

SUMMARY
Targeting defects in metabolism is an underutilized strategy for the treatment of cancer. Arginine auxotrophy resulting from the silencing of argininosuccinate synthetase 1 (ASS1) is a common metabolic alteration reported in a broad range of aggressive cancers. To assess the metabolic effects that arise from acute and chronic arginine starvation in ASS1-deficient cell lines, we performed metabolite profiling. We found that pharmacologically induced arginine depletion causes increased serine biosynthesis, glutamine anaplerosis, oxidative phosphorylation, and decreased aerobic glycolysis, effectively inhibiting the Warburg effect. The reduction of glycolysis in cells otherwise dependent on aerobic glycolysis is correlated with reduced PKM2 expression and phosphorylation and upregulation of PHGDH. Concurrent arginine deprivation and glutaminase inhibition was found to be synthetic lethal across a spectrum of ASS1-deficient tumor cell lines and is sufficient to cause in vivo tumor regression in mice. These results identify two synthetic lethal therapeutic strategies exploiting metabolic vulnerabilities of ASS1-negative cancers.
In Brief
Using global metabolomics analysis and stable isotope tracing, Kremer et al. show that arginine starvation of ASS1-deficient tumors causes an increase in serine biosynthesis, glutamine anaplerosis, and oxidative phosphorylation with a simultaneous decrease in aerobic glycolysis. Pharmacological inhibition of escape pathways to arginine deprivation exhibits a synthetic lethal interaction.
INTRODUCTION
Metabolic reprogramming is a well established hallmark of cancer that remains to be fully exploited therapeutically (DeBerardinis et al., 2008; Hanahan and Weinberg, 2011). Common metabolic alterations in malignancies include the loss of argininosuccinate synthetase 1 (ASS1) expression and the energetic phenotype known as the Warburg effect (Dillon et al., 2004; Warburg, 1956). The Warburg effect refers to the preferential generation of ATP through aerobic glycolysis as opposed to mitochondrial oxidative phosphorylation (OxPhos) despite adequate oxygen levels (Warburg, 1956). The Warburg effect results in increased generation of glucose-derived biomass required for unrestrained cellular proliferation and tumor growth (DeBerardinis et al., 2008; Icard and Lincet, 2012; Lunt and Vander Heiden, 2011). Recent studies have also shown that the Warburg effect is important in providing rapidly proliferating cells with an electron acceptor in the form of pyruvate, necessary in the oxidative biosynthesis of aspartate (Birsoy et al., 2015). Aspartate is utilized for the biosynthesis of proteins, purines, and pyrimidines, and insufficient aspartate levels inhibit proliferation (Birsoy et al., 2015; Sullivan et al., 2015).
Meeting the specific energy needs of cancer cells often requires differential regulation of one or more metabolic enzymes. Pyruvate kinase muscle isozymes (PKMs), especially the embryonic splice isoform PKM2, are critical to rapidly dividing cells and are expressed exclusively in cells undergoing high levels of proliferation, such as embryonic and tumor cells (Chaneton and Gottlieb, 2012). Although the alternatively spliced isoform PKM1 is expressed in healthy cells, tumor formation coincides with dramatic increases in PKM2 expression (Chen et al., 2010; Christofk et al., 2008; Gumińska et al., 1997; Hitosugi et al., 2009; Macintyre and Rathmell, 2011). Further highlighting the tumorigenic consequences of altered PKM isoform expression, PKM2 expression has been shown to be sufficient, but not necessary, for induction of the Warburg effect, whereas overexpression of PKM1 results in inhibition of growth and proliferation of tumor cells (Chen et al., 2010; Grüning et al., 2011; Icard and Lincet, 2012; Marín-Hernández et al., 2009). PKM2 has been shown to be degraded through acetylation and subsequent chaperone-mediated autophagy under certain cellular contexts, prompting one approach to therapeutically target cancer metabolism by manipulation of PKM2 activity and protein levels (Cheong et al., 2012; Lv et al., 2011; Macintyre and Rathmell, 2011).
Phosphorylation of PKM2 decreases enzymatic activity, resulting in a lower rate of conversion of phosphoenolpyruvate (PEP) to pyruvate and altered glycolytic kinetics (Christofk et al., 2008; Hitosugi et al., 2009). A decreased pyruvate level reduces the flux of glycolytic metabolites into the tricarboxylic acid (TCA) cycle. However, the generation of lactate or acetyl-coenzyme A (CoA) from pyruvate ultimately depends on the regulation of cellular lactate dehydrogenases (LDH) and pyruvate dehydrogenases (PDH), respectively (Fan et al., 2011; Rardin et al., 2009).
