

Abstract
Long non-coding RNAs (lncRNAs) represent a significant population of the human transcriptome. Many lncRNAs exhibit cell and/or tissue/tumor specific expression making them excellent candidates for therapeutic applications. In this review we discuss examples of lncRNAs that demonstrate the diversity of their function in various cancer types. We also discuss recent advances in nucleic acid drug development with a focus on oligonucleotide-based therapies as a novel approach to inhibit tumor progression. The increased success rates of nucleic acid therapeutics provides an outstanding opportunity to explore lncRNAs as viable therapeutic targets to impact various aspects of cancer progression.
Long non-coding RNAs as novel players in tumorigenesis
Large-scale cancer genomics projects such as The Cancer Genome Atlas (TCGA) revealed that many of the mutations and copy number changes found in cancer do not overlap with protein-coding genes [1,2], but are frequently located in non-coding DNA; including both regulatory DNA elements such as enhancers as well as non-coding RNA genes [3]. MicroRNAs (miRNAs) were among the first non-coding RNAs to be investigated in the context of cancer, and their role as therapeutic targets or biomarkers in cancer has been previously reviewed [4]. Here, we focus on the clinical relevance of long non-coding RNAs (lncRNAs), representing the largest and most diverse class of non-coding transcripts, with up to 60,000 lncRNA genes present in the human genome [5]. LncRNAs are defined by length (>200 nt), are transcribed by RNA polymerase II, and commonly originate from intergenic regions [5,6]. LncRNAs can be capped, spliced, and polyadenylated, but lack a significant open reading frame. Members of this class of non-coding transcripts have been implicated as molecular scaffolds, architectural RNAs, or as regulatory molecules in a variety of cellular functions: including epigenetic gene regulation, splicing, mRNA stability and translation, as well as acting as decoys or “sponges” for miRNAs or transcription factors [7,8] (Figure 1).
Figure 1. Proposed molecular functions of mammalian lncRNAs.
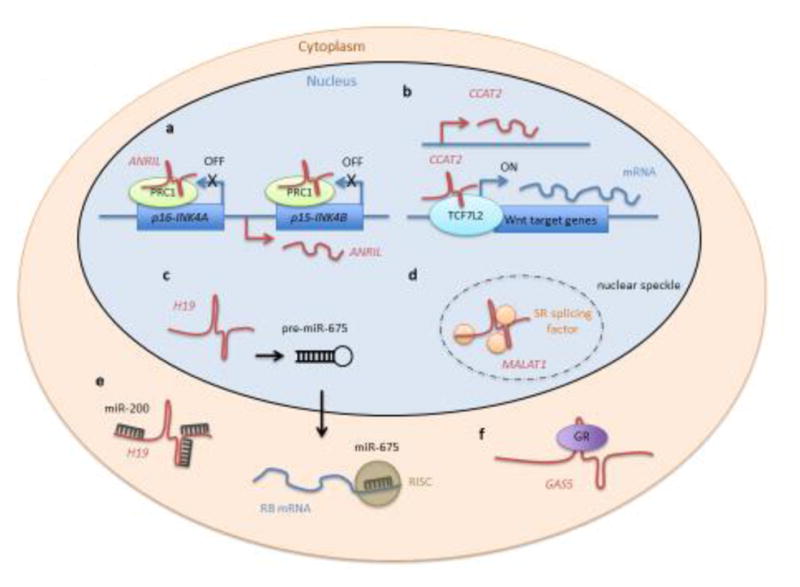
The schematic depicts examples of proposed lncRNA (red) molecular functions and their location in corresponding cellular compartments.
a) ANRIL represses gene transcription of the INK4 locus in cis by binding to polycomb repressive complex 1 (PRC1, green).
b) CCAT2 activates expression of WNT target genes including MYC, possibly by directly interacting with the transcription factor TCF7L2 (light blue).
c) H19 is a precursor for miR-675. The lncRNA is processed to pre-miR-675, which is exported into the cytoplasm and further processed to miR-675. The mature miRNA assembles with RNA-induced silencing complex (RISC, dark green). The tumor suppressor RB is one of the targets of miR-675.
d) MALAT1 associates with SR splicing factors (orange) and is located in nuclear speckles.
e) H19 also acts as a sponge for many different miRNAs, including members of the miR-200 family.
f) GAS5 functions as a decoy for glucocorticoid receptor (GR, purple), preventing GR-dependent gene activation.
While the first cancer-related lncRNAs were identified based on their altered expression in cancer cells or tumor tissue (see examples in Table 1), the function of a number of lncRNAs is beginning to be revealed. Many lncRNAs play a critical role in one or several hallmarks of cancer such as uncontrollable proliferation, evasion of cell death or metastasis [9,10], and can directly act as oncogenes or tumor suppressors, or indirectly by interacting with well-known oncogenes or tumor suppressors such as MYC or p53, on both a transcriptional or post-transcriptional level [11,12]. Here, we highlight several well-studied examples of lncRNAs involved in the acquired capabilities of cancer cells, and selected based on clinical data as well as in vitro and in vivo evidence. The majority of lncRNAs are expressed in a highly tissue- and cell-type specific manner [13,14], making them potential highly eficacious targets for systemic cancer treatment. We discuss examples of lncRNAs that highlight diversity of function in various types of cancer. We also outline recent advances in drug development aimed at targeting cancer-associated lncRNAs, with a focus on oligonucleotide-based therapies as a novel approach to inhibit tumor progression.
Table 1.
Selected human lncRNAs and their expression in tumorigenesis
| Name | Function | Hallmarks | References |
|---|---|---|---|
| aHIF | Up-regulated | 5 | 18 |
| ANCR | Down-regulated | 6 | 185 |
| ANRIL | Up-regulated | 1, 2, 3, 6, 9 | 86,89 |
| BCAR4 | Up-regulated | 6 | 186 |
| CCAT1 | Up-regulated | 1, 2, 6 | 73,74,75 |
| CCAT2 | Up-regulated | 1, 6, 9 | 76 |
| ECONEXIN/MaTAR1 | Up-regulated | 1 | 57 |
| FILNC1 | Down-regulated | 7 | 187 |
| GAS5 | Down-regulated | 2 | 77,78,79 |
| H19 | Up-regulated/Down-regulated | 1, 5, 6 | 11,12,19 |
| HOTAIR | Up-regulated | 6 | 29,32 |
| HOTTIP | Up-regulated | 1,6 | 188 |
| HULC | Up-regulated | 1, 5, 7 | 189 |
| Lilam | Up-regulated | 1 | 121 |
| Linc-PINT | Down-regulated | 6 | 97 |
| Linc-ROR | Up-regulated | 6 | 190 |
| lincRNA-EPS/MaTAR18 | Up-regulated | 10 | 55 |
| lincRNA-p21 | Up-regulated | 3, 5, 6, 7, 9 | 91,95 |
| lncRNA-ATB | Up-regulated | 6 | 191 |
| lncRNA-LET | Down-regulated | 6 | 192 |
| lncRNA-MIAT | Up-regulated | 5, 6 | 193 |
| LUNAR1 | Up-regulated | 1 | 34 |
| MALAT1 | Up-regulated | 6 | 36,37,48 |
| MaTARs | Up-regulated | 1, 6 | 55 |
| MEG3 | Down-regulated | 1, 3 | 194 |
| MVIH | Up-regulated | 1, 6 | 195 |
| NBAT1 | Down-regulated | 1,6 | 196 |
| NBR2 | Down-regulated | 7 | 197 |
| ncRAN | Up-regulated | 1, 6 | 198 |
| NEAT1 | Up-regulated/Down-regulated | 3 | 49,50,51 |
| NORAD | Down-regulated | 9 | 82 |
| PANDA | Up-regulated/Down-regulated | 4, 9 | 199 |
| PCAT-1 | Up-regulated | 1, 6 | 66,68 |
| PCAT-29 | Down-regulated | 1,6 | 200 |
| PCGEM1 | Up-regulated | 2, 3, 7 | 201 |
| PTENP1 | Down-regulated | 2 | 202 |
| PVT1 | Up-regulated | 1, 3, 6 | 60 |
| SAMMSON | Up-regulated | 7 | 203 |
| SChLAP1 | Up-regulated | 6 | 204 |
| TERRA | Down-regulated | 4 | 205 |
| TUG1 | Up-regulated | 1, 6 | 206 |
| THOR | Up-regulated | 1, 7 | 207 |
| u-Eleanor | Up-regulated | 1 | 208 |
| UCA1 | Up-regulated | 2 | 209 |
Hallmarks of cancer:
(1) sustained proliferative signaling, (2) insensitivity to growth suppressors, (3) evasion of apoptosis, (4) replicative immortality, (5) induced angiogenesis, (6) tissue invasion and metastasis, (7) abnormal metabolic pathways, (8) immune evasion, (9) genomic instability, (10) inflammation [16,17].
LncRNAs as Oncogenes
H19
H19, one of the first lncRNAs described to be overexpressed in a wide range of cancer types including hepatocellular carcinoma, colorectal cancer and breast cancer, is a paternally imprinted gene, initially found to be expressed in embryonic tissues during mouse development and silenced in most tissues at birth [15,16]. Loss of imprinting leads to H19 re-expression which correlates with many steps of tumorigenesis as shown using mouse models and human cell lines [17,18]. Transcription of H19 is controlled in part by the tumor suppressor and master cell cycle regulator p53, as well as by the ubiquitous oncogene MYC. Loss of functional p53 or up-regulation of MYC in various cancers correlates with increased H19 expression [11,19]. In the absence of wild type p53, H19 can also be up-regulated by hypoxic stress through hypoxia-induced factor 1α (HIF1-α) [20]. Analysis of TCGA data has revealed increased levels of H19 in colorectal and stomach cancer, but not in other types of cancer [21].
However, overexpression of H19 cDNA has also been shown to lead to decreased tumorigenicity of human rhabdoid tumor cell lines in vivo [22]. In addition, in an Apc mouse model of colorectal cancer, H19 knockout mice were reported to develop more polyps and a faster onset of tumorigenesis than wild type mice, revealing a tumor suppressive role [23,24]. Such divergent outcomes based on whether H19 can act as an oncogene or as a tumor suppressor might be explained by the heterogeneity in the genetic dependencies of tumors, the use of different model systems, (i.e. transgenic mouse models compared to human cancer cell lines), or alternatively, might potentially reflect a dual, context-dependent role of H19 in tumorigenesis, although this remains to be tested.
Several different pathways have been proposed to explain how H19 can impact tumor progression. H19 is a precursor for microRNA (miR)-675 [24], shown to target the tumor suppressor retinoblastoma (RB) protein in colorectal cancer. In this model, elevated levels of H19 lead to increased expression of miR-675 and decreased expression of RB, in turn promoting cell proliferation in colorectal cancer cell lines [25]. There is mounting evidence that H19, in addition to being a precursor for miR-675, can function as a competitive endogenous RNA (ceRNA) for several different miRNAs [26]. In general, ceRNAs are lncRNAs that can compete with mRNAs for binding of common miRNAs, sequestering miRNAs from the cellular pool [26]. H19 has been proposed to act as a “sponge” for many different miRNAs, such as let-7 family members, miR-200 family members (including miR-141), as well as miR-138, miR-630, miR-138 and miR200a [25,27]. This model of action is consistent with the predominantly cytoplasmic localization of H19 [28].
