

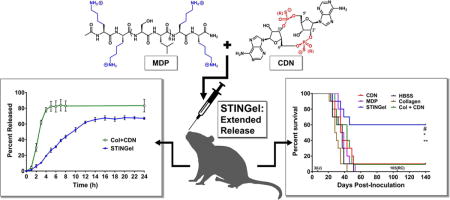
Abstract
Recent advancements in the field of immunotherapy have yielded encouraging results for the treatment of advanced cancers. Cyclic dinucleotides (CDNs) are a powerful new class of immunotherapy drugs known as STING (Stimulator of Interferon Genes) agonists, currently in clinical trials. However, previous studies of CDNs in murine cancer models have required multiple injections, and improve survival only in relatively nonaggressive tumor models. Therefore, we sought to improve the efficacy of CDN immunotherapy by developing a novel biomaterial we call “STINGel.” STINGel is an injectable peptide hydrogel that localizes and provides controlled release of CDN delivery, showing an 8-fold slower release rate compared to a standard collagen hydrogel. The carrier hydrogel is a positively charged, MultiDomain Peptide (MDP) which self-assembles to form a nanofibrous matrix and is easily delivered by syringe. The highly localized delivery of CDN from this nanostructured biomaterial affects the local histological response in a subcutaneous model, and dramatically improves overall survival in a challenging murine model of head and neck cancer compared to CDN alone or CDN delivered from a collagen hydrogel. This study demonstrates the feasibility of biomaterial-based immunotherapy platforms like STINGel as strategies for increasing the efficacy of CDN immunotherapies.
Keywords: STINGel, Immunotherapy, Extended Drug Release, Peptide Hydrogel, Intratumoral Injection
Graphical abstract
1. Introduction
Essential to modern medicine is the effective delivery of drug therapies. Injectable biomaterials have been established as powerful methods of therapy administration due to their ability to control the release profile of loaded agents and influence the local biological reponse.1–3 Such biomaterial-based delivery systems have been used for a host of useful applications, including tissue engineering, growth factor release, DNA delivery, and vaccine incorporation.4–7 Recent interest has focused on biomaterials as platforms for cancer immunotherapy, which can provide extended and localized drug release, and also allow for the intelligent modulation of immune cells in situ.
Cancer immunotherapy has arisen as an exciting treatment modality for several advanced-stage cancers, with the potential to generate specific and durable anti-tumor responses through the use of the host’s natural immune reponse.8–11 Significant progress has been made in this field, generating encouraging results in the treatment of cancers such as metastatic melanoma, for which the 5-year overall survival rate has increased from less than 10%, to almost 40%.12–14 However, in the case of other types of cancer such as head and neck squamous cell carcinoma (HNSCC) survival rates have failed to improve at a comparable rate, remaining relatively stagnant for the past few decades and emphasizing the need for new treatment options.15
HNSCC is the sixth most common cancer worldwide, with more than 60,000 men and women in the U.S. expected to develop HNSCC this year.16–17 Etiologic risk factors include exposure to carcinogens such as tobacco and alcohol, and viral infections such as high-risk types of human papilloma viruses (HPVs).18–20 Given the well-known co-morbidities and recurrence rates associated with conventional treatments such as surgery, radiation therapy, and chemotherapy, there is a real need for innovative approaches to treat HNSCC. While some HNSCC patients undergoing immune checkpoint inhibitor antibody therapy experience durable complete remissions, the majority (80–85%) of patients derive no benefit.21 Improving the rate of success in HNSCC immunotherapy is therefore a highly desirable goal.
Recently, a novel class of immunotherapeutics based on synthetic cyclic dinucleotides (CDNs) have been found to induce strong anti-tumor responses in preclinical models through the Stimulator of Interferon Genes (STING) pathway.22–24 The STING pathway has emerged as a key mechanism linking the detection of cytosolic tumor DNA to downstream activation of innate immune cells.25–26 The rationally-designed synthetic Cyclic Dinucleotide dithio-(RP,RP)-[cyclic[A(2′,5′)pA(3′,5′)p]] (abbreviated as ML RR-S2 CDA or just CDN, see Fig. 1A) is a promising candidate molecule in clinical trials (see NCT02675439) that has shown efficacy in murine cancer models, promoting the specific rejection of several types of tumors.22 However, to date CDN monotherapy has shown poor efficacy in preclinical models of HNSCC, requiring multiple injections and concomitant administration of immune checkpoint antibodies.27–28 Current clinical trials are evaluating intratumoral injections of CDN as monotherapy, a strategy that may prove to be insufficient.27 Thus, novel approaches to improve the efficacy of CDN in challenging, treatment-refractory tumor models are warranted.
Fig. 1.
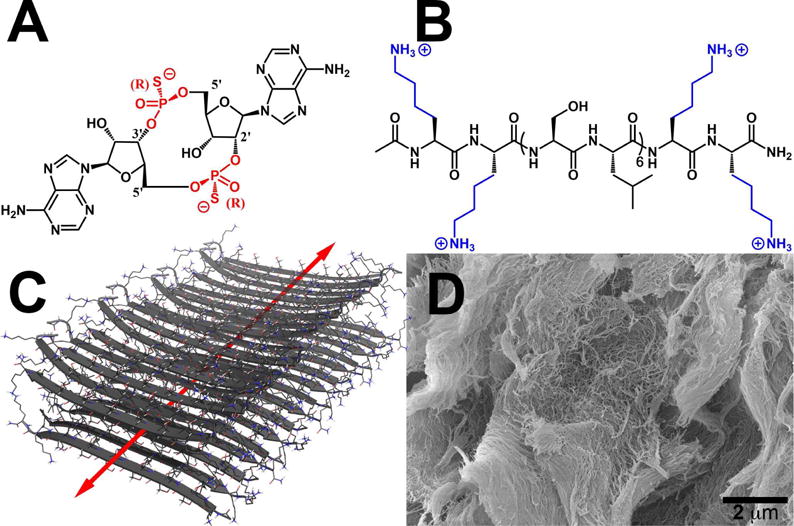
Chemical structures of (A) ML RR-S2 CDA synthetic STING agonist (CDN), (B) K2(SL)6K2 multidomain peptide (MDP), showing charge-pair complementarity of positive lysine termini and negative thiophosphate linkages. (C) Model of anti-parallel β-sheet nanofiber formed by the MDP in solution. The red arrow indicates the axis of the nanofiber and orientation of hydrogen bonding. (D) Scanning Electron Microscopy image of the MDP gel showing a wide field image of the self-assembled nanofibers.
