independent experiments with n = 4 per group. Data are mean ± SEM or shown from individual mice. Statistics were analyzed using t-test or two-way ANOVA with Sidak correction, *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.0001.
Since levels of T-bet, Eomes, and Helios were not different in both mouse groups, (Supplementary Fig. 2), we tested the possibility that augmented IFNγ response in the absence of NCR1 is mediated via Dok-1 (Fig. 3S). By recruiting enzymes transducing negative signals such as RasGAP and SHIP1, Dok-1 inhibits Ras/ERK and PI3K/Akt signaling pathways resulting in negative regulation of leukocyte activation (14). In T cells, Dok-1 is tyrosine phosphorylated resulting in negative regulation of TCR-induced T cell activation. Overexpression of Dok-1 decreased IL-2 production (15), and loss of Dok-1 enhanced TCR-mediated signaling (16). Likewise, overexpression of Dok-1 in NK cells reduced IFNγ production while Dok-1 gene ablation augmented IFNγ response (17). Consistent with these results, we found that the loss of NCR1 in NK cells was sufficient to prevent Dok-1 activation downstream of Ly49H engagement, resulting in increased MAP Kinase activation and augmented IFNγ response in NK cells (Fig. 3S). Although it is tempting to speculate that NCR1 may serve as a regulator of Dok-1 activation downstream of NKR engagement, further studies are required to elucidate the specific nature of the interplay between NCR1 and Dok-1 in control of NK cell IFNγ.
Given the frequent use of CD45.1 strain as a tool for tracking lymphocyte development, our finding raised one important question: did the Ncr1C14R mutation identified in our congenic mouse colony originate from the vendor or did it occur in our animal facility as a result of genetic drifting? Using flow cytometry data from our archives, we were able to trace this mutation backward and have identified the loss of NCR1 in CD45.1 mice from experiments dated from 2009 (Fig. 4). Conversely, analysis of recent studies performed with CD45.1 mice purchased after 2014 showed normal expression of NCR1 (Fig. 4). Colony maintenance information from The Jackson Laboratory indicates that the CD45.1 strain (Stock # 002014) had been imported into The Jackson Laboratory in 1990 from Dr. Edward Boyse at generation N22. From that time until 2009, the strain was maintained by pedigrees and filial matings. Between 2009-2010, the B6 CD45.1 congenic mouse colony underwent 3 backcrosses to the inbred mouse strain C57BL/6J (N25). It is possible that this backcrossing may have removed the Ncr1C14R mutation identified in mice obtained prior to 2010, however this has not been directly tested. Notably, as the Ncr1C14R point mutation results in loss of NCR1 expression, determining the status of your congenic mouse colony will be easily achievable by flow cytometry. Otherwise, using these mice, as a tool for tracking lymphocyte development under the presumption that they differ from B6 strain only at the CD45 locus, will certainly be conducive to confounding experimental interpretation.
Figure 4. Tracing the Ncr1C14R mutation in CD45.1 “congenic” mice.
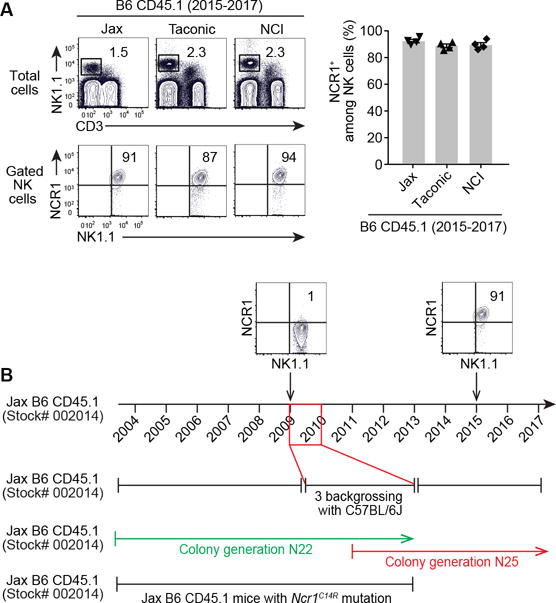
(A) NCR1 expression from CD45.1 mice purchased from commercial vendors between 2015-2017. Data are representative of three independent experiments with n = 4. (B) NCR1 expression from CD45.1 mice purchased from The Jackson Laboratories before 2009 versus after 2015. (C) The colony generation of Jax B6 CD45.1 strain over time. (D) Mice from N22 colony generation could be the source of the Ncr1C14R mutation.
Supplementary Material
Acknowledgments
We thank Dr. Low-Marchelli from the Jackson Laboratories for providing helpful information on the colony generation of CD45.1 congenic strain.
This work was supported by the National Institutes of Health (R01 AI102893 and R01 CA179363 to S.M.; R01 HL119682 to B.M.; and R01 AI083642 to Y.L.); MACC Fund (to S.M.); Nicholas Family Foundation (to S.M.); and AAI Careers in Immunology (N023607 to Y.L.)
Abbreviation
- NCR
Natural Cytotoxicity Receptor
- ILC
Innate Lymphoid Cells
- NK
Natural Killer cells
- NKR
NK cell Receptors
- IFNγ
Interferon gamma
- Dok
Downstream of kinase
- GzmB
Granzyme B
- MCMV
Murine Cytomegalovirus
Footnotes
ORCID: 0000-0002-8865-4791 (Y.L.)