It is reasonable to speculate that elevated levels of H19/miR-675 in various cancers likely contribute to increased cancer cell proliferation and successful tumor metastasis. Hence, targeting H19 may be an interesting therapeutic approach in a distinct set of cancer types where this lncRNA may act as an oncogene.
HOTAIR
HOX antisense intergenic RNA (HOTAIR) is transcribed from the HOXC locus during normal development. Early studies showed that HOTAIR acts in trans by recruiting the Polycomb repressive complex 2 (PRC2), the LSD1 and the CoREST/REST H3K4 demethylase complex to their target genes [29]. However, recent studies raise controversy with regard to its function and target genes during this time period [30]. HOTAIR can function as an oncogene in multiple tumor types including breast, lung, liver, pancreas, and colorectal promoting tumor invasion and metastasis, but can also act as an independent predictor of patient survival rates [31–33]. Therefore, HOTAIR may prove to be a valuable therapeutic cancer target, but this remains to be tested.
LUNAR1
Another example of a potential oncogenic lncRNA is the Leukemia-induced Non-coding Activating RNA 1 (LUNAR1), originally identified in ten T-ALL (T cell acute lymphoblastic leukemia) patients harboring a mutation in NOTCH1 [34]. The LUNAR1 gene is located adjacent to the insulin-like growth factor receptor 1 (IGF1R) gene, previously implicated in T-ALL and is a NOTCH1 target itself [35]. Knockdown of the nuclear-retained LUNAR1 using either shRNAs or antisense oligonucleotides (ASOs) has been found to lead to repression of IGF1R as well as reduced cell growth of human T-ALL cells, both in vitro and in a mouse xenograft model [34]. Ectopic expression of IGF1R rescued the growth defect, indicating that LUNAR1 could regulate T-ALL cell growth by activating IGF1R expression in cis [34]. LUNAR1 can bind the intronic IGF1R enhancer element, recruiting the Mediator complex, and resulting in full activation of the IGF1R promoter [34]. Thus, targeting LUNAR1 therapeutically may be a viable alternative to targeting IGF1R, when aiming to attenuate the downstream effects of aberrant NOTCH signaling in T-ALL cells [34], but further testing will be required.
MALAT1
The highly conserved Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1) lncRNA was originally identified as being over-expressed in human lung tumors exhibiting a higher propensity for metastasis [36] and loss of MALAT1 in a lung cancer xenograft mouse model resulted in reduced metastasis [37], establishing MALAT1 as an oncogene. Subsequently, increased MALAT1 expression has been shown to be associated with tumorigenesis in different types of cancer (reviewed in [38]) including breast cancer. Human MALAT1 expression has been found to be up-regulated 3–4 fold in breast tumors, as well as in different human breast tumor cell lines, compared to normal tissues/cells, respectively [39]. In addition, MALAT1 gene mutations frequently occur in luminal type breast tumors [1,40].
In normal tissues, MALAT1 is an abundant ubiquitously expressed lncRNA, post-transcriptionally processed into a long ~6.7kb nuclear retained transcript and a tRNA-like small RNA, and which translocates into the cytoplasm [41]. Multiple mechanisms of action have been proposed for MALAT1 including regulation of pre-mRNA splicing, and its knockdown in human cells can result in cell cycle arrest [42,43] or activation of E2F target genes by relocating them from polycomb bodies to transcriptionally active nuclear sites in a serum-dependent manner [44].
Malat1 knockout mice exhibit a normal phenotype: they are fertile and no major changes in gene expression or pre-mRNA splicinghave been noted, indicating that Malat1 is dispensible for normal mouse development and viability [45–47]. However, genetic knockout or ASO knockdown of Malat1 in the MMTV-PyMT mouse model of breast cancer, has resulted in highly differentiated primary tumors and nearly 80% reduction in lung metastasis [48]. Therefore, Malat1 likely functions in a context-dependent manner to play a critical role in regulating gene expression at both the transcriptional and post-transciptional levels in various cancers. The lack of a phenotype upon loss or knockdown of Malat1 in “normal” tissues/cells is likely due to its redundancy, which may be absent or “weak” in a cancer context, although this has not been directly demonstrated. Nevertheless, together, these studies indicate that MALAT1 may constitute a critical player in tumor progression, and its therapeutic knockdown may represent an excellent option to impact metastatic disease [48].
NEAT1
The Nuclear enriched abundant transcript 1 (NEAT1) is another lncRNA that has been implicated in tumorigenesis [49–51]. NEAT1 is required for paraspeckle formation in the nucleus and has been shown to mediate nuclear retention of Interleukin 8 (Il8) mRNA in virus infected mouse cells [47,52–54]. Several groups have now shown that NEAT1 is a p53 target gene and enables tumorigenesis in vivo by promoting the survival of oncogene expressing cells such as activated Kras in genetically engineered mouse models [51]. Furthermore, high NEAT1 expression has been reported to correlate with poor prognosis in different types of human cancers including glioma, ovarian cancer, hepatocellular carcinoma, melanoma and prostate cancer [50,51,54]. NEAT1 upregulation in human prostate cancer cells also confers resistance to androgen receptor (AR) antagonists, leading to a poorer disease outcome [50]. However, a recent report suggested that NEAT1 may also suppress transformation in p53 mutant cells [49]. Specifically, loss of NEAT1 increased tumorigenicity in a p53 null/mutant Kras mouse model of pancreatic ductal adenocarcinoma [49], suggesting that similar to H19, NEAT1 can elicit a context-specific function in either promoting or suppressing tumor formation; thus, future assessment is warranted.
MaTARs
Our laboratory recently performed an RNA-seq screen comparing mammary tumor cells to normal mammary epithelial cells from mouse models of mammary cancer. We identified and characterized 30 lncRNAs dubbed Mammary Tumor Associated RNAs (MaTARs) [55]. 21 MaTARs were significantly overexpressed in human breast cancer based on TCGA data analysis, often dependent on hormone receptor status [55]. Independent ASO-mediated knockdown of 20 MaTARs resulted in reduced mouse mammary tumor cell proliferation, invasion and/or tumor organoid branching [55]. The process of organoid branching is driven by both collective cell migration and cell proliferation -- two essential characteristics of tumor progression; thus, organoids represent a powerful 3D ex vivo model system to study potential oncogenes [56]. In addition, some MaTARs seem to play important roles in other cancer types: MaTAR1 (aka LINC00461 or ECONEXIN) has been found to act as an oncogene in glioma [57]. Human EXONEXIN/MaTAR1 was proposed to act as a sponge for miR-411-5p, in turn regulating topoisomerase 2 alpha in human glioma cell lines [57]. Inhibition of the putative oncogene using siRNAs resulted in decreased cell proliferation [57]. Ongoing studies aim to reveal the molecular mechanism(s) of action of this and additional MaTARs, investigating their potential as candidate therapeutic targets for various cancers.
LncRNAs regulating oncogenes
The most frequent somatic copy-number amplification in cancer is the gain of the oncogenic transcription factor MYC [58]. Several lncRNAs have been implicated in the regulation of MYC expression, such as PVT1, PCAT1, CCAT1 and CCAT2. As attempts to target MYC directly in tumors have not been successful in the past, these regulatory lncRNAs may provide exciting new targets to modulate MYC expression or MYC activity indirectly.
PVT1
The Plasmacytoma Variant Translocation 1 (PVT1) gene is located adjacent to MYC on human chromosome 8q24 and is co-amplified with MYC in 98% of cancers [59]. PVT1 lncRNA stabilizes MYC protein in a post-transcriptional manner, resulting in increased cell proliferation and tumorigenicity in cells with MYC amplifications [60]. Recent studies suggest that the generation of PVT1 fusion proteins resulting from DNA rearrangements that can contribute to tumorigenesis, as in the case of the chimeric gene PVT1-WWOX in multiple myeloma [61]. WWOX (WW Domain Containing Oxidoreductase) has been described as a tumor suppressor implicated in mediating apoptosis in several cell lines [62]. PVT1 fusion transcripts are frequently found in other cancer types, such as medulloblastoma, where the fusion genes result from a chromothripsis-like mechanism [63]. The PVT1 locus also harbors a cluster of six miRNAs [64,65]; reduced expression of one of them, miR-1204 in a medulloblastoma cell line, decreased cell proliferation to similar levels to those achieved via siRNA-mediated knockdown of MYC. Of note, this anti-proliferative effect was only observed in cell lines with PVT1-MYC fusions [63]. The exact relationship between PVT1 and c-MYC remains to be further investigated for devising optimal therapeutic strategies targeting PVT1.
PCAT-1
The Prostate Cancer Associated Transcript 1 (PCAT-1) was identified as an activator of cell proliferation in prostate cancer [66] and subsequently detected in several other types of cancer. For example, high levels of PCAT-1 have been found to correlate with distant metastasis and lower patient survival rates in colorectal cancer [67]. PCAT-1 in human prostate cancer cell lines modulates the transcription of cell cycle related genes, potentially acting as a transcriptional repressor in a complex with the polycomb repressive complex 2 (PRC2) [66]. PCAT-1 has also been shown to function in the cytoplasm, where, it acts as a ceRNA, abrogating the binding of miR34-1 to MYC transcripts, which results in increased levels of MYC protein in human prostate cancer cells [68]. In this regard, PCAT-1 may act similarly to PVT1 by stabilizing MYC protein post-transcriptionally, albeit presumably, via a different mechanism. PCAT-1 has also been implicated in DNA damage repair by reducing the RNA levels of BRCA2-- an important tumor suppressor-- in a post-transcriptional manner [69]. The same study also demonstrated increased sensitivity of PCAT-1 over-expressing cells to Poly ADP Ribosyl Phosphatase (PARP) inhibitors, commonly used to treat tumors harboring mutations in BRCA1 or BRCA2 [69]. Thus, PCAT-1 expression might serve not only as prognostic marker but also potentially, as an indicator of PARP sensitivity. These possibilities merit further investigation.
CCAT1
The expression of Colon Cancer Associated Transcript 1 (CCAT1) [70], also known as CARLo-5 [71] or onco-lncRNA-40 [72]) correlates with the cancer-associated variant rs6983267, located within the human 8q24 MYC super-enhancer region, and associated with increased cancer susceptibility [71]. CCAT1 knockdown in vitro using siRNAs resulted in decreased colon cancer cell proliferation due to G1 cell cycle arrest, caused by up-regulation of CDKN1A/p21. Injection of these siRNA-transfected cells resulted in prolonged tumor-free survival in mouse xenografts, implicating CCAT1 in tumorigenesis [71]. The exact mechanism of action is not well understood for this lncRNA. Of note, CCAT1 bears potential as a colorectal cancer biomarker as it can be detected in all stages of tumorigenesis from pre-malignant lesions to metastasis and is detectable in the peripheral blood of 40% of patients with colorectal cancer [70,73]. In addition to colon cancer, CCAT1 is also up-regulated in prostate and lung cancer [71,72]. Recently, CCAT1 has been described to be highly sensitive to BET (bromodomain and extraterminal) protein inhibitors such as JQ1, and was proposed to be a clinically relevant biomarker for patients who might benefit from treatment with BET inhibitors in colorectal cancer [74].