In response to this challenge, we have developed a novel peptide hydrogel-based platform for intratumoral CDN delivery, which we call “STINGel.” This localizable drug delivery vehicle, utilizing the power of immunotherapy, is based on prior work in our laboratory studying the utility of multidomain peptides (MDPs) as unique supramolecular biomaterials. These self-assembling peptides mimic the extracellular matrix of cells through the formation of a nanofibrous network, and can act as biofunctional delivery platforms that allow for an immense diversity of functionality to be introduced.29–30 Rationally engineered MDPs have been shown to form anti-parallel β-sheets of peptide nanofibers in solution, which when electrostatically crosslinked with multivalent ions form extended nanofiber networks to create robust hydrogels (see Fig. 1B–D).30
MDPs possess a number of attractive characteristics as biomaterials. 1) The peptide hydrogels are thixotropic, allowing them to be easily delivered by syringe and yet remain localized for applications such as intratumoral injections. 2) The design of the peptide sequence can allow for the incorporation of charged small molecule drugs by nanofiber crosslinking to achieve extended release, or can allow for the encapsulation and gradual release of small hydrophobic drugs such as daunorubicin, etodolac, levofloxacin, and others.29, 31–32 3) The hydrogels undergo complete cellular infiltration within three days post injection in vivo and are not fibrously encapsulated, maximizing matrix-tissue interaction.
In this study, we sought to use the MDP hydrogel with sequence K2(SL)6K2 to deliver a promising STING agonist CDN, by taking advantage of favorable electrostatic interactions between the CDN’s negative thiophosphate linkages and the positive lysine residues at the peptide termini (Fig. 1). We hypothesized that a combination of controlled release and favorable local environment would result in improved tumor treatment efficacy in vivo.
2. Materials and Methods
2.1 Peptide synthesis
Peptide synthesis reagents were purchased from EMD Chemicals (Philadelphia, PA). An Apex Focus XC (Aapptec) synthesizer was used to synthesize the multidomain peptide K2(SL)6K2 (MW = 1773.171 g/mol) according to a standard synthetic method previously published to yield pure peptide with acetylated N-terminus and C-terminal amide.30, 32–33 All peptides were analyzed by Autoflex MALDI-TOF MS (Bruker Instruments, Billerica, MA) for purity and confirmation of successful synthesis (Fig. S1).
2.2. Hydrogel preparation and loading
All chemicals not otherwise specified were purchased from Sigma-Aldrich (St. Louis, MO). For preparation of sterile MDP stock solutions, 2 wt. % (11 mM) peptide solutions were dissolved in 298 mM sucrose to support cytocompatibility. Stock ML RR-S2 CDA (CDN) (InvivoGen, San Diego, CA and MedChem Express, Monmouth Junction, NJ) was prepared at 2.67 μg/μL in endotoxin-free H2O (3.64 mM, 4X the final dose of 0.67 ug/uL, or 20 μg in 30 μL of gel), with concentration confirmed by UV-Vis. For preparation of control collagen gel formulations, a stock was prepared at 6.67 μg/μL CDN (10X). Collagen gel formulations were prepared according to the provided kit protocol (ECM675, EMD Millipore, Temecula, CA) with the substitution of CDN stock in DPBS (Thermo Scientific, Rockford, IL). The collagen stock solutions were provided at a concentration of 3.5–5.0 mg/mL varying by batch. Final collagen formulations were 1X DPBS, 80% v/v collagen stock solution (approximately 3–4 mg/mL), and 1X CDN (20 μg/30 μL). Collagen formulations were kept on ice prior to injections to maintain liquid state (gelation occurring at in vivo temperatures). STINGel formulations were prepared by diluting 4X CDN stock in an equal volume of 2X HBSS (Fisher Scientific, Hampton, NH), which was mixed with sterile 2 wt. % MDP in 298 mM sucrose to induce gelation. After mixing, final STINGel concentrations were 0.5X HBSS, 149 mM sucrose, 1 wt. % peptide (5.6 mM), and 1X CDN (20 μg/30 μL). All other controls were prepared with the same concentrations of HBSS and sucrose. For in vivo studies, prepared controls and gel formulations were loaded into Monoject 300 μL Insulin syringes (Covidien, Mansfield, MA) and allowed to equilibrate for ~1 hour before injection.
2.3. CDN drug release kinetics
To study the kinetics of CDN release from STINGel vs. collagen gel, 30 μL CDN-loaded gel aliquots were deposited into Falcon® 96 well flat bottom polystyrene plates (Becton Dickinson Labware, Franklin Lakes, NJ). For each experiment, cylindrical pucks of the 30 μL gel aliquots were created in each well, allowing the pipetted gels to shear recover for at least 5 min before adding 200 μL of buffer (1X HBSS for STINGel, 1X DPBS for collagen, according to gelation requirements) to the top of the gels at what was defined as t = 0 for the start of kinetics experiments. The buffer height was approximately 2.5 mm, the gel thickness approximately 0.5 mm, and the gel’s solvent exposed surface area approximately 12.6 mm2.
A Thermo Scientific Nanodrop 2000C Spectrophotometer was used to measure UV absorbance at 259 nm, the characteristic wavelength of maximum absorbance for the CDN’s adenine nucleobases. An extinction coefficient of 24,000 M−1cm−1 at 259 nm was used. Absorbance measurements were taken by removal of 1 μL aliquots from the surface of the buffers, measuring the increase in absorbance over time. Release of CDN was measured over the course of the first 24 hours and converted to total percent released, with additional measurements also made at 48 and 72 hours to confirm that the samples had reached equilibrium.
2.4. Cell culture
MOC2-E6E7, the murine oral cancer cell line used in this study, was generated by retroviral transduction of HPV16 E6E7 in MOC2 cells.34–35 MOC2 cells were provided by Dr. Ravindra Uppaluri, Dana-Farber Cancer Institute, Harvard University, Boston. MOC2-E6E7 cells were maintained in presence of 4 mg/mL puromycin dihydrochloride in medium routinely used for maintaining MOC2.36 Cells were used at 80–95% confluency for all experiments.
2.5. Preparation of cell–laden hydrogel for in vitro viability
MDP gel formulations with MOC2-E6E7 cells were prepared by diluting a cell suspension of 2,000,000 cells/mL in 2X HBSS with an equal volume of 4X CDN. The resulting suspension was then added to an equal volume of sterile 2 wt. % peptide in 298 mM sucrose and mixed to produce final samples of approximately 35,000 cells per 70 μL of 1 wt. %, 149 mM sucrose gel. Gels were then transferred into 0.4 cm2 wells of Lab-Tek 16 well glass chamber slides (Thermo Fischer, Rochester, NY), pipetted to produce 70 μL pucks with flat profiles. Samples were allowed to shear recover for 5–10 min before adding 200 μL cell media on top of each gel. The cell media was changed every 2 days, taking care to not dislodge the hydrogel material.