References
- 1.Ryan MA, Nattamai KJ, Xing E, Schleimer D, Daria D, Sengupta A, Kohler A, Liu W, Gunzer M, Jansen M, Ratner N, Le Cras TD, Waterstrat A, Van Zant G, Cancelas JA, Zheng Y, Geiger H. Pharmacological inhibition of EGFR signaling enhances G-CSF-induced hematopoietic stem cell mobilization. Nat Med. 2010;16:1141–1146. doi: 10.1038/nm.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan XC, Kabakchiev B, Waldron L, Tyler AD, Tickle TL, Milgrom R, Stempak JM, Gevers D, Xavier RJ, Silverberg MS, Huttenhower C. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome biology. 2015;16:67. doi: 10.1186/s13059-015-0637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martoglio B, Dobberstein B. Signal sequences: more than just greasy peptides. Trends Cell Biol. 1998;8:410–415. doi: 10.1016/s0962-8924(98)01360-9. [DOI] [PubMed] [Google Scholar]
- 4.Henderson JE, Amizuka N, Warshawsky H, Biasotto D, Lanske BM, Goltzman D, Karaplis AC. Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol Cell Biol. 1995;15:4064–4075. doi: 10.1128/mcb.15.8.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnold A, Horst SA, Gardella TJ, Baba H, Levine MA, Kronenberg HM. Mutation of the signal peptide-encoding region of the preproparathyroid hormone gene in familial isolated hypoparathyroidism. The Journal of clinical investigation. 1990;86:1084–1087. doi: 10.1172/JCI114811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster CE, Colonna M, Sun PD. Crystal structure of the human natural killer (NK) cell activating receptor NKp46 reveals structural relationship to other leukocyte receptor complex immunoreceptors. J Biol Chem. 2003;278:46081–46086. doi: 10.1074/jbc.M308491200. [DOI] [PubMed] [Google Scholar]
- 7.Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, Moretta L, Moretta A. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med. 1997;186:1129–1136. doi: 10.1084/jem.186.7.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nature immunology. 2006;7:517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]
- 9.Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, Mandelboim O. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol. 2012;188:2509–2515. doi: 10.4049/jimmunol.1102461. [DOI] [PubMed] [Google Scholar]
- 10.Narni-Mancinelli E, Jaeger BN, Bernat C, Fenis A, Kung S, De Gassart A, Mahmood S, Gut M, Heath SC, Estelle J, Bertosio E, Vely F, Gastinel LN, Beutler B, Malissen B, Malissen M, Gut IG, Vivier E, Ugolini S. Tuning of natural killer cell reactivity by NKp46 and Helios calibrates T cell responses. Science. 2012;335:344–348. doi: 10.1126/science.1215621. [DOI] [PubMed] [Google Scholar]
- 11.Glasner A, Simic H, Miklic K, Roth Z, Berhani O, Khalaila I, Jonjic S, Mandelboim O. Expression, Function, and Molecular Properties of the Killer Receptor Ncr1-Noe. Journal of immunology. 2015;195:3959–3969. doi: 10.4049/jimmunol.1501234. [DOI] [PubMed] [Google Scholar]
- 12.Glasner A, Isaacson B, Mandelboim O. Expression and function of NKp46 W32R: the human homologous protein of mouse NKp46 W32R (Noe) Sci Rep. 2017;7:40944. doi: 10.1038/srep40944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spallanzani RG, Torres NI, Avila DE, Ziblat A, Iraolagoitia XL, Rossi LE, Domaica CI, Fuertes MB, Rabinovich GA, Zwirner NW. Regulatory Dendritic Cells Restrain NK Cell IFN-gamma Production through Mechanisms Involving NKp46, IL-10, and MHC Class I-Specific Inhibitory Receptors. Journal of immunology. 2015;195:2141–2148. doi: 10.4049/jimmunol.1403161. [DOI] [PubMed] [Google Scholar]
- 14.Acuto O, Di Bartolo V, Michel F. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat Rev Immunol. 2008;8:699–712. doi: 10.1038/nri2397. [DOI] [PubMed] [Google Scholar]
- 15.Nemorin JG, Laporte P, Berube G, Duplay P. p62dok negatively regulates CD2 signaling in Jurkat cells. J Immunol. 2001;166:4408–4415. doi: 10.4049/jimmunol.166.7.4408. [DOI] [PubMed] [Google Scholar]
- 16.Dong S, Corre B, Foulon E, Dufour E, Veillette A, Acuto O, Michel F. T cell receptor for antigen induces linker for activation of T cell-dependent activation of a negative signaling complex involving Dok-2, SHIP-1, and Grb-2. J Exp Med. 2006;203:2509–2518. doi: 10.1084/jem.20060650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Celis-Gutierrez J, Boyron M, Walzer T, Pandolfi PP, Jonjic S, Olive D, Dalod M, Vivier E, Nunes JA. Dok1 and Dok2 proteins regulate natural killer cell development and function. The EMBO journal. 2014;33:1928–1940. doi: 10.15252/embj.201387404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.