CCAT2
Like CCAT1, the Colon Cancer Associated Transcript 2 (CCAT2) is overexpressed in colorectal cancer, located upstream of MYC and is associated with the single nucleotide polymorphism (SNP) rs6983267 [75]. CCAT2 overexpression has been found to be growth promoting in xenografted tumors, leading to a higher number of liver metastases in mice relative to controls [75]. CCAT2, predominantly located in the nucleus, has also been shown to induce chromosomal instability [75]. Furthermore, this lncRNA has been reported to increase MYC expression by enhancing Wnt signaling through the transcription factor TCF7L2, possibly due to direct interactions between CCAT2 and TCF7L2 [75]. Lastly, up-regulation of CCAT2 has also been detected in other types of cancer such as human breast, lung, gastric and esophageal tumors [76] suggesting that this lncRNA might have pro- tumorigenic properties in several cancers.
LncRNAs as tumor suppressors
Not all cancer-associated lncRNAs are genomically amplified or over-expressed. Loss-of-function models have identified an increasing number of lncRNAs that can act as tumor suppressors, whereby inactivation contributes to tumor onset and/or drives tumor progression. While these lncRNAs may not be immediate targets for currently available therapeutic interventions, it is important to understand their function in the context of major cancer signaling pathways, as well as their value as putative prognostic markers. Future therapeutic advances may enable re-activation of tumor suppressors including lncRNAs.
GAS5
The Growth Arrest-Specific Transcript 5 (GAS5) was first identified as one of six genes preferentially expressed in growth-arrested mammalian cells [77]. GAS5 is one of the most highly expressed lncRNAs, present in all human tissues and, similarly to H19, is involved in embryogenesis [78]. Its expression is significantly reduced in a variety of cancer types such as breast, prostate, bladder, gastric, colorectal, pancreatic and cervical cancer [79]. In addition, GAS5 expression is inversely correlated with clinico-pathological characteristics such as tumor size, staging or metastasis [79]. Furthermore, overexpression of GAS5 in xenografted breast cancer cell lines in nude mice has been reported to inhibit breast tumor growth in vivo by inducing cell cycle arrest and apoptosis, further supporting its role as a tumor suppressor [79]. On a molecular level, GAS5 acts as a decoy for the glucocorticoid receptor (GR), thereby suppressing GR-dependent gene regulation in HeLa and HepG2 cell lines [80]. GAS5 can bind other steroid hormone receptors such as androgen and progesterone receptors which play important roles in hormone-dependent cancers [81].
NORAD
The Non-coding RNA Activated by DNA damage (NORAD) transcript is induced in response to DNA damage, and was originally identified in a doxorubicin treatment screen in colon cancer cell lines [82,83]. This lncRNA is expressed in a p53-dependent manner upon DNA damage in colon cancer cells [82,83]. NORAD is ubiquitously expressed, very abundant and highly conserved [82]. As a cytoplasmic lncRNA it functions as a decoy for the RNA-binding proteins PUMILIO 2 (PUM2) and, to a lesser extent, PUMILIO 1 (PUM1) in colorectal carcinoma cells [82,83]. These proteins stimulate deadenylation and decapping of target mRNAs, leading to post-transcriptional gene repression [84] and loss of NORAD releases PUMILIO proteins, thus causing repression of their target mRNAs, many of them encoding proteins involved in mitosis, DNA replication and DNA repair [84]. Hence, hyperactivity of PUM1/2 in NORAD knockout cells leads to chromosomal instability, a phenotype frequently observed in the course of tumor progression [82,85]. Furthermore, the increased aneuploidy observed upon NORAD loss suggested that this lncRNA could act as a tumor suppressor [82].
LncRNAs that regulate tumor suppressors
Several lncRNAs can impact important regulators of the cell cycle, such as p21 or p53. These lncRNAs can thus act as oncogenes by regulating essential tumor suppressors in the cell.
ANRIL
The INK4B-ARF-INK4A locus on human chromosome 9p21 is a cluster of well-known tumor suppressor genes, frequently deleted in cancer [86,87]. This locus encodes p15INK4B and p16INK54A, two cyclin-dependent kinase inhibitors, as well as ARF, a regulator of the p53 pathway. This genomic region also harbors the Antisense Noncoding RNA in the INK4 locus” (ANRIL, also known as p15-AS or CDKN2B-AS1), originally identified in familial melanoma, and has since been reported as an oncogene in gastric, breast, lung, bladder and hepatocellular cancer, among others [88,89]. Expression of the INK4B-ARF-INK4A locus is tightly regulated by Polycomb group protein complexes (PcG) (reviewed in [89]). ANRIL recruits chromobox 7 (CBX7), a component of the Polycomb repressive complex 1 (PRC1), to the INK4B-ARF-INK4A locus, contributing to its epigenetic repression in prostate cancer cells [90]. Thus, the oncogenic activity of ANRIL might be explained by its transcriptional repression of tumor suppressor genes in cis. Knockdown of ANRIL resulted in reduced cell proliferation, migration and/or invasion and promoted apoptosis in vitro in many epithelial cancers. But in addition, therapeutic inhibition of ANRIL released repression of the INK4B-ARF-INK4A locus, thus re-activating the expression of the tumor suppressor [89].
LincRNA-p21
Similarly to NORAD, expression of lincRNA-p21 can be induced upon DNA damage in a p53-dependent manner [91]. LincRNA-p21, located 16.7 kb upstream of the tumor suppressor gene p21 (CDKN1A), is transcribed in an antisense direction [12]. LincRNA-p21 expression has been associated with tumor stage and invasion status in colorectal cancer [92]. Moreover, it can act as a transcriptional repressor in trans by binding to hnRNP-K [12], and it can also enhance p53 transcriptional activity by binding to Mouse Double Minute (MDM2) in cancer cells [93]. Early studies of lincRNA-p21 did not detect an effect of lincRNA-p21 expression on its neighboring gene p21, or on the cell cycle in general [12]. However, a recent study using LincRNA-P21 LoxP conditional knock-out mice revealed that it could predominantly functionby activating p21 in cis and compromising the G1/S cell cycle checkpoint; however, it did not seem to affect apoptosis or to regulate target genes in trans [91]. This difference might be based on the use of RNAi-based knockdown methods in earlier studies compared to a genetic deletion, highlighting the importance of complementary approaches to alter and assess lncRNA function. In addition, lincRNA-p21 has been reported to interact with HuR and repress the translation of β-catenin (CTNNB1) and JunB in breast cancer cells [94]. Regulation at both transcriptional and post-transcriptional levels is consistent with the localization of lincRNA-p21, found in the nucleus as well as the cytoplasm [94]. Furthermore, lincRNA-p21 has been shown to be induced by hypoxia and involved in hypoxia-induced glycolysis in mouse embryonic fibroblasts, suggesting a role of this lncRNA in the regulation of the Warburg effect in cell metabolism [95]. In summary, current data suggests that lincRNA-p21, among various roles, is involved in fine-tuning the transcriptional regulatory network of p21 and/or p53, two well-studied tumor suppressors.
Several other lncRNAs, such as PANDA (Table 1) and PINT are induced by p53 and can either enhance the p53 transcriptional response in the cell or antagonize it [96,97]. Consequently, therapeutic targeting of lncRNAs within these transcriptional networks rather than the cell cycle regulators themselves may provide a viable alternative to overcome the limitations of targeting those proteins.
Therapeutic targeting of lncRNAs
Noncoding RNAs play a significant role in cancer pathogenesis [98]. Given the diversity in their potential modes of action, as described earlier, lncRNAs can be targeted by multiple approaches (Key Figure, Figure 2): i) Post-transcriptional RNA degradation pathways can knockdown pathogenic RNAs. This can be achieved by using siRNAs that will invoke a Dicer and Argonaute dependent cleavage pathway. Alternatively, antisense oligonucleotides (ASOs) with chemical modifications can be used to target the RNA of interest for degradation via an RNAse H dependent mechanism.; ii) Modulation of lncRNA genes by steric blockade of the promoter or by using genome-editing techniques; iii) Finally, one can also achieve loss of function by creating steric inhibition of RNA-protein interactions or preventing secondary structure formation. RNA binding small molecules, or ASOs, can be used in this case. Here, we describe examples of current advances in each of the above-mentioned strategies to target lncRNAs, as well as their potential therapeutic applications.
Key Figure, Figure 2.

Therapeutic targeting of human lncRNAs
The schematic summarizes different approaches to target lncRNAs in the nucleus and cytoplasm.
a) Transcriptional inhibition can be attained through classical CRISPR/Cas9 to delete regions of interest in lncRNA loci. Alternatively, dead-Cas9 fused to a repressor complex can inhibit transcription of lncRNA genes.
b) Transcriptional upregulation of tumor suppressors can be attained through knockdown of the corresponding Natural Antisense Transcripts (NATs). NATs can be targeted post-transcriptionally using ASOs.
c) ASOs can also be used to post-transcriptionally knockdown lncRNAs that are overexpressed in cancers.
d) Post-transcriptional silencing can also be achieved by siRNAs targeting lncRNAs. SiRNAs stimulate Dicer activity in the cytoplasm and recruit RISC complex (RNA Induced Silencing Complex) to post-transcriptionaly degrade target RNAs
e) Steric inhibition of lncRNA-protein interactions can be achieved using small molecules, morpholinos, or uniformly-modified ASOs that cannot stimulate an RNA degradation pathway.
Post-transcriptional targeting of lncRNAs
Post-transcriptional targeting provides a straightforward means of impacting any RNA of interest. The highly selective nature of RNA-RNA or RNA-DNA duplex formation has enabled investigators to explore the potential of oligonucleotide-based therapeutics [99,100]. These nucleic acid based drugs can target any unique region of the human transcriptome and importantly, provide the ability to impact the ‘undruggable’ portions of the genome. This is currently impossible with small molecules or antibody based drugs, which have a much smaller repertoire of molecules and targets [101,102]. Currently there are two major approaches employing nucleic acid therapeutics; double stranded RNA-mediated interference (RNAi) and single stranded antisense oligonucleotides (ASOs) (see below). Newer generation nucleic acid therapeutics have demonstrated improved stability, higher efficacy and result in significantly reduced off-target effects, leading to the generation of drugs at various stages of clinical development for a number of diseases, including malignancies [103–105]
RNAi
The discovery of the RNA interference (RNAi) pathway in Cenorhabditis elegans followed by the demonstration of specific knockdown of target RNAs using exogenous double-stranded RNA ignited the field of RNAi [106–108]. Addition of a double stranded small interfering RNA engages a degradation pathway that involves Dicer, an RNaseIII enzyme and a multiprotein complex RISC (RNA induced silencing complex) along with the endonuclease Argonaute 2 (Ago2) [109]. RNAi is also active in human cells and this has led to investigating the use of synthetic siRNAs to target RNAs in human cells and mouse models [110,111]. The first report of a successful in vivo study used unmodified siRNAs to knockdown FAS mRNA in a mouse model of fulminant hepatitis [112]. Since then, numerous studies have demonstrated the successful use of siRNAs against target mRNAs for different pathological conditions such as cancer, neurological diseases, and metabolic disorders [113]. Several pharmaceutical companies including Alnylam Pharmaceuticals, siRNA Therapeutics, and miRNA Therapeutics have pioneered the development of siRNA and miRNA based therapies [114]. Since double stranded RNAs are inherently susceptible to nucleases, they require additional chemical modification to prevent them from serving as substrates for subsequent enzymatic degradation pathways [115]. Advances in chemical modifications such as 2′-O methyl (2′-O-Me) sugar residues and phosphothioate linkages in the 3′-end of the RNA have improved the pharmacological properties of siRNA-based drugs [116].