Cell viability was determined at each desired time point by performing live/dead assays as described below. Live/dead staining solution was prepared in DPBS with 2 μM Calcein AM for live cells (Life Technologies), 4 μM Ethidium homodimer for dead cells (Life Technologies), and 5 μg/mL Hoechst 33342 for nuclei (MP Biomedicals, Solon, OH). In many cases significant background staining of the hydrogels resulted in reduced resolution of the blue Hoechst channel in confocal images. Cell media was removed and the gels were washed with PBS. Samples were then stained with 100 μL of prepared solution by incubating at RT for 15–30 min. Following staining, gels were placed in PBS for immediate analysis by confocal microscopy. Gels were analyzed by z-stack imaging (100 μm) using a Nikon A1 Confocal Microscope with 20X air and 40X water objectives (405 nm blue channel laser, 488 nm green channel laser, 561 nm red channel laser). Image processing was done using NIS Elements, and live/dead cell counting was performed using the cell identification tool in Imaris 3D/4D Image Processing software, manually verified with nuclear counts when resolution allowed.
2.6. Subcutaneous experiments and histology
Male/female mice (C57BL/6 strain) of age 8–12 weeks were obtained for subcutaneous experiments, which were conducted with Rice IACUC approval and according to NIH guidelines. Mice were injected with 40 μL CDN-loaded hydrogel (910 μM CDN) or 100 μL unloaded hydrogel in each of four separate sites in the subcutaneous space of the dorsal flank. At days 3 and 7 the mice were euthanized and the dorsal skin around the entire implant was removed, fixed overnight in 10% neutral buffered formalin, processed and paraffin embedded by the Baylor College of Medicine Pathology Core, and finally sectioned at 5 μm thickness for Masson’s trichrome staining and hematoxylin and eosin (H&E) staining (Fig S4 and S6).
2.7. Tumor growth and survival study
For tumor experiments, 6–8 week old wild-type C57BL/6 female mice were maintained in a pathogen-free environment for the study. All protocols were in accordance with the guidelines for humane treatment of laboratory animals by the National Institutes of Health, the Animal Welfare Committee and the Center for Laboratory Animal Medicine and Care (CLAMC) at the University of Texas Health Science Center at Houston. Mice were injected with MOC2-E6E7 tumor cells on day 0 into the maxillary oral vestibule (30,000 cells in 30 μL volume), followed by controls or gel injections loaded with or without CDN on day 3 in the same oral cavity location (20 μg CDN per 30 μL injection). Tumor growth was measured using calipers and body weight was taken in all subjects two to three times per week. Photographs were also obtained. Kaplan-Meier survival and tumor growth curve analyses were performed from data obtained. Mice that maintained tumor clearance for 100 days were re-challenged with MOC2-E6E7 tumor cells again at day 105 and monitored similarly. All endpoints in tumor growth curve and survival data are a result of euthanasia due to excessive tumor burden, defined as tumor reaching 12 mm, tumor ulceration, or a weight loss of greater than 20%. We observed no signs of unexpected disease or discomfort in the mice over the course of the experiments.
2.8. Statistical methods
Statistical analyses for Kaplan-Meier survival curves were performed using the log-rank/Mantel-Cox test with GraphPad Prism (GraphPad, San Diego, CA). For tumor growth curves, the Wilcoxon rank sum test was used to compare tumor size between STINGel and other groups. No adjustment for multiple testing was made because the study is novel and exploratory. All p values are two-sided and p values less than 0.05 were considered as significant. All statistical analyses were performed using the SAS software (version 9.4, the SAS Institute, Cary, NC).
3. Results
3.1. Hydrogel properties and drug release kinetics
The MDP hydrogel K2(SL)6K2, extensively characterized in past studies in terms of peptide secondary structure, nanostructure, rheological properties, and biocompatibility, was examined in the context of this experiment for its ability to encapsulate and deliver the small molecule CDN.29–31 Ionic crosslinking of MDP fibers to achieve drug loading was previously accomplished with the negatively charged anti-parasitic compound suramin, and thus our work with CDN was a natural extension.31 As shown in Fig. 2, release profiles of CDN from MDP hydrogel and a collagen control hydrogel were obtained by detecting the released drug concentration via UV-Vis. Over time the solvent-exposed surface area of the hydrogels allowed for free exchange of CDN with the surroundings until an equilibrium had been achieved. This equilibrium was observable when a maximum and unchanging concentration of CDN was recorded over multiple time points.
Fig. 2.
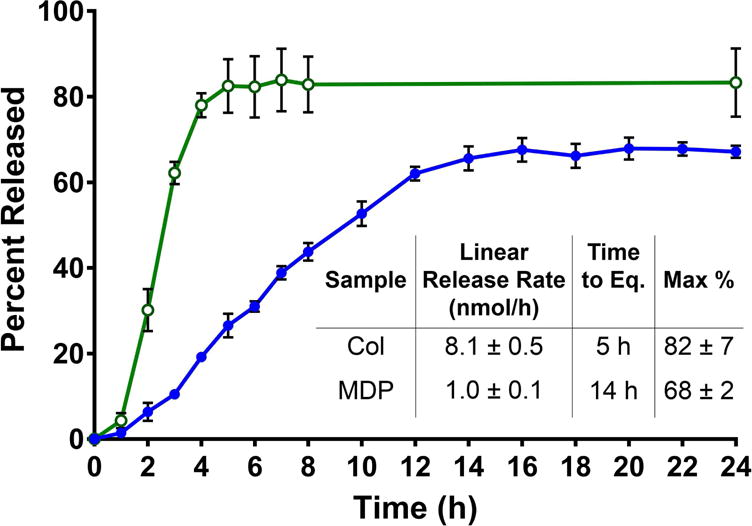
CDN drug release kinetics profiles of MDP hydrogels (blue, closed circles) compared to collagen control hydrogels (green, open circles). Samples are 30 μL gels in 96 well plates, loaded with 910 μM CDN and placed under 200 μL HBSS. Absorbance was measured at 259 nm and converted to total percent released for 24 h to monitor release rate and time until equilibrium. Values represent the mean and standard deviation in all plots (n = 3).
Fig. 2 shows that controlled release of CDN was achieved, with a factor of 8 decrease in release rate for the highly positively charged MDP hydrogel compared to collagen control gels. Collagen gels showed a release rate of approximately 8.1 nmol CDN/hour in the initial linear release phase, compared to only 1.0 nmol CDN/hour from MDP gels. Indeed, the release profiles show that in this system, within 4–5 hours collagen gels have fully released CDN and reached the maximum theoretical equilibrium with the surrounding buffer (approximately 85%). In contrast, not only do the kinetics profiles of MDP hydrogel show 14–15 hour continuous release in this system, but they also plateau at a significantly lower maximum release of loaded CDN at approximately 68%. This suggests that the material, presumably as a result of ion-ion charge-pair interactions between negative drug molecules and positive peptide nanofibers, reaches a different chemical equilibrium with the surrounding buffer system, thereby withholding more CDN until the equilibrium is disturbed. This equilibrium can be disturbed by exchanging the buffer in long release studies, or by fluid flow in vivo. As this is the basis of the designed controlled release mechanism, a lower equilibrium is an expected result, and supports our conclusion that extended release was successfully achieved in STINGel (MDP+CDN).