Several lncRNAs have been knocked down using traditional siRNAs in cell lines [115,116]. However, in vivo experiments using siRNAs have been quite challenging. This is in part due to the lack of efficient delivery methods and limited bioavailability of siRNAs in animals [117,118]. MicroRNAs were the first non-coding RNAs that were pharmacologically targeted using RNA based therapeutics (reviewed in [119]). However, pre-clinical studies using siRNAs/shRNAs to target lncRNAs are very limited. siRNAs directed against MALAT1 in human prostate cancer cell lines resulted in inhibition of cell growth, invasion, migration, and induced cell cycle arrest [120]. siRNA-mediated knockdown of HOTAIR inhibited matrix invasion in human breast cancer cell lines [32]. Furthermore, subcutaneous injection of human gastric cancer cell lines transfected with HOTAIR shRNA, inhibited engraftment efficiency in nude mice [31]. In a recent shRNA-based screen in a mouse model of leukemia, several lncRNA species were identified as essential for leukemia maintenance, and revealed that a number of lncRNAs can act by promoting leukemia stem cell signatures [121].
Antisense oligonucleotides (ASOs)
ASOs bind to RNA via standard Watson-Crick base pairing. Zamecnik and Stephenson first demonstrated that a short 13mer single-stranded DNA targeting Rous sarcoma virus RNA could inhibit viral replication in cell lines [122,123]. Currently, ASOs targeting different mRNAs have entered clinical trials for several diseases including cancer [124]. They are also emerging as a promising therapeutic approach for targeting lncRNAs [48]. Upon binding to their target RNA, ASOs can inhibit or alter gene expression via steric hindrance, splicing alterations, initiation of target degradation, and/or other events. The evolution of ASO chemistry has clearly contributed to their clinical success in various scenarios. The newer generation of ASOs consist of 15–20 nucleotides with a backbone modification containing phosphothioate linkages [125]. The phosphothioate increases the resistance to degradation by cellular nucleases [126,127]. However, upon binding to their target, these ASOs are able to invoke an RNase H dependent cleavage mechanism resulting in an endo-nucleolytic cleavage of the target RNA [127]. In addition to the phosphothioate modification, chemical modification of the sugar backbone especially in the 2′ position (2′-O-Methoxyethyl) has conferred drug-like properties to ASOs [128]. These second generation ASOs show improved binding affinity and sustained pharmacokinetics [129]. While all the sugars in the backbone can be modified resulting in ‘uniformly modified ASOs’, these ASOs demonstrate very low or no RNAse H activity and cannot be utilized for RNA knockdown as the modified bases are resistant to RNAse H cleavage [130]. However, these are effective as splice switching ASOs and can be used to modulate splicing patterns of target RNAs by blocking splicing enhancers or repressor binding sites [103]. The first clinically approved splice switching ASO drug Spinraza™ (nusinersen, Biogen Inc) is based on this technology and is used for correcting a splice switch in the SMN2 (survival of motor neuron 2) gene in the CNS of patients suffering from Spinal Muscular Atrophy (SMA) [103,131].
In order to achieve RNAse H-mediated knockdown of target RNAs, chimeric ASOs referred to as ‘gapmers’ are used. Gapmer ASOs are RNA-DNA-RNA hybrids, where RNA residues contain a 2′-O-MOE modified sugar backbone [130]. Kynamro® (mipomersen sodium) is the first FDA approved drug used to knockdown APOB-100 mRNA to treat familial hypercholesterolemia [132,133]. However, there have been reports of liver toxity associated with Kynamro® usage and it is not clear if this is sequence dependent [132]. The gapmer chemistry coupled with various sugar modifications have given rise to much improved drug-like attributes to ASOs [134]. One of the major advances in ASO chemistry is the development of locked nucleic acids (LNA) and S-constrained ethyl (cEt) modifications, which have demonstrated improved potency and promising pharmacokinetic profiles that have advanced to clinical trials for various cancer related and neurological diseases [135–137]. cEt gapmers for STAT3 and Androgen receptor developed by Ionis Pharmaceuticals have entered phase II clinical trials [104,138]. While ASO-mediated knockdown works for cytoplasmic RNAs, ASOs also function effectively in the cell nucleus. This is due to the fact that the major effector nuclease RNase H is enriched in the nucleus [127]. A large number of lncRNAs are enriched in the nucleus [8], hence, ASOs represent an ideal approach for achieving significant lncRNA knockdown.
Our laboratory has pioneered the use of cEt gapmers to target Malat1 lncRNA in various models of breast cancer. Subcutaneous delivery of Malat1 ASO in the MMTV-PyMT mouse model of luminal B breast cancer resulted in differentiation of primary tumors and a nearly 80% reduction in metastasis relative to nonspecific ASO treated control mice [48]. Further, Malat1 knockdown using ASOs also reduced branching morphogenesis in a 3D organoid model derived from MMTV-PyMT tumors and a Her2 amplified mouse mammary tumor model [48]. Apart from breast tumors, MALAT1 ASOs have also been shown to elicit a potent anti-metastatic response in a lung cancer xenograft model. Specifically, systemic knockdown of MALAT1 in nude mice intravenously injected with human lung cancer cells reduced homing of the cells to the lungs by more than 70% relative to control ASO injected mice [37]. These results suggest that MALAT1 ASO may emerge as a potential therapeutic for metastatic disease in several cancer types, but further assessment will be needed [139]. In addition, as discussed earlier, numerous MaTARs, are also susceptible to ASO mediated knockdown in ex vivo organoid models and show a significant anti-tumor response [55,56]. Ongoing pre-clinical studies in patient-derived xenograft models as well as in patient-derived tumor organoids are focusing on moving these studies towards possible clinical trials (Box 1).
Box 1. Useful Pre-clinical Models for LncRNA Research.
The biggest challenge in lncRNA research is the lack of conservation for many lncRNA species. It can occur at different levels: primary sequence conservation, small blocks of sequence conservation, syntenic conservation, and in some instances, structural conservation. Some cancer-associated lncRNAs such as MALAT1[45], NEAT1 [49], H19 [18] are well conserved. However, most human cancer associated lncRNAs do not exhibit sequence conservation across mammalian species. This lack of conservation impedes the translation of lncRNAs that are identified in human cancer to pre-clinical mouse studies for therapeutic targeting. Thus, models can be used for pre-clinical studies assessing lncRNA targeting.
Genetically Engineered Mouse Models (GEMMs)
GEMMs can be used to study all aspects of tumor progression including transformation and tumor initiation in a genetically defined context (e.g. activation of an oncogene or knockout of a tumor suppressor). The lncRNA of interest is required to be evolutionarily conserved in mouse. Genetic knockouts of lncRNAs such as H19[22], MALAT1 [48], and NEAT1 [49,51] have been used in combination with other oncogene targeted GEMMs. RNA therapeutics targeting lncRNAs can also be used in combination with GEMM models [48].
Patient-Derieved Xenograft Models (PDX)
For lncRNAs that are restricted to humans or for which human orthologs have been identified, PDX models are a valuable resource for pre-clinical evaluation. PDX tumors transplanted into nude mice provide a means to evaluate tumor growth rate and therapeutic response upon targeting lncRNAs. PDXs have been used to translate lncRNA basic research to pre-clinical studies: HOTAIR [31,32] SAMMSON [203] etc. have been targeted using siRNAs or ASOs in breast and melanoma models, respectively. The caveat of this method is the inability to study tumor initiation and earlier events leading to transformation. In addition, most PDX models do not metastasize limiting the insight they provide.
Xenografted Human Cell Lines
Human cancer cell lines can be xenografted into nude mice in pre-clinical experiments. Cancer cell lines are readily accessible and can be simultaneously tested in a high throughput fashion. The potential caveat is that cell lines remain in culture for prolonged periods of time, bearing distinct genomic and transcriptomic profiles that can potentially introduce artifacts in readouts. LncRNAs such as HOTAIR [32],LUNAR[34], MALAT1 [37] etc., have been knocked down in xenografted human cell lines in breast cancer, T-ALL and lung cancer, respectively.
Patient-Derived Tumor Organoids
Derived from surgically resected tumors, they can serve as a powerful tool to manipulate lncRNA levels in a patient-specific manner. These organoids maintain tumor heterogeneity and grow in 3D culture on an extracellular matrix scaffold that preserves tumor-extracellular matrix characteristics. They can be used for both ex-vivo knockdown studies and can also be orthotropically transplanted into nude mice for in vivo therapeutic validation. An advantage is that they can be developed from corresponding normal tissue from the same patients for patient-specifc comparisons. A caveat of this method is that the culture conditions for patient-derived tumor organoids is rather complex and not all tumor types have thus far been efficientely established into 3D organoids. Tumor organoids derived from mouse mammary tumor models have been used to knockdown Malat1 and MATARsusing ASOs to study branching morphogenesis [48, 55].
Zebrafish Models
Recently, the use of zebrafish to study cancer progression has gained interest. The role of the lncRNA THOR in melanoma progression has been evaluated in a Zebrafish model [208]. It remains to be determined whether other lncRNAs can be similarly targeted.
Morpholinos
Morpholino oligonucleotides are nonionic DNA analogs originally used in developmental biology for loss of function studies in zebrafish [140,141]. They have also been used to modulate miRNA activity in a variety of organisms [142]. Morpholinos are generally 25 nucleotides long and are used to prevent translation or promote splice switching upon binding to target mRNAs/pre-mRNAs [141,143]. Morpholino ASOs can be chemically modified with methylenemorpholine rings replacing sugar moieties and contain non-ionic phosphorodiamidate linkages that substitute for the anionic phosphates of DNA or RNA [144]. The first clinically approved morpholino, Exondys 51 (eteplirsen, Sarepta Therapeutics, Inc) has been used to achieve splicing modulation of the dystrophin mRNA (DMD) in patients with Duchenne Muscular Dystropy [105,145,146]. However, pre-clinical studies targeting lncRNAs using morpholinos are very limited. Nevertheless, given the clinical success and antisense binding to targets, morpholinos may prove useful for steric interference between lncRNAs and their protein or DNA targets in other pathological conditions.