3.2. In vitro cell viability
In order to determine the cytocompatibility of our STINGel material with the murine oral cancer cell line MOC2-E6E7, we assessed in vitro cell viability by live/dead staining to visualize and quantify cell survival in our gels. MOC2-E6E7 cells suspended in 3-D gels of unloaded MDP hydrogel controls showed results consistent with all our previous work: cells are able to survive and proliferate within the 3-D hydrogel matrix, exchanging waste and nutrients with the surrounding buffer and remodeling the peptide hydrogel to allow for larger colonies.29 Control experiments are shown in Fig. 3A surveyed at 20× magnification by confocal microscopy, where the cancer cells are seeded as small clumps within hydrogel pucks and placed under media for 7 days. We observed the cells grow from discrete groups of around 3–10 cells distributed throughout the hydrogel matrix, to larger clumps of hundreds of cells by day 7 in the absence of CDN (similar proliferation results shown for MDP controls in Fig. S2). Fig. 4 shows the mean cell counts and quantification of cell density within the gels.
Fig. 3.
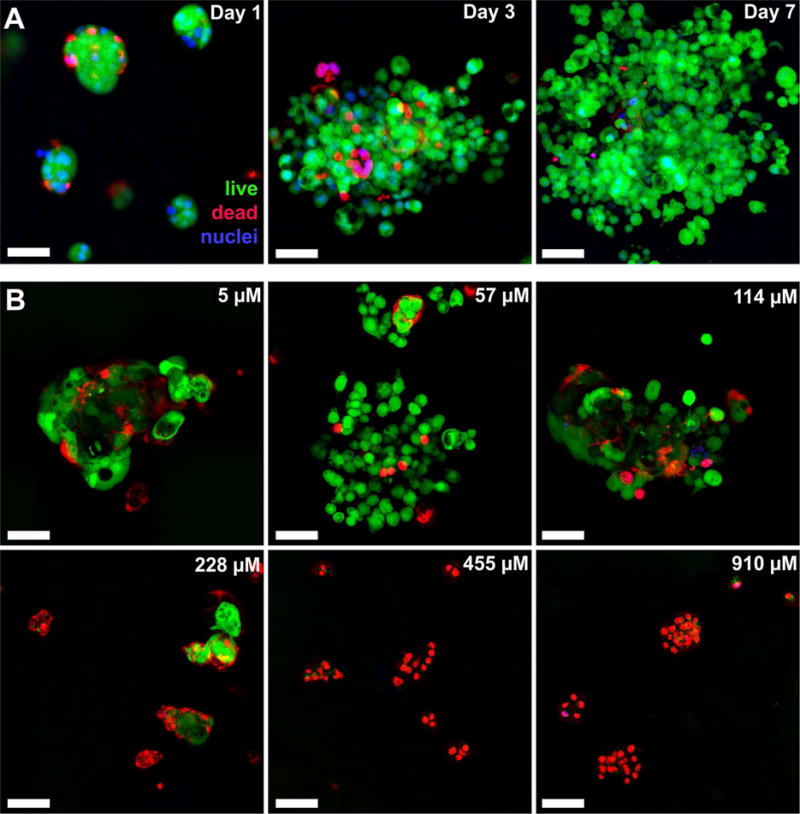
Cell viability in unloaded and loaded MDP hydrogel. MOC2-E6E7 cells were seeded at a density of approximately 35,000 cells within 70 μL of gel under 200 μL of media (changed every two days) and processed under Live/Dead viability assays (green- live cells; red- dead cells; blue- nuclei). (A) Unloaded hydrogel control showing cell viability over time from small clumps into large spreading masses throughout the peptide hydrogel. (B) CDN dose response assays with images shown from day 3 time point, at which time cells had either died or survived past the initial stage of exposure to CDN. All scale bars are 50 μm, and z-stacks are 100 μm in thickness.
Fig. 4.
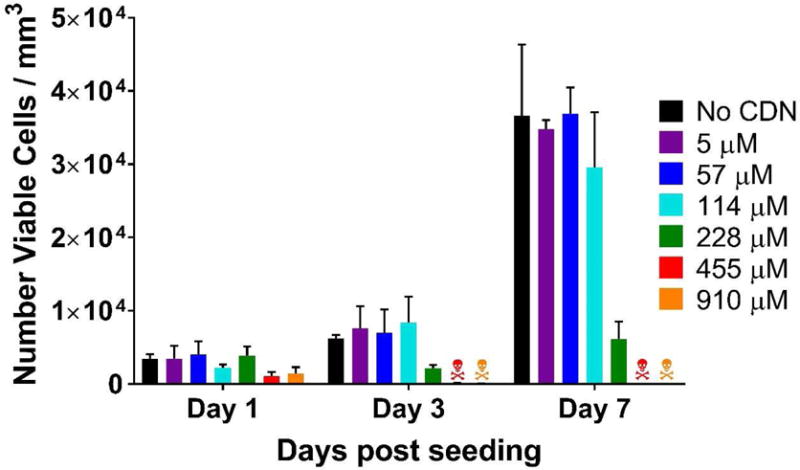
Live/dead viability assay quantification used to assess CDN toxicity to MOC2-E6E7 cells. The graph shows the number of viable cells per mm3 of hydrogel over days 1–7 post seeding with cells, testing increasing concentrations of CDN loaded into the MDP hydrogel. The
 symbols refers to >99% cell death. Values represent the mean and standard deviation in all plots (n = 3).
symbols refers to >99% cell death. Values represent the mean and standard deviation in all plots (n = 3).
In contrast, increasing STINGel formulations (MDP loaded with 228, 455 and 910 μM CDN), resulted in significant cell death (Fig. 3B). While no difference in cell growth (Fig. 4) or percent viability (Fig. S3) was observed in samples loaded with 5 μM and 57 μM CDN (typical in vitro concentrations for induction of STING pathway and Type 1 INF expression28), observable cell stress, seen as a difference in percent cell viability shown in Fig. S3, was observed even at 114 μM CDN at the day 1 timepoint. However, cells seeded in 114 μM CDN gels recover to normal levels of viability and growth by days 3 and 7 (Fig. 4 and S3). Significant cell death and inhibition of cell growth was observed at 228 μM, with cell counts largely reduced from controls even as late as day 7 in Fig. 4. Massive cell death was observed at 455 and 910 μM (the concentration of in vivo injections) such that cell viability approached 0% by day 3 post seeding. In summary, while unloaded MDP gel is fully biocompatible, CDN loaded MDP gel exhibited inherent cytotoxicity at high drug concentrations in vitro.
3.3 In vivo subcutaneous characterization
In order to assess the properties of the STINGel material in vivo, the host response was studied using subcutaneous injections in mice. At specific timepoints the mice were euthanized and the implants removed for histological analysis (Fig. 5, Fig. S4–S6). Injection volumes were chosen to be 40 μL for CDN-loaded hydrogels and 100 μL for unloaded hydrogels. In past work, 100 μL or greater was an ideal injection volume as it aided in the location and removal of the hydrogel implants.37–38 However, in the case of drug-loaded injections we had to make a compromise between the implant being large enough to be visible by 3–7 days post injection, but also small enough for the total CDN dose to be well below potentially toxic levels. Therefore 40 μL was chosen and found to be sufficient for our purposes, with no signs of systemic toxicity observed in any experiments.