Limitations of nucleic acid based therapies
While there is immense enthusiasm surrounding nucleic acid based therapies for various disorders including cancers, there are some caveats associated with such strategies. Crossing the cellular plasma membrane is the first and foremost issue. Additionally, the presence of cellular nucleases and the innate immune response to foreign RNAs, such as via Toll-like receptor (TLR) and retinoic acid inducible gene-I like (RIG-I) RNA helicase pathways, may pose a substantial block in effective cellular uptake of these molecules [147,148]. Additionally, entrapment of synthetic ASOs in the endosomal compartment can significantly reduce the bioavailability of these molecules [117,149,150]. Finally, it is critical to ensure that the oligonucleotides possess minimal to no off-target effects or toxicity. Due to the aforementioned issues it has taken more than 20 years from the initial discovery of antisense therapeutics [122, 123] to the initial therapeutic successes [103, 132]. The pharmacological evolution of newer generation chemistries detailed above has played a critical role in overcoming many of these barriers including the ability to achieve “free-uptake”, increased stability, nuclease resistance, and extended pharmacokinetics. To overcome innate immune responses triggered by TLRs, ASOs are extensively screened to identify sequences that are well tolerated, and also by avoiding CpG motifs that might provoke an immune response [151,152]. A major advance in targeting has been the development of N-Acetylgalactosamide (GalNAc) conjugated siRNAs to achieve liver specific targeting (Alnylam Pharmaceuticals) [153]. Asialoglycoproteins such as GalNAc bind to the asialoglycoprotein receptor (ASGPR), and because hepatocytes express numerous copies of ASGPR on their cell membrane, effective uptake of such conjugated siRNAs can occur in the hepatic tissue at a very low dose [153]. Other conjugation methods include lipids such as cholesterol, Peptide Nucleic Acids (PNAs) and antibodies, among others [154,155]. Oligonucleotide conjugation is emerging as an exciting area of research aiming to extend the specific targeting obtained in hepatic tissues to other tissue types. However, it may require identification of tissue- or tumor-specific receptors or cell surface markers and substrates amenable for conjugation. Off target effects remain a potential issue that also needs to be thoroughly addressed. One must employ bioinformatics analyses to eliminate sequences with potential off-target matches. Additionally, with advancements in gene editing methods, one can score for off target effects by performing knockdowns in cells that are null for the target gene and evaluate any gene expression changes induced in the absence of the target RNA.
Modulation of lncRNA-expressing loci using CRISPR-Cas9
LncRNA genes, similar to protein-coding genes, are transcribed by RNA polymerase II. With recent advances in genome editing methods such as CRISPR-Cas9, it is possible to achieve transcriptional silencing of lncRNA expressing loci using CRISPR-interference (CRISPRi) [156,157]. In this approach dead-Cas9 is fused to transcriptional repressors and this fusion protein is targeted to a specific gene promoter by guide RNAs to achieve transcriptional silencing [158]. In a genome wide CRISPRi study, guide RNAs were developed to target the promoters of more than 16,000 lncRNAs in the human genome [159]. CRISPRi was used to selectively inactivate lncRNA genes in an array of seven human cell lines, including six cancer cell types and a line of induced pluripotent stem cells (iPSCs); approximately 500 lncRNAs were found to be essential for cancer cell growth. Many of these were essential in only one cell type, underscoring the fact that lncRNA function is highly specific for a given cell type [159]. These experiments indicate that transcriptional silencing of lncRNAs using CRISPR based approaches is feasible and will likely be used in the future for therapeutic targeting of these molecules at the transcriptional level [156,160]. Recently, the RNA targeting CRISPR-Cas13 system was identified [161], representing another promising approach to knockdown lncRNAs and which may prove to be useful for therapeutic development. Thus, lncRNAs implicated in blood related cancers such as PVT1, and LUNAR1, may be targeted more easily if CRISPR based approaches are considered [34,60]. While several pre-clinical studies are taking advantage of CRISPR-editing methods, it remains to be seen how well they will translate into clinical scenarios for cancer related illness.
Transcriptional upregulation by targeting Natural Antisense RNAs
The majority of current therapeutic strategies operate via inhibition or knockdown of target molecules. Yet, there is a considerable lack of therapeutic strategies to upregulate gene expression. Such up-regulation is critical for activating tumor suppressors in cancers and other critical growth factors or transcription factors whose expression confers a growth disadvantage to proliferating cells. Natural Antisense RNAs (NATs) are a subset of noncoding RNAs that overlap with protein coding genes and are transcribed in an antisense direction [162]. Overlapping NATs are found in a number of protein coding loci in the human genome [162]. The majority of such transcripts affect the transcription of their overlapping genes, thus acting in cis [163,164] Inhibition of cis acting antisense transcripts can result in up-regulation of the neighboring/overlapping protein-coding genes, suggesting that a significant number of NATs can act as repressors of the sense coding transcripts [163,164]. NATs are present near many critical tumor suppressor genes such as CDKN2B (ANRIL) [165] and CDKN1A (P21-AS) [91]. In several cancers, elevated expression of NATs suppress the expression of the corresponding tumor suppressor genes [166]. Thus, therapeutic inhibition of NATs using ASOs present a potential to upregulate tumor suppressors. These ASOs are known as ‘AntagoNATs’ which are single-stranded, chemically modified LNAs or other ASOs [164]. Currently, OPKO-CURNA and RaNA Therapeutics hold several AntagoNATs in various stages of pre-clinical and clinical development. These examples provide exciting possibilities of modulating lncRNA expression to exploit multiple facets of gene regulation and therapeutic interference.
Steric inhibition of lncRNA function
Accumulating evidence suggests that many lncRNAs manifest their function by binding to proteins or to integral components of protein complexes [167,168]. Many nuclear lncRNAs are bound to chromatin-modifying complexes such as members of the PRC complex, as described above. In these cases, the RNA will dock onto the protein surface which can be sequence or structure dependent [169]. Morpholinos and uniformly modified ASOs that cannot stimulate RNAse H activity can be used in such cases to bind specifically to the RNA, and block the RNA-protein interface resulting in loss of function [142].
Small molecules
LncRNAs, like other non-coding RNAs, can potentially form stable secondary and tertiary structures [170]. While computational predictions based on a covariance model suggest that very few lncRNAs have conserved structural features [171], in vivo chemical crosslinking studies have demonstrated secondary and/or tertiary structures for several lncRNAs [170,172,173]. With the development of RNA structure determination assays such as SHAPE and PARIS it is possible to precisely map the secondary and tertiary structure of lncRNAs [174–176]. Clinical trials are underway to target highly structured bacterial or viral riboswitches using small molecule inhibitors for bacterial and viral infections respectively [177]. Several human genetic disorders including Huntington’s disease, fragile X syndrome and myotonic dystrophy are caused by trinucleotide repeat expansions in RNA transcripts, which fold into stable RNA hairpin structures and result in impaired splicing and/or translation [172,178,179]. These disease-causing RNA elements represent a completely novel class of drug targets. It is thus reasonable to speculate that similar structured RNA elements might also be present in numerous pathogenic lncRNAs [175]. MALAT1 and NEAT1 are classic examples where the 3′ end of the transcripts fold into a unique triple helical structure [172,173,180,181]. Small molecule inhibitors targeting these unique structural elements in lncRNAs could potentially destabilize the transcript or allosterically interfere with protein binding to confer a therapeutic effect, although this remains to be tested.
Therapeutic manipulation of lncRNA promoters
Aside from the fact that lncRNAs themselves could serve as potential therapeutic targets, recent findings from clinical trials utilizing lncRNA promoters are providing encouraging insights. BC-819 (DTA-H19), is a double-stranded DNA plasmid carrying the gene for the A subunit of diphtheria toxin under the regulation of the H19 gene promoter, administered to elicit an anti-tumor response in various solid tumors (BioCancell Therapeutics Inc) [182]. Originally tested in a mouse bladder cancer model, BC-819 is now also being tested in non-small cell lung carcinoma, colon, pancreatic and ovarian cancers [183,184]. A phase I/IIa clinical trial in patients with invasive bladder cancer receiving intravesical BC-819 has reported mild, local toxicity along with complete and partial response rates of 22 and 44%, respectively [183]. Highly selective tissue expression of several lncRNAs such as H19, combined with the therapeutic responses in early stage clinical trials with BC-819, have created significant interest among scientists and clinicians, prompting the investigation of using tissue or cell-type specific lncRNA promoters to drive the expression of toxins to stimulate cytotoxic effects in disease-related cells.
Concluding Remarks
LncRNAs are emerging as important players in tumorigenesis. A detailed understanding of their expression and mechanisms of action is critical in order to appreciate the importance of this unique class of molecules. The fact that lncRNAs are generally expressed in a cell- or tissue-specific manner makes them exceptional therapeutic targets. Further, sequence-based nucleic acid therapeutics are evolving at a rapid pace. Identification and evaluation of a lncRNA target in the context of cancer may be translated into clinical scenarios in a reasonable time window. Although many questions and challenges remain to be addressed (see Outstanding Questions and Box 2), the increased success rate of nucleic acid therapeutics provides an exciting opportunity to explore lncRNAs as viable candidate therapeutic targets in cancer and other pathologies.
Outstanding Questions.
How many lncRNAs are functionally and clinically relevant for specific cancers? Is there a functional interplay between these lncRNAs?
How can we develop systematic genomic and functional approaches to better understand the role of lncRNAs in tumor initiation, progression and metastasis?
How can we best address the off-target effects of systemic nucleic acid based therapies?
What are the best preclinical models to study human lncRNAs?
How can we best integrate patient genomic and transcriptomic data to establish a lncRNA discovery pipeline to drive preclinical studies in animal models?
Box 2. Clinician’s Corner.
LncRNAs are emerging as critical players in cancer pathogenesis. Deep sequencing of patient tumor DNA and RNA have identified many lncRNAs that are up- or down-regulated in tumor tissues. Many mutations and recurrent CNVs in cancers map to non-coding regions of the genome.
An understanding of the molecular functions of several lncRNAs in different cancers is just emerging. Many lncRNAs have been shown to play a direct role in cancer cell proliferation, cancer progression and/or metastasis.
Tissue-specific expression of lncRNAs renders these exciting candidates for diagnostic marker development or as putative therapeutic targets for systemic treatment.
The ability to target lncRNAs at various functional levels provides a wide range of therapeutic options. Targeting approaches may include nucleic acid based drugs, small molecule inhibitors, and gene editing methods.
Nucleic acid based RNA targeting approaches are evolving at a rapid pace to potentially treat various pathologies. Recent clinical success and FDA approval of antisense drugs for Spinal Muscular Atrophy and Duchenne Muscular Dystrophy have led to various pre-clinical studies targeting lncRNAs using nucleic acid based therapies.
Further functional studies using appropriate pre-clinical models will validate the importance of several lncRNA species in cancer pathogenesis leading to a broad exploration of this class of molecules as viable therapeutic targets in many cancer types.
HIghlights.
Approximately 27% of annotated human genes encode lncRNAs.
Recent tumor genome sequencing efforts have identified several lncRNA loci that are deleted, amplified and/or mutated in various cancers.
Many lncRNAs are up- or down-regulated in cancers compared to respective normal tissues. Several lncRNAs have been shown to play a critical role in various aspects of cancer progression.
Tissue-specific expression of lncRNAs positions them as interesting potential therapeutic targets for a variety of pathologies.
Nucleic acid based therapeutics are emerging as a promising approach to target pathogenic lncRNAs. RNA targeting therapies have been clinically approved for several diseases.
Nucleic acid based therapeutics have shown success in several pre-clinical studies targeting lncRNAs in cancers.
Acknowledgments
Research in the Spector lab is funded by NCI 5P01CA013106 and NIGMS 42694. G.A. and S.D.D. are supported by Susan G. Komen postdoctoral fellowships. S.D.D. is also supported by NCI 1K99CA215362-01. We thank Mona S. Spector for helpful discussions and editorial assistance. D.L.S. is a consultant to, and receives research support from, Ionis Pharmaceuticals.
Glossary
- Aneuploidy
the presence of an abnormal number of chromosomes in a cell; a common feature of tumor cells.
- AntagoNATs
Short, chemically-modified, single-stranded oligonucleotides that interfere with the function of natural antisense transcripts (NATs).
- Apc mouse model
Mouse model of colorectal cancer, where the Apc (Adenomatous Polyposis Cancer) gene is mutated.
- Argonaute
Belongs to a family of RNaseH domain containing proteins. Is an essential component of the RNA-induced silencing complex (RISC), the key mediator of RNA interference.
- BET Inhibitors
A class of drugs that reversibly bind the bromodomains of Bromodomain and Extra-Terminal motif (BET) proteins and prevent their interaction with acetylated histones.
- Branching Morphogenesis
A developmental process that involves formation of tree-like structures directed by a gene expression program and tissue remodeling.
- Chromothripsis
Large scale genomic rearrangements occurring in a single event in confined genomic regions in one or a few chromosomes.
- Competing Endogenous RNA (CeRNA)
class of lncRNAs that regulate other RNAs by competing for shared miRNA binding sites and acting as a sponge to sequester miRNAs.
- CRISPR/Cas9
A genome editing technology that uses the bacterial nuclease Cas9 to direct specific editing events in mammalian genomes, guided by a short complementary RNA.
- Doxorubicin
Chemotherapeutic agent used for treatment against various cancers.
- Dicer
An RNAse III enzyme involved in cleavage of double-stranded RNAs and pre-miRNAs into short double-stranded RNA fragments.
- Duchenne Muscular Dystrophy
X- linked recessive disorder characterized by severe muscular weakness.
- Guide RNA
short stretch of RNA that can direct Cas9 nucleases to cut at a specific genomic location.
- Imprinted Gene
gene expressed from either the maternal or paternal allele
- Locked Nucleic Acid (LNA)
modified RNA nucleotide. The ribose sugar moiety of an LNA nucleotide is modified with an extra bridge connecting the 2′ oxygen and 4′ carbon.
- Mouse double minute 2 homolog (MDM2)
E3 ubiquitin-protein ligase; important negative regulator of the p53 tumor suppressor.
- Natural Antisense RNA (NATs)
subclass of non-coding RNAs that are transcribed antisense to a protein coding gene.
- Paraspeckle
dynamic sub-nuclear compartment thought to be involved in nuclear RNA retention.
- PARIS (Psoralen Analysis of RNA Interactions and Structures)
method to determine RNA-RNA and RNA- protein interactions at high resolution.
- Peptide Nucleic Acid (PNA)
synthetic chemically modified oligonucleotide analog where the nucleotides are linked to the backbone composed of repeating amino ethyl glycines.
- Polycomb Bodies
sub-nuclear compartment formed by accumulation of polycomb proteins that remodel chromatin and cause epigenetic silencing of genes.
- RNA-Seq (RNA sequencing)
Transcriptome-wide shotgun sequencing that uses a next-generation sequencing (NGS) approach to determine RNA complexity and abundance in cells.
- S-Constrained Ethyl (cET)
A modified RNA nucleotide similar to LNA containing a constrained ethyl substitution in the 4′ Carbon of the ribose sugar.
- SHAPE (Selective 2′-hydroxyl acylation analyzed by primer extension)
chemical assay to determine RNA secondary structure
- Somatic Copy Number Amplification
process by which clustered regions of the genome are repeated and the number of repeats in the genome varies between normal and diseased somatic cells.
- Spinal muscular atrophy (SMA)
rare genetic disorder characterized by loss of motor neurons and progressive loss of neuromuscular function.
- Toll-like receptors (TLRs)
class of proteins that play a critical role in the innate immune system, recognizing bacterial or viral RNAs.
- Uniformly modified ASO
Phosphothioate oligonucleotides in which the sugar residues in all the nucleosides are chemically modified to resist nuclease cleavage.
- Warburg effect
High rate of aerobic glycolysis accompanied by lactic acid fermentation in tumor cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nik-Zainal S, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khurana E, et al. Integrative Annotation of Variants from 1092 Humans: Application to Cancer Genomics. Science. 2013;342:1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143–159. doi: 10.1002/emmm.201100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer MK, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Research. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rinn JL, Chang HY. Genome Regulation by Long Noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergmann JH, Spector DL. Long non-coding RNAs: modulators of nuclear structure and function. Current Opinion in Cell Biology. 2014;26:10–18. doi: 10.1016/j.ceb.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Barsyte-Lovejoy D, et al. The c-Myc Oncogene Directly Induces the H19 Noncoding RNA by Allele-Specific Binding to Potentiate Tumorigenesis. Cancer Res. 2006;66:5330–5337. doi: 10.1158/0008-5472.CAN-06-0037. [DOI] [PubMed] [Google Scholar]
- 12.Huarte M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nature Publishing Group. 2013;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 15.Davis RL, et al. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- 16.Bartolomei MS, et al. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–155. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- 17.Berteaux N, et al. H19mRNA-like Noncoding RNA Promotes Breast Cancer Cell Proliferation through Positive Control by E2F1. Journal of Biological Chemistry. 2005;280:29625–29636. doi: 10.1074/jbc.M504033200. [DOI] [PubMed] [Google Scholar]
- 18.Gabory A, et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development. 2009;136:3413–3421. doi: 10.1242/dev.036061. [DOI] [PubMed] [Google Scholar]
- 19.Dugimont T, et al. The H19 TATA-less promoter is efficiently repressed by wild-type tumor suppressor gene product p53. Oncogene. 1998;16:2395–2401. doi: 10.1038/sj.onc.1201742. [DOI] [PubMed] [Google Scholar]
- 20.Matouk IJ, et al. The oncofetal H19 RNA connection: hypoxia, p53 and cancer. Biochimica et biophysica acta. 2010;1803:443–451. doi: 10.1016/j.bbamcr.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Weidle UH, et al. Long Non-coding RNAs and their Role in Metastasis. CGP. 2017;14:143–160. doi: 10.21873/cgp.20027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao Y, et al. Tumour-suppressor activity of H19 RNA. Nature. 1993;365:764–767. doi: 10.1038/365764a0. [DOI] [PubMed] [Google Scholar]
- 23.Gabory A, et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development. 2009;136:3413–3421. doi: 10.1242/dev.036061. [DOI] [PubMed] [Google Scholar]
- 24.Yoshimizu T, et al. The H19 locus acts in vivo as a tumor suppressor. Proceedings of the National Academy of Sciences. 2008;105:12417–12422. doi: 10.1073/pnas.0801540105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsang WP, et al. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis. 2009;31:350–358. doi: 10.1093/carcin/bgp181. [DOI] [PubMed] [Google Scholar]
- 26.Poliseno L, et al. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kallen AN, et al. The Imprinted H19 LncRNA Antagonizes Let-7 MicroRNAs. Molecular Cell. 2013;52:101–112. doi: 10.1016/j.molcel.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brannan CI, et al. The product of the H19 gene may function as an RNA. Mol Cell Biol. 1990;10:28–36. doi: 10.1128/mcb.10.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rinn JL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selleri L, et al. A Hox-Embedded Long Noncoding RNA: Is It All Hot Air? PLoS Genet. 2016;12:e1006485. doi: 10.1371/journal.pgen.1006485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Endo H, et al. Enhanced Expression of Long Non-Coding RNA HOTAIR Is Associated with the Development of Gastric Cancer. PLoS ONE. 2013;8:e77070. doi: 10.1371/journal.pone.0077070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta RA, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kogo R, et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71:6320–6326. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 34.Trimarchi T, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. 2014;158:593–606. doi: 10.1016/j.cell.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medyouf H, et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. The Journal of Experimental Medicine. 2011;208:1809–1822. doi: 10.1084/jem.20110121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji P, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 37.Gutschner T, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutschner T, et al. MALAT1 — a paradigm for long noncoding RNA function in cancer. Journal of Molecular Medicine. 2013;91:791–801. doi: 10.1007/s00109-013-1028-y. [DOI] [PubMed] [Google Scholar]
- 39.Guffanti A, et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163. doi: 10.1186/1471-2164-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellis MJ, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilusz JE, et al. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell. 2008;135:919–932. doi: 10.1016/j.cell.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tripathi V, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9:e1003368. doi: 10.1371/journal.pgen.1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tripathi V, et al. The Nuclear-Retained Noncoding RNA MALAT1 Regulates Alternative Splicing by Modulating SR Splicing Factor Phosphorylation. Molecular Cell. 2010;39:925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L, et al. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011;147:773–788. doi: 10.1016/j.cell.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2:111–123. doi: 10.1016/j.celrep.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eissmann M, et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012;9:1076–1087. doi: 10.4161/rna.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakagawa S, et al. Malat1 is not an essential component of nuclear speckles in mice. RNA. 2012;18:1487–1499. doi: 10.1261/rna.033217.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arun G, et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1lncRNA loss. Genes Dev. 2016;30:34–51. doi: 10.1101/gad.270959.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mello SS, et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017;31:1095–1108. doi: 10.1101/gad.284661.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakravarty D, et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun. 2014;5:5383. doi: 10.1038/ncomms6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adriaens C, et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nature Medicine. 2016;22:861–868. doi: 10.1038/nm.4135. [DOI] [PubMed] [Google Scholar]
- 52.Sunwoo H, et al. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Research. 2009;19:347–359. doi: 10.1101/gr.087775.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasaki YTF, et al. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc Natl Acad Sci U S A. 2009;106:2525–2530. doi: 10.1073/pnas.0807899106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imamura K, et al. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Molecular Cell. 2014;53:393–406. doi: 10.1016/j.molcel.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 55.Diermeier SD, et al. Mammary Tumor-Associated RNAs Impact Tumor Cell Proliferation, Invasion, and Migration. CellReports. 2016;17:261–274. doi: 10.1016/j.celrep.2016.08.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diermeier S, Spector D. Antisense Oligonucleotide-mediated Knockdown in Mammary Tumor Organoids. BIO-PROTOCOL. 2017:7. doi: 10.21769/BioProtoc.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deguchi S, et al. Oncogenic effects of evolutionarily conserved noncoding RNA ECONEXIN on gliomagenesis. Oncogene. 2017;36:4629–4640. doi: 10.1038/onc.2017.88. [DOI] [PubMed] [Google Scholar]