Fig. 5.
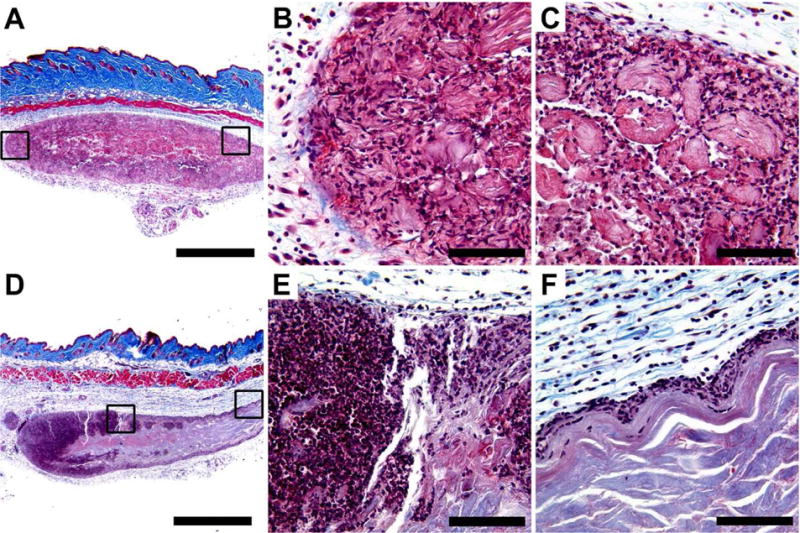
Masson’s trichrome stained MDP hydrogel implants unloaded and loaded with CDN, injected subcutaneously in the dorsal flank of mice. Time point shown is 3 days post injection, at which time hydrogel implant was removed and processed for histology. Scale bars in panels A and D = 1 mm; scale bars in panels B, C, E, and F = 0.1 mm. (A-C) MDP unloaded control implant at 4× magnification showing even infiltration of cells, with boxes drawn around chosen areas whose 40X counterparts are shown in panels B and C, respectively. (D-F) MDP implant loaded with 910 μM CDN (STINGel) at 4× magnification showing uneven infiltration of cells across the implant. Boxes drawn around chosen areas in panel D again have 40X counterparts shown in panels E and F, respectively.
Masson’s trichrome staining shows the hydrogel implants as purple-red oblong ovals in the mouse hypodermis, contrasting to the blue natural collagen and dark red muscle (Fig. 5 and Fig. S5), while H&E staining shows the hydrogel as well-defined eosinophilic (pink) material in the subcutaneous space (Fig. S4 and Fig S6). Uniform cellular infiltration of the control unloaded MDP implant without fibrous encapsulation was observed, a result consistent with all prior subcutaneous experiments performed with MDP hydrogels (Fig. 5AC).29, 38 At day 3 almost the entirety of the unloaded implant is observed to be infiltrated by inflammatory cells that rim the hydrogel material, resembling histologic features commonly seen in a foreign body reaction. By visual evaluation, many of these cells are likely monocytes trying to engulf the foreign hydrogel material. However, in contrast are CDN drug-loaded implants, which show substantially increased inflammatory condensation along the periphery of the hydrogel (Fig. 5D–F). Unlike the unloaded hydrogel, the infiltrating immune cells in CDN-loaded implants are much denser and encompass a high proportion of what appear to be lymphocytes (Fig. 5E–F). By day 7, cell infiltration of CDN-loaded hydrogels increases, presumably as CDN concentration in the implants fell below the toxic thresholds observed in cell culture (Fig. S5–S6). However, CDN-loaded implants still did not reach the uniform cellular infiltration observed in unloaded controls, providing evidence of continuous immune-cell chemotaxis that is sustained by our slow release system.
3.4. In vivo murine experiments: STINGel induced rejection of MOC2-E6E7 tumors
We examined the growth of MOC2-E6E7 tumors in mice treated with HBSS, CDN alone, MDP gel, STINGel (MDP+CDN), collagen or collagen+CDN to determine antitumor efficacy. We observed no signs of systemic toxicity after treatment administration, nor signs of unexpected disease or discomfort in the mice over the course of the experiments. Treatments with STINGel show a significant decrease in tumor growth, or a complete tumor clearance compared to other groups (Fig. 6A). Median (Fig. 6A) and individual (Fig. 6B-E) tumor growth curves for treatment groups compared to HBSS control show that treatments with STINGel extend the period of progression-free disease (Fig. 6D). Although tumor growth in mice treated with collagen+CDN (Fig. 6F) show similar initial periods of progression-free disease as STINGel, 90% of mice develop tumors. The data represented in Fig. 6 has been determined from tumor dimensions collected from day 10-day 35 as median tumor size. Mice that maintained tumor clearance were rechallenged with a second tumor cell inoculation of MOC2-E6E7 cells at day 105 after initial inoculation. 6 out of 10 STINGel mice were able to reach this reinoculation point. On the contrary, only 1 in 10 CDN and collagen+CDN mice reached this point. All animals surviving to day 105 survived a second tumor inoculation to day 140 without showing any signs of tumor growth.
Fig. 6.
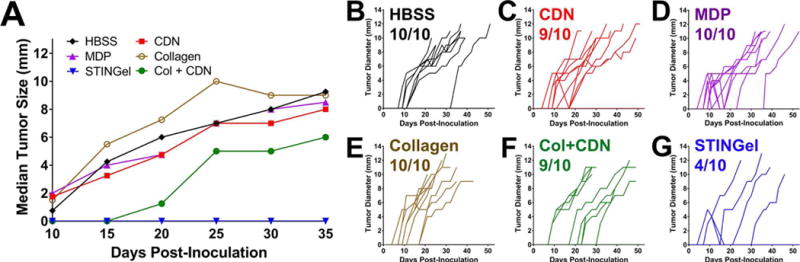
Tumor growth curves in controls and STINGel treated animals (n=10 per treatment group). (A) Median primary tumor growth for each group, showing significantly smaller median tumor size in CDN treated groups and a complete delayed growth in STINGel (MDP+CDN) C57BL/6 mice. (B–G) Individual tumor size growth data for tumor bearing mice in each group (number of tumor bearing mice above each plot), showing a clear improvement in progressive tumor free survival for the STINGel treated mice relative to controls and collagen+CDN. (B) HBSS, (C) CDN-alone, (D) MDP gel, (E) collagen gel, (F) collagen+CDN, (G) STINGel.
The Kaplan-Meier survival curve (Fig. 7) shows survival of mice from all six treatment groups, HBSS, CDN, MDP gel, STINGel, collagen or collagen+CDN based upon the time of euthanasia defined as tumors reaching 12 mm and a weight loss of 20%. In some instances, tumors reached up to 14 mm and did not have a weight loss of 20%. Treatments with STINGel resulted in a prolonged-disease free survival state and had the most number of survivors, 6/10 mice. Thus, we conclude that treatment with STINGel shows considerable antitumor efficacy.
Fig. 7.
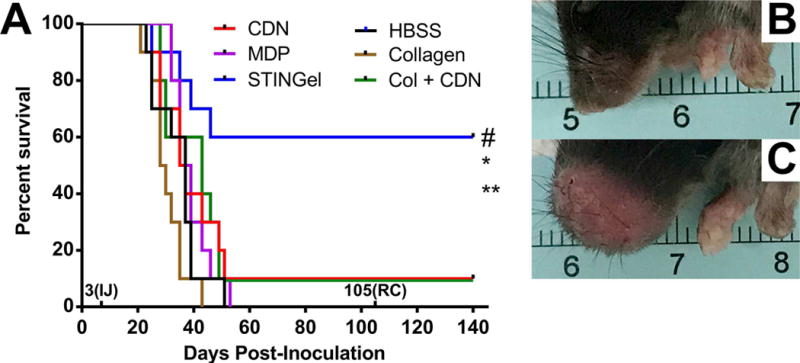
(A) Survival of the different experimental groups based on euthanasia timepoints resulting from excessive tumor burden. The total experimental period was 140 days post-tumor cell inoculation. The 3(IJ) on the x-axis refers to timepoint for intratumoral injection, and 105(RC) refers to timepoint for survivor rechallenge. Whereas 60% of the STINGel-treated C57BL/6 mice survived until the endpoint of the study, nearly all control group (HBSS, MDP gel, and collagen gel) mice were euthanized prior to reaching the endpoint due to excessive tumor burden. Only 10% of CDN alone and collagen+CDN treated mice survived (lines overlaid on plot). *p < 0.0282 vs. CDN, **p < 0.0064 vs. MDP gel, #p < 0.0498 vs. Collagen + CDN. (B) Representative image of STINGel treated mouse that maintained tumor clearance at day 37. (C) Representative image of CDN-only treated mouse growing tumor at day 37.
4. Discussion
The ability of biomaterials to allow for spatiotemporal control over payload delivery means diverse factors can be released in a controlled manner within a specific volume, reducing off-target toxicity while also enabling localized improvements in efficacy. Materials able to exploit such capabilities to release factors that can intelligently direct and modulate immune cells in situ are highly attractive, and thus we sought to develop STINGel as such a platform to improve current immunotherapies.
Through the use of favorable electrostatic interactions between the positive lysine termini and negative thiophosphate linkages, controlled and extended release of CDN was achieved in MDP compared to a collagen control gel (Fig. 2). The data show that the MDP releases its payload significantly slower than a collagen hydrogel, thereby increasing the length of time spent at higher concentrations of CDN.
Cell viability obtained in vitro, suggests that the initial concentration injected in vivo (910 μM) is strongly cytotoxic. This concentration was directly taken from the literature and previous studies that use CDN in vivo.28 The results suggest that one fourth of the initial concentration (228 μM) may be the max tolerable dose cells can experience in their local environment without the massive cell death observed at 455 μM and above (Fig. 3B). Even at 114 μM cells experience some initial stress and loss of viability, though at this concentration it is recoverable (Fig. 4). Thus, the in vitro data suggest we are injecting the STINGel material at a concentration that is initially toxic, and must decrease by almost a factor of 10 before it is no longer a direct threat to cell viability. CDN injected without gel or other delivery agent presumably diffuses away very rapidly and does not promote a lasting cytotoxic effect.
This aids in the interpretation of the subcutaneous histology, which shows a dramatic difference in cellular response to biomaterial loaded with CDN. At day 3, unloaded MDP shows an even dispersion of immune cells both rimming the hydrogel and thoroughly scattered in the inter-gel space (Fig. 5A). However, as described previously, CDN-loaded hydrogels show uneven cellular infiltration, and what appear to be dense pools of lymphocytes. Thus CDN-loading appears to substantially increase the chemotactic recruitment of a mixed population of inflammatory cells. Indeed, while large areas of the implant are non-infiltrated, with cells lining the edges, other areas of the hydrogel structure seem to be heavily disrupted by dense inflammatory infiltrate with substantial necrosis and nuclear debris (Fig. 5E). This observation is consistent with what was observed in vitro, that high CDN concentration resulted in significant cell death. Very high levels of Type I IFN activation is known to launch a transcriptional program that promotes cell death, which probably underpins CDN-induced cytotoxicity.39–40 Cell death may well be a beneficial aspect of this system, for nuclear debris will release danger-associated molecular patterns (DAMPs) that can further exacerbate inflammation by recruiting more immune cells to the area.41–42 Although some cytotoxicity of immune cells is well tolerated, which is also evidenced by the significant survival improvement upon STINGel treatment, further optimization of dosing and slow release kinetics may further enhance the therapeutic potency.
The STINGel material appears to create and prolong a period of high, localized CDN concentration, discouraging cellular infiltration into a cytotoxic implant while also directly causing cell necrosis. Our data indicates the material is injected at a high concentration and maintains this concentration for a significantly longer period of time than a collagen gel, due to the extended release of CDN and the lower equilibrium reached with the surrounding environment until diffusion carries CDN away. This promotion of cell death and lack of infiltration may be maintained until the concentration of CDN falls below a certain threshold (possibly around 114–228 μM). It is possible that at this point massive immune cell infiltration occurs (Fig. 5E). One possible explanation for this phenomenon is stimulation by CDN STING signaling23, 43 and further exacerbation by DAMP release.42 These results shed light on the anti-tumor efficacy of STINGel seen in this murine oral cancer model (Fig. 6–7).
We have hypothesized that the mechanism of this system’s efficacy is primarily spatiotemporal control over CDN delivery, in combination with exacerbation of the inflammation response by the inherent local cytotoxity of STINGel treatment. However, other explanations for the success of this system are possible. For example, a recent study with a similar CDN molecule showed that treatment efficacy was improved by combination with positively charged poly-arginine cell penetrating peptides, increasing cellular uptake and CDN drug internalization.44 CDNs are known to suffer from poor membrane permeability due to their negative phosphate linkages, often requiring high dosages or even viral transfection to aid internalization.45 Thus a reasonable hypothesis is that the complexation of the negative drug molecules with the highly positive MDP hydrogel in this study achieves not only controlled release, but also enhances cellular uptake.44 Further studies will investigate the mechanism of STINGel in detail.
A significant advantage of the delivery system used in our study is that anti-tumor efficacy is seen after only single injection of MDP biomaterial given at day 3 post tumor cell inoculation. In previous studies, sufficient activation of STING pathway required multiple CDN injections, and led only to a subset of MOC1 tumor rejection and unsuccessful MOC2 tumor rejection.28 Thus, as expected a single injection of CDN alone remained ineffective in preventing tumor growth. However a single injection of MDP biomaterial loaded with the same concentration of CDN (STINGel) is highly effective in maintain tumor clearance and rejecting tumor growth.