- 58.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shtivelman E, Bishop JM. The PVT gene frequently amplifies with MYC in tumor cells. Mol Cell Biol. 1989;9:1148–1154. doi: 10.1128/mcb.9.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tseng YY, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagoshi H, et al. Frequent PVT1 Rearrangement and Novel Chimeric Genes PVT1-NBEA and PVT1-WWOX Occur in Multiple Myeloma with 8q24 Abnormality. Cancer Res. 2012;72:4954–4962. doi: 10.1158/0008-5472.CAN-12-0213. [DOI] [PubMed] [Google Scholar]
- 62.Aqeilan RI, et al. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci U S A. 2004;101:4401–4406. doi: 10.1073/pnas.0400805101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Northcott PA, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beck-Engeser GB, et al. Pvt1-encoded microRNAs in oncogenesis. Retrovirology. 2008;5:4. doi: 10.1186/1742-4690-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huppi K, et al. The Identification of MicroRNAs in a Genomically Unstable Region of Human Chromosome 8q24. Molecular Cancer Research. 2008;6:212–221. doi: 10.1158/1541-7786.MCR-07-0105. [DOI] [PubMed] [Google Scholar]
- 66.Prensner JR, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29:742–749. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie X, et al. Long non-coding RNAs in colorectal cancer. Oncotarget. 2016;7:5226–5239. doi: 10.18632/oncotarget.6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prensner JR, et al. The Long Non-Coding RNA PCAT-1 Promotes Prostate Cancer Cell Proliferation through cMyc. Neoplasia. 2014;16:900–908. doi: 10.1016/j.neo.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prensner JR, et al. PCAT-1, a Long Noncoding RNA, Regulates BRCA2 and Controls Homologous Recombination in Cancer. Cancer Res. 2014;74:1651–1660. doi: 10.1158/0008-5472.CAN-13-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nissan A, et al. Colon cancer associated transcript-1: A novel RNA expressed in malignant and pre-malignant human tissues. Int J Cancer. 2012;130:1598–1606. doi: 10.1002/ijc.26170. [DOI] [PubMed] [Google Scholar]
- 71.Kim T, et al. Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proceedings of the National Academy of Sciences. 2014;111:4173–4178. doi: 10.1073/pnas.1400350111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cabanski CR, et al. Pan-cancer transcriptome analysis reveals long noncoding RNAs with conserved function. RNA Biol. 2015;12:628–642. doi: 10.1080/15476286.2015.1038012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alaiyan B, et al. Differential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence. BMC Cancer. 2013;13:196. doi: 10.1186/1471-2407-13-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCleland ML, et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. Journal of Clinical Investigation. 2016;126:639–652. doi: 10.1172/JCI83265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ling H, et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Research. 2013;23:1446–1461. doi: 10.1101/gr.152942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang D, et al. Long noncoding RNA CCAT2 promotes breast tumor growth by regulating the Wnt signaling pathway. OncoTargets and Therapy. 2015 doi: 10.2147/OTT.S90485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schneider C, et al. Genes specifically expressed at growth arrest of mammalian cells. Cell. 1988;54:787–793. doi: 10.1016/s0092-8674(88)91065-3. [DOI] [PubMed] [Google Scholar]
- 78.Hudson WH, et al. Conserved sequence-specific lincRNA-steroid receptor interactions drive transcriptional repression and direct cell fate. Nat Commun. 2014;5:5395. doi: 10.1038/ncomms6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pickard M, Williams G. Molecular and Cellular Mechanisms of Action of Tumour Suppressor GAS5 LncRNA. Genes. 2015;6:484–499. doi: 10.3390/genes6030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kino T, et al. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Science Signaling. 2010;3:ra8. doi: 10.1126/scisignal.2000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hudson WH, et al. Conserved sequence-specific lincRNA-steroid receptor interactions drive transcriptional repression and direct cell fate. Nat Commun. 2014;5:5395. doi: 10.1038/ncomms6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee S, et al. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell. 2016;164:69–80. doi: 10.1016/j.cell.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tichon A, et al. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat Commun. 2016;7:1–10. doi: 10.1038/ncomms12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miller MA, Olivas WM. Roles of Puf proteins in mRNA degradation and translation. Wiley Interdisciplinary Reviews: RNA. 2010;2:471–492. doi: 10.1002/wrna.69. [DOI] [PubMed] [Google Scholar]
- 85.Rajagopalan H, et al. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 86.Nobori T, et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 87.Popov N, Gil J. Epigenetic regulation of the INK4b-ARF-INK4alocus. Epigenetics. 2014;5:685–690. doi: 10.4161/epi.5.8.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pasmant E, et al. Characterization of a Germ-Line Deletion, Including the Entire INK4/ARF Locus, in a Melanoma-Neural System Tumor Family: Identification of ANRIL, an Antisense Noncoding RNA Whose Expression Coclusters with ARF. Cancer Res. 2007;67:3963–3969. doi: 10.1158/0008-5472.CAN-06-2004. [DOI] [PubMed] [Google Scholar]
- 89.Gil J, Peters G. Regulation of the INK4b--ARF--INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 90.Yap KL, et al. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Molecular Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dimitrova N, et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Molecular Cell. 2014;54:777–790. doi: 10.1016/j.molcel.2014.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhai H, et al. Clinical Significance of Long Intergenic Noncoding RNA-p21 in Colorectal Cancer. Clinical Colorectal Cancer. 2013;12:261–266. doi: 10.1016/j.clcc.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 93.Wu G, et al. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation. 2014;130:1452–1465. doi: 10.1161/CIRCULATIONAHA.114.011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoon JH, et al. LincRNA-p21 Suppresses Target mRNA Translation. Molecular Cell. 2012;47:648–655. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang F, et al. Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Molecular Cell. 2014;53:88–100. doi: 10.1016/j.molcel.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 96.Hung CL, et al. A long noncoding RNA connects c-Myc to tumor metabolism. Proceedings of the National Academy of Sciences. 2014;111:18697–18702. doi: 10.1073/pnas.1415669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marín-Béjar O, et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013;14:R104. doi: 10.1186/gb-2013-14-9-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huarte M. The emerging role of lncRNAs in cancer. Nature Medicine. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 99.Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stein CA, Castanotto D. FDA-Approved Oligonucleotide Therapies in 2017. Molecular Therapy. 2017;25:1069–1075. doi: 10.1016/j.ymthe.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nero TL, et al. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14:248–262. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- 102.Beck A, et al. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–337. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 103.Hua Y, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hong D, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7:314ra185. doi: 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goemans NM, et al. Systemic Administration of PRO051 in Duchenne’s Muscular Dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 106.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 107.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 108.Brummelkamp TR. A System for Stable Expression of Short Interfering RNAs in Mammalian Cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 109.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 110.Beronja S, et al. RNAi screens in mice identify physiologicalregulators of oncogenic growth. Nature. 2013;501:185–190. doi: 10.1038/nature12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Luo J, et al. A Genome-wide RNAi Screen Identifies Multiple Synthetic Lethal Interactions with the Ras Oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Song E, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nature Medicine. 2003;9:347–351. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 113.Petrocca F, et al. A Genome-wide siRNA Screen Identifies Proteasome Addiction as a Vulnerabilityof Basal-like Triple-Negative Breast Cancer Cells. Cancer Cell. 2013;24:182–196. doi: 10.1016/j.ccr.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nature Rev Genet. 2015;16:543–552. doi: 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vickers TA, et al. Efficient Reduction of Target RNAs by Small Interfering RNA and RNase H-dependent Antisense Agents. Journal of Biological Chemistry. 2003;278:7108–7118. doi: 10.1074/jbc.M210326200. [DOI] [PubMed] [Google Scholar]
- 116.Morrissey DV, et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- 117.Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222–229. doi: 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- 118.Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov. 2004;3:318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- 119.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 120.Ren S, et al. Long Noncoding RNA MALAT-1 is a New Potential Therapeutic Target for Castration Resistant Prostate Cancer. Journal of Urology. 2013;190:2278–2287. doi: 10.1016/j.juro.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 121.Delás MJ, et al. lncRNA requirements for mouse acute myeloid leukemia and normal differentiation. eLife. 2017;6:e25607. doi: 10.7554/eLife.25607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bennett CF, et al. Pharmacology of Antisense Drugs. Annual Review of Pharmacology and Toxicology. 2017;57:81–105. doi: 10.1146/annurev-pharmtox-010716-104846. [DOI] [PubMed] [Google Scholar]
- 125.Allerson CR, et al. Fully 2′-Modified Oligonucleotide Duplexes with Improved in Vitro Potency and Stability Compared to Unmodified Small Interfering RNA. J Med Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 126.Stein CA, et al. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988;16:3209–3221. doi: 10.1093/nar/16.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu H, et al. Principles, Strategies, and Applications, Second Edition. CRC Press; 2010. The RNase H Mechanism; pp. 47–74. [Google Scholar]
- 128.Yoo BH, et al. 2′-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004;32:2008–2016. doi: 10.1093/nar/gkh516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Crooke ST. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017;27:70–77. doi: 10.1089/nat.2016.0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Monia BP, et al. Evaluation of 2 “-modified oligonucleotides containing 2-”deoxy gaps as antisense inhibitors of gene expression. Journal of Biological Chemistry. 1993;268:14514–14522. [PubMed] [Google Scholar]
- 131.Finkel RS, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. The Lancet. 2016;388:3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 132.Geary RS, et al. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (kynamro((R))): a second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin Pharmacokinet. 2015;54:133–146. doi: 10.1007/s40262-014-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rader DJ, Kastelein JJP. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022–1032. doi: 10.1161/CIRCULATIONAHA.113.001292. [DOI] [PubMed] [Google Scholar]
- 134.Bennett CF, Swayze EE. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annual Review of Pharmacology and Toxicology. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 135.Obika S, et al. Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C3, -endo sugar puckering. Tetrahedron Letters. 38:8735–8738. [Google Scholar]
- 136.Burel SA, et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther. 2013;23:213–227. doi: 10.1089/nat.2013.0422. [DOI] [PubMed] [Google Scholar]
- 137.Pandey SK, et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J Pharmacol Exp Ther. 2015;355:329–340. doi: 10.1124/jpet.115.226969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yamamoto Y, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin Cancer Res. 2015;21:1675–1687. doi: 10.1158/1078-0432.CCR-14-1108. [DOI] [PubMed] [Google Scholar]