In summary, we demonstrate successful rejection of challenging MOC2-E6E7 tumors in wild type C57BL/6 mice with single injection of STINGel, establishing that 60% of STINGel treated mice exhibit complete anti-tumor response and acquired immunity. All mice that were rechallenged with a secondary inoculation exhibited no tumor growth, demonstrating that our MDP biomaterial enables a persistent immunological memory and promising durability of response. We believe that our injectable material creates and prolongs a period of high, localized CDN concentration, and thus can overcome the limitations associated with CDN monotherapy that have required higher and repeated doses to be administered for effective treatment to be observed.28
In principle, one might expect that CDN loaded into any hydrogel may have the same effect as observed here. In order to understand the differences in delivery material, we also evaluated an off-the-shelf material commonly used, a collagen hydrogel. Notably, our data show that CDN loaded collagen had no effect on improving survival over CDN alone. This suggests that the specific design criteria of the MDP is important for the drug delivery kinetics, supported by the fact that the release data from collagen hydrogels is significantly inferior to the MDP. Therefore, we believe that we have developed a uniquely effective biomaterial, a delivery platform capable of revitalizing CDN immunotherapy’s future in the lab and the clinic.
5. Conclusions
We have developed a cyclic dinucleotide-loaded multidomain peptide hydrogel we call STINGel, which dramatically improves overall survival in a challenging murine oral cancer model compared to CDN monotherapy injection. In addition to a six-fold improvement in survival, 100% of surviving mice demonstrate immunological memory and reject a secondary challenge of cancer cells. STINGel shows extended release kinetics compared to a CDN loaded collagen gel. This translates into superior survival demonstrating that the chemistry and structure of the MDP hydrogel plays a critical role which is not duplicated by collagen. The controlled release of CDN provided by STINGel creates a high local CDN concentration which is observed to affect the immediate vicinity of the hydrogel in vivo for at least seven days, including high immune cell recruitment and cytotoxicity. This suggests a mechanism for the improvement in survival observed over CDN alone or CDN poorly delivered by a collagen hydrogel, in which the local CDN concentration is rapidly depleted. Future studies will address these and other mechanistic issues in greater detail as we explore the scope of STINGel immunotherapy.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (grants DE021798, DE023577 and DE024173), the Oral & Maxillofacial Surgery Foundation (Research Support Grant), and the Welch Foundation (C1557). DGL was supported by the NSF Graduate Research Fellowship Program under Grant No. 1450681. The authors would also like to thank Nicole Carrejo, and I-Che Li for scientific discussions and Xu Zhang for help with statistical analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
DGL and JDH designed the kinetics, in vitro viability, and histological studies and DGL conducted the studies. TLLS provided additional histological data and interpretation. YLL provided pathological assessment of the histology data. ND, SLP, JDH, and SY designed the in vivo studies, and ND and SLP conducted the studies. DGL and ND analyzed the data, and DGL, JDH, ND, and SY interpreted the data. DGL, JDH, ND and SY wrote the manuscript.
Supplementary Data
Supplementary data relevant to this article is included in a separate document.
References
- 1.Langer R. New methods of drug delivery. Science. 1990;249(4976):1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 2.Koshy ST, Ferrante TC, Lewin SA, Mooney DJ. Injectable, porous, and cell-responsive gelatin cryogels. Biomaterials. 2014;35(8):2477–2487. doi: 10.1016/j.biomaterials.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bae KH, Wang LS, Kurisawa M. Injectable biodegradable hydrogels: Progress and challenges. J Mater Chem B. 2013;1(40):5371–5388. doi: 10.1039/c3tb20940g. [DOI] [PubMed] [Google Scholar]
- 4.Langer R, Vacanti JP. Tissue Engineering. Science. 1993;260(5110):920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 5.Silva EA, Mooney DJ. Effects of VEGF temporal and spatial presentation on angiogenesis. Biomaterials. 2010;31(6):1235–1241. doi: 10.1016/j.biomaterials.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shea LD, Smiley E, Bonadio J, Mooney DJ. DNA delivery from polymer matrices for tissue engineering. Nat Biotechnol. 1999;17(6):551–554. doi: 10.1038/9853. [DOI] [PubMed] [Google Scholar]
- 7.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nat Mater. 2009;8(2):151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Modena A, Ciccarese C, Iacovelli R, Brunelli M, Montironi R, Fiorentino M, Tortora G, Massari F. Immune Checkpoint Inhibitors and Prostate Cancer: A New Frontier? Oncol Rev. 2016;10(1):293. doi: 10.4081/oncol.2016.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Margolin K. The Promise of Molecularly Targeted and Immunotherapy for Advanced Melanoma. Curr Treat Options Oncol. 2016;17(9):48. doi: 10.1007/s11864-016-0421-5. [DOI] [PubMed] [Google Scholar]
- 10.Economopoulou P, Kotsantis I, Psyrri A. Checkpoint Inhibitors in Head and Neck Cancer: Rationale, Clinical Activity, and Potential Biomarkers. Curr Treat Options Oncol. 2016;17(8):40. doi: 10.1007/s11864-016-0419-z. [DOI] [PubMed] [Google Scholar]
- 11.Atkins MB, Larkin J. Immunotherapy Combined or Sequenced With Targeted Therapy in the Treatment of Solid Tumors: Current Perspectives. JNCI: J Natl Cancer I. 2016;108(6):djv414–djv414. doi: 10.1093/jnci/djv414. [DOI] [PubMed] [Google Scholar]
- 12.Hodi F, Kluger KM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma (MEL) who received nivolumab (NIVO) monotherapy in a phase I trial: Abstract CT001. 107th Annual Meeting of the American Association for Cancer Research: AACR; New Orleans, LA. 2016. [Google Scholar]
- 13.Errico A. CheckMate 067–frontline nivolumab improves PFS alone or in combination with ipilimumab. Nat Rev Clin Oncol. 2015;12:435. doi: 10.1038/nrclinonc.2015.112. [DOI] [PubMed] [Google Scholar]
- 14.Hauschild A, Garbe C. Combined immunotherapy–a new standard in metastatic melanoma? Nat Rev Clin Oncol. 2015;12 doi: 10.1038/nrclinonc.2015.118. [DOI] [PubMed] [Google Scholar]
- 15.Méry B, Rancoule C, Guy J-B, Espenel S, Wozny A-S, Battiston-Montagne P, Ardail D, Beuve M, Alphonse G, Rodriguez-Lafrasse C, Magné N. Preclinical models in HNSCC: A comprehensive review. Oral Oncol. 2017;65:51–56. doi: 10.1016/j.oraloncology.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 16.SEER Surveillance Epidemiology and End Results Fast Facts [Internet] http://seer.cancer.gov/faststats/selections.php?#Output (accessed June 06 2017).
- 17.SEER Cancer of the Oral Cavity and Pharynx - SEER Fast Fact Sheets. http://seer.cancer.gov/statfacts/html/oralcav.html (accessed June 06 2017).
- 18.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence M, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortés ML, Auclair D, Berger MF, Saksena G, Guiducci C, Onofrio R, Parkin M, Romkes M, Weissfeld JL, Seethala RR, Wang L, Rangel-Escareño C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Winckler W, Ardlie K, Gabriel SB, Meyerson M, Lander ES, Getz G, Golub TR, Garraway LA, Grandis JR. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science (New York, NY) 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gooi Z, Chan JYK, Fakhry C. The epidemiology of the human papillomavirus related to oropharyngeal head and neck cancer. The Laryngoscope. 2016;126(4):894–900. doi: 10.1002/lary.25767. [DOI] [PubMed] [Google Scholar]
- 20.Maxwell JH, Grandis JR, Ferris RL. HPV-Associated Head and Neck Cancer: Unique Features of Epidemiology and Clinical Management. Annu Rev Med. 2016;67:91–101. doi: 10.1146/annurev-med-051914-021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, Cheng JD, Chow LQ. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17(7):956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 22.Corrales L, Glickman Laura H, McWhirter Sarah M, Kanne David B, Sivick Kelsey E, Katibah George E, Woo S-R, Lemmens E, Banda T, Leong Justin J, Metchette K, Dubensky Thomas W, Jr, Gajewski Thomas F. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11(7):1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadkaree SK, Fu J, Sen R, Korrer MJ, Allen C, Kim YJ. Induction of tumor regression by intratumoral STING agonists combined with anti-programmed death-L1 blocking antibody in a preclinical squamous cell carcinoma model. Head Neck-J Sci Spec. 2017;16(Suppl 1):3. doi: 10.1002/hed.24704. -n/a. [DOI] [PubMed] [Google Scholar]
- 25.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478(7370):515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg M-C, Picard C, Neven B, Fischer A, Amigorena S, Rieux-Laucat F, Manel N. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med. 2017;214(6):1769. doi: 10.1084/jem.20161674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.AduroBiotech, NCT02675439: Study of the Safety and Efficacy of MIW815 (ADU-S100) in Patients With Advanced/Metastatic Solid Tumors or Lymphomas. National Institute of Health; Feb 5, 2016. ClinicalTrails.gov 2017. [Google Scholar]
- 28.Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T Cell–Inflamed Tumors Rejected after Adaptive Resistance Was Reversed by Combination STING Activation and PD-1 Pathway Blockade. Cancer Immunol Res. 2016;4(12):1061. doi: 10.1158/2326-6066.CIR-16-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore AN, Hartgerink JD. Self-Assembling Multidomain Peptide Nanofibers for Delivery of Bioactive Molecules and Tissue Regeneration. Accounts of Chem Res. 2017;50(4):714–722. doi: 10.1021/acs.accounts.6b00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aulisa L, Dong H, Hartgerink JD. Self-Assembly of Multidomain Peptides: Sequence Variation Allows Control over Cross-Linking and Viscoelasticity. Biomacromolecules. 2009;10(9):2694–2698. doi: 10.1021/bm900634x. [DOI] [PubMed] [Google Scholar]
- 31.Kumar VA, Shi S, Wang BK, Li I-C, Jalan AA, Sarkar B, Wickremasinghe NC, Hartgerink JD. Drug-triggered and cross-linked self-assembling nanofibrous hydrogels. J Am Chem Soc. 2015;137(14):4823–4830. doi: 10.1021/jacs.5b01549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li IC, Moore AN, Hartgerink JD. “Missing Tooth” Multidomain Peptide Nanofibers for Delivery of Small Molecule Drugs. Biomacromolecules. 2016;17(6):2087–2095. doi: 10.1021/acs.biomac.6b00309. [DOI] [PubMed] [Google Scholar]
- 33.Dong H, Paramonov SE, Aulisa L, Bakota EL, Hartgerink JD. Self-Assembly of Multidomain Peptides: Balancing Molecular Frustration Controls Conformation and Nanostructure. J Am Chem Soc. 2007;129(41):12468–12472. doi: 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- 34.Young SW, Dharmaraj N, Piotrowski S, Koshy ST, Li WA, Mooney DJ, Sikora AG. Overcoming Resistance to Checkpoint Inhibitors through a Rationally-Designed Combinatorial Immunotherapy Approach. J Oral Maxil Surg. 2017;75(10):e331–e332. [Google Scholar]
- 35.Young S, Dharmaraj N, Piotrowski S, Koshy ST, Li WA, Mooney DJ, Sikora AG. Overcoming Resistance to Checkpoint Inhibitors through a Rationally-Designed Combinatorial Immunotherapy Approach. J Dent Res. 2017;96(A) [Google Scholar]
- 36.Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS, Dunn GP, Bui JD, Sunwoo JB, Uppaluri R. ERK1/2 Regulation of CD44 Modulates Oral Cancer Aggressiveness. Cancer Res. 2012;72(1):365–374. doi: 10.1158/0008-5472.CAN-11-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar VA, Liu Q, Wickremasinghe NC, Shi S, Cornwright TT, Deng Y, Azares A, Moore AN, Acevedo-Jake AM, Agudo NR, Pan S, Woodside DG, Vanderslice P, Willerson JT, Dixon RA, Hartgerink JD. Treatment of hind limb ischemia using angiogenic peptide nanofibers. Biomaterials. 2016;98:113–119. doi: 10.1016/j.biomaterials.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar VA, Taylor NL, Shi S, Wang BK, Jalan AA, Kang MK, Wickremasinghe NC, Hartgerink JD. Highly Angiogenic Peptide Nanofibers. ACS Nano. 2015;9(1):860–868. doi: 10.1021/nn506544b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF Family Transcription Factors in Immunity and Oncogenesis. Annu Rev Immunol. 2008;26(1):535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 40.Lei Y, Moore CB, Liesman RM, O’Connor BP, Bergstralh DT, Chen ZJ, Pickles RJ, Ting JPY. MAVS-Mediated Apoptosis and Its Inhibition by Viral Proteins. PLoS ONE. 2009;4(5):e5466. doi: 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: Endogenous Danger Signaling. Mol Med. 2008;14(7–8):476–484. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukocyte Biol. 2007;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 43.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nature immunol. 2013;14(1):19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 44.Yildiz S, Alpdundar E, Gungor B, Kahraman T, Bayyurt B, Gursel I, Gursel M. Enhanced immunostimulatory activity of cyclic dinucleotides on mouse cells when complexed with a cell-penetrating peptide or combined with CpG. Eur J Immunol. 2015;45(4):1170–1179. doi: 10.1002/eji.201445133. [DOI] [PubMed] [Google Scholar]
- 45.McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, Fitzgerald KA, Hayakawa Y, Vance RE. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med. 2009;206(9):1899–1911. doi: 10.1084/jem.20082874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.