- 139.Mendell JT. Targeting a Long Noncoding RNA in Breast Cancer. N Engl J Med. 2016;374:2287–2289. doi: 10.1056/NEJMcibr1603785. [DOI] [PubMed] [Google Scholar]
- 140.Nasevicius A, Ekker SC. Effective targeted gene “knockdown” in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 141.Moulton JD. Using Morpholinos to Control Gene Expression. Curr Protoc Nucleic Acid Chem. 2017;68:4.30.1–4.30.29. doi: 10.1002/cpnc.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kloosterman WP, et al. Targeted Inhibition of miRNA Maturation with Morpholinos Reveals a Role for miR-375 in Pancreatic Islet Development. PLoS Biol. 2007;5:e203. doi: 10.1371/journal.pbio.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Staton AA, Giraldez AJ. Use of target protector morpholinos to analyze the physiological roles of specific miRNA-mRNA pairs in vivo. Nat Protoc. 2011;6:2035–2049. doi: 10.1038/nprot.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Blum M, et al. Morpholinos: Antisense and Sensibility. Developmental Cell. 35:145–149. doi: 10.1016/j.devcel.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 145.Aartsma-Rus A. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Human Molecular Genetics. 2003;12:907–914. doi: 10.1093/hmg/ddg100. [DOI] [PubMed] [Google Scholar]
- 146.Alter J, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nature Medicine. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 147.Crooke ST, et al. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 35:230 EP. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 148.Thompson AJV, Locarnini SA. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunology And Cell Biology. 85:435–445. doi: 10.1038/sj.icb.7100100. [DOI] [PubMed] [Google Scholar]
- 149.Wittrup A, et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat Biotech. 33:870–876. doi: 10.1038/nbt.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sahay G, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31:653–658. doi: 10.1038/nbt.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kandimalla ER, et al. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 2013;41:3947–3961. doi: 10.1093/nar/gkt078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vollmer J, et al. Oligodeoxynucleotides lacking CpG dinucleotides mediate Toll- like receptor 9 dependent T helper type 2 biased immune stimulation. Immunology. 2004;113:212–223. doi: 10.1111/j.1365-2567.2004.01962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nair JK, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 154.Aldrian-Herrada G, et al. A peptide nucleic acid (PNA) is more rapidly internalized in cultured neurons when coupled to a retro-inverso delivery peptide. The antisense activity depresses the target mRNA and protein in magnocellular oxytocin neurons. Nucleic Acids Res. 1998;26:4910–4916. doi: 10.1093/nar/26.21.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Khan T, et al. Silencing Myostatin Using Cholesterol-conjugated siRNAs Induces Muscle Growth. Mol Ther Nucleic Acids. 2016;5:e342. doi: 10.1038/mtna.2016.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Koch L. Functional genomics: Screening for lncRNA function. Nat Rev Genet. 18:70–70. doi: 10.1038/nrg.2016.168. [DOI] [PubMed] [Google Scholar]
- 157.Gilbert LA, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thakore PI, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Meth. 12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu SJ, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017:355. doi: 10.1126/science.aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhu S, et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR Cas9 library. Nat Biotechnol. 34:1279–1286. doi: 10.1038/nbt.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Abudayyeh OO, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Katayama S, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 163.Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10:637–643. doi: 10.1038/nrm2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Modarresi F, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yap KL, et al. Molecular Interplay of the Non-coding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Molecular Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Modarresi F, et al. Natural Antisense Inhibition Results in Transcriptional De-Repression and Gene Upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Brockdorff N. Noncoding RNA and Polycomb recruitment. RNA. 2013;19:429–442. doi: 10.1261/rna.037598.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hendrickson D, et al. Widespread RNA binding by chromatin- associated proteins. Genome Biol. 2016;17:28. doi: 10.1186/s13059-016-0878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kaneko S, et al. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Molecular Cell. 2014;53:290–300. doi: 10.1016/j.molcel.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Pegueroles C, Gabaldón T. Secondary structure impacts patterns of selection in human lncRNAs. BMC Biology. 2016 doi: 10.1186/s12915-016-0283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rivas E, et al. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Meth. 2017;14:45–48. doi: 10.1038/nmeth.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wilusz JE, et al. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012;26:2392–2407. doi: 10.1101/gad.204438.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Brown JA, et al. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nature Struct Mol Biol. 2014;21:633–640. doi: 10.1038/nsmb.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wilkinson KA, et al. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- 175.Lu Z, et al. RNA Duplex Map in Living Cells Reveals Higher- Order Transcriptome Structure. Cell. 2016;165:1267–1279. doi: 10.1016/j.cell.2016.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Novikova IV, et al. 3S: shotgun secondary structure determination of long non-coding RNAs. Methods. 2013;63:170–177. doi: 10.1016/j.ymeth.2013.07.030. [DOI] [PubMed] [Google Scholar]
- 177.Howe JA, et al. Selective small-molecule inhibition of an RNA structural element. Nature. 2015;526:672–677. doi: 10.1038/nature15542. [DOI] [PubMed] [Google Scholar]
- 178.Disney MD, et al. A Small Molecule That Targets r(CGG) expand Improves Defects in Fragile X-Associated Tremor Ataxia Syndrome. ACS Chem Biol. 2012;7:1711–1718. doi: 10.1021/cb300135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rzuczek SG, et al. Precise small-molecule recognition of a toxic cug rna repeat expansion. Nat Chem Biol. 2017;13:188–193. doi: 10.1038/nchembio.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang, et al. Identification and Characterization of a Class of MALAT1-like Genomic Loci. Cell Rep. 2017;19:1723–1738. doi: 10.1016/j.celrep.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Brown JA, et al. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc Natl Acad Sci U S A. 2014;113:14013–14018. doi: 10.1073/pnas.1614759113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Amit D, et al. Development of targeted therapy for bladder cancer mediated by a double promoter plasmid expressing diphtheria toxin under the control of H19 and IGF2-P4 regulatory sequences. J Transl Med. 2010;8:134. doi: 10.1186/1479-5876-8-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sidi AA, et al. Phase I/II Marker Lesion Study of IntravesicalBC-819 DNA Plasmid in H19 Over Expressing Superficial Bladder Cancer Refractory to Bacillus Calmette-Guerin. Journal of Urology. 2008;180:2379–2383. doi: 10.1016/j.juro.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 184.Gofrit ON, et al. DNA Based Therapy with Diphtheria Toxin-A BC-819: A Phase 2b Marker Lesion Trial in Patients with Intermediate Risk Nonmuscle Invasive Bladder Cancer. Journal of Urology. 2014;191:1697–1702. doi: 10.1016/j.juro.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 185.Li Z, et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2016;24:59–71. doi: 10.1038/cdd.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xing, et al. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. 2014;159:1110–1125. doi: 10.1016/j.cell.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xiao Z-D, et al. Energy stress-induced lncRNA FILNC1 repressesc-Myc-mediated energy metabolism and inhibitsrenal tumor development. Nat Commun. 2017;8:783. doi: 10.1038/s41467-017-00902-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lin C, et al. Transcriptional and posttranscriptional regulation ofHOXA13 by lncRNA HOTTIP facilitates tumorigenesis and metastasis in esophageal squamous carcinoma cells. Oncogene. 2017;36:5392–5406. doi: 10.1038/onc.2017.133. [DOI] [PubMed] [Google Scholar]
- 189.Panzitt K, et al. Characterization of HULC, a Novel Gene With Striking Up-Regulation in Hepatocellular Carcinoma, as Noncoding RNA. Gastroenterology. 2007;132:330–342. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 190.Hou P, et al. LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. 2014;5:e1287–10. doi: 10.1038/cddis.2014.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yuan JH, et al. A Long Noncoding RNA Activated by TGF-b Promotes the Invasion-Metastasis Cascade in Hepatocellular Carcinoma. Cancer Cell. 2014;25:666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 192.Yang F, et al. Repression of the Long Noncoding RNA-LET by Histone Deacetylase 3 Contributesto Hypoxia-Mediated Metastasis. Molecular Cell. 2013;49:1083–1096. doi: 10.1016/j.molcel.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 193.Zhang H-Y, et al. The long non-coding RNA MIAT regulates zinc finger E-box binding homeobox 1 expression by sponging miR-150 and promoteing cell invasion in non-small-cell lung cancer. 2017 doi: 10.1016/j.gene.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 194.Zhou Y, et al. MEG3 noncoding RNA: a tumor suppressor. Journal of Molecular Endocrinology. 2012;48:R45–R53. doi: 10.1530/JME-12-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nie FQ, et al. Long non-coding RNA MVIH indicates a poor prognosis for non- small cell lung cancer and promotes cell proliferation and invasion. Tumor Biol. 2014;35:7587–7594. doi: 10.1007/s13277-014-2009-7. [DOI] [PubMed] [Google Scholar]
- 196.Pandey, et al. The Risk-Associated Long Noncoding RNA NBAT-1 Controls Neuroblastoma Progression by Regulating Cell Proliferation and Neuronal Differentiation. 2014;26:722–737. doi: 10.1016/j.ccell.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 197.Liu X, et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol. 2016;18:431–442. doi: 10.1038/ncb3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhu Y, et al. ncRAN, a Newly Identified Long Noncoding RNA, Enhances Human Bladder Tumor Growth, Invasion, and Survival. Urology. 2011;77:510.e1–510.e5. doi: 10.1016/j.urology.2010.09.022. [DOI] [PubMed] [Google Scholar]
- 199.Puvvula PK, et al. Long noncoding RNA PANDA and scaffold-attachment-factor SAFA controlsenescence entry and exit. Nat Commun. 2014;5:1–16. doi: 10.1038/ncomms6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Malik, et al. The lncRNA PCAT29Inhibits Oncogenic Phenotypes in Prostate Cancer. 2014;12:1081–1087. doi: 10.1158/1541-7786.MCR-14-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tay Y, et al. Coding-Independent Regulation of the Tumor Suppressor PTEN by Competing Endogenous mRNAs. Cell. 2011;147:344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Petrovics G, et al. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene. 2004;23:605–611. doi: 10.1038/sj.onc.1207069. [DOI] [PubMed] [Google Scholar]
- 203.Leucci E, et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016;531:518–522. doi: 10.1038/nature17161. [DOI] [PubMed] [Google Scholar]
- 204.Prensner, et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nature. 2013;45:1392–1398. doi: 10.1038/ng.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Redon S, et al. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010;38:5797–5806. doi: 10.1093/nar/gkq296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li Z, et al. TUG1: a pivotal oncogenic long non-coding RNA of human cancers. Cell Proliferation. 2016;49:471–475. doi: 10.1111/cpr.12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hosono Y, et al. Oncogenic Role of THOR, a Conserved Cancer/Testis Long Non-coding RNA. Cell. 2017;171(7):pp.1559–1572. doi: 10.1016/j.cell.2017.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tomita S, et al. A cluster of noncoding RNAs activates the ESR1 locus during breast cancer adaptation. Nature Commun. 2015;6:6966. doi: 10.1038/ncomms7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Huang J, et al. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1) Cell Death & Disease. 2014;5:e1008–10. doi: 10.1038/cddis.2013.541. [DOI] [PMC free article] [PubMed] [Google Scholar]