The inhibition of the terminal step of glycolysis by PKM2 phosphorylation leads to the buildup of glycolytic intermediates that are shunted into the pentose phosphate pathway (PPP) and other biosynthetic pathways to facilitate the synthesis of nucleic acids, amino acids, lipids, and other cellular building blocks needed for cell growth and division (Grüning et al., 2011; Gumińska et al., 1997; Hitosugi et al., 2009; Icard and Lincet, 2012; Locasale et al., 2011; Macintyre and Rathmell, 2011). The amino acid serine acts as a natural allosteric activator of PKM2 in a regulatory feedback loop (Chaneton et al., 2012; Kung et al., 2012). PKM2 functions as a serine biosynthetic rheostat by increasing the rate of serine biosynthesis when enzymatically inactive until serine levels reach a minimal threshold and allosterically activate the enzymatic activity of PKM2 (Ye et al., 2012). This increase in PKM2 activity leads to decreased concentrations of glycolytic metabolites and subsequent decreases in the flux of glucose-derived carbon into serine biosynthesis. Therefore, PKM2 plays an important role in maintaining serine levels sufficient for cellular growth and proliferation (Chaneton et al., 2012; Kung et al., 2012; Locasale et al., 2011; Ye et al., 2012).
Another common metabolic alteration observed in many cancers is the silencing of the ASS1 gene that encodes the rate-limiting enzyme in the urea cycle, the metabolic pathway responsible for the clearance of nitrogenous waste and the biosynthesis of arginine (Delage et al., 2010). Because ASS1 catalyzes the formation of argininosuccinate from citrulline and aspartate, recent studies have found that the loss of ASS1 protein expression results in increased cellular abundance of aspartate and subsequent aspartate utilization in nucleotide and protein biosynthesis (Rabinovich et al., 2015). Additional studies have shown that loss of ASS1 expression is a prognostic biomarker of reduced metastasis-free survival in a variety of cancers (Allen et al., 2014; Changou et al., 2014; Kobayashi et al., 2010; Mok et al., 2007; Nicholson et al., 2009; Szlosarek et al., 2006). Loss of ASS1 expression leads to dependence upon extracellular arginine for continued cell growth, proliferation, and survival, known as arginine auxotrophy. Exploiting this auxotrophy through arginine starvation induces autophagy to maintain cell survival (Bean et al., 2016; Kim et al., 2009; Nicholson et al., 2009; Syed et al., 2013).
Preliminary studies, including clinical trials, have targeted ASS1 loss with the pegylated form arginine deiminase (ADI-PEG20), an enzyme that degrades arginine to citrulline, causing significant tumor growth inhibition. These studies have investigated certain types of sarcoma, melanoma, hepatocellular carcinoma, prostate cancer, leukemia, lymphoma, and pancreatic cancer (Bean et al., 2016; Delage et al., 2010, 2012; Miraki-Moud et al., 2015). Prolonged treatment with ADI-PEG20 has been shown to induce re-expression of ASS1 and subsequent resistance to arginine starvation (Feun et al., 2012; Shen et al., 2003). Acquired resistance to ADI-PEG20 has been an obstacle in the long-term efficacy of this biomarker-driven therapy (Izzo et al., 2004; Ott et al., 2013).
We hypothesized that understanding the metabolic response to arginine starvation would allow for the identification of emergent vulnerabilities that, when targeted, convert a growth arrest phenotype to a synthetic lethality. Therefore, we performed capillary electrophoresis mass spectrometry (CE-MS) on metabolite extractions from cell lines acutely and chronically treated with ADI-PEG20. Treatment caused dramatic alterations in cellular metabolism, including glucose, glutamine, and serine metabolism. We identify that arginine deprivation diverts glucose into the serine biosynthetic pathway and renders ASS1-deficient cells more sensitive to the inhibition of serine biosynthesis and downstream folate-dependent enzymes, reinforcing the previously documented importance of serine biosynthesis in cancer (Cantor and Sabatini, 2012; Locasale et al., 2011; Mattaini et al., 2015). Finally, treatment also caused a switch from dependence on aerobic glycolysis (the Warburg effect) to glutamine anaplerosis and OxPhos, thus identifying inhibition of glutamine metabolism as a synthetic lethal target in arginine-starved cells. Taken together, numerous metabolic alterations induced by arginine starvation can be exploited therapeutically to induce synthetic lethality in ASS1-deficient cancers.
RESULTS
Determining the Effects of Acute and Chronic Arginine Deprivation on Metabolism
The immediate response to arginine starvation in ASS1-deficient tumors is inhibition of cellular proliferation and induction of autophagy (Figures S1A and S1B; Bean et al., 2016; Delage et al., 2012; Kim et al., 2009; Syed et al., 2013). During the period of growth arrest, ASS1 is re-expressed in a c-Myc-dependent manner, which allows cells to escape the effects of arginine deprivation, overcome autophagy, and resume proliferation (Figure 1A; Figure S1A; Long et al., 2013; Tsai et al., 2012). To identify synthetic lethal interactions with arginine starvation, we determined the metabolic alterations that occur when cells adapt to acute and chronic arginine deprivation. To model the long-term effects of arginine deprivation and identify potential synthetic lethal interactions, we continuously cultured three cell lines that lack functional expression of ASS1 in the presence of ADI-PEG20 for at least 3 months. This included two leiomyosarcoma cell lines (SKLMS1 and SKUT1) and a melanoma cell line (SKMEL2) (Figure 1A). A melanoma line was included to investigate whether the metabolic alterations identified were a sarcoma-specific phenomenon or a general characteristic of cellular metabolism in ASS1-deficient cancer cells. The long-term ADI-PEG20-treated (LTAT) cell lines are resistant to the growth-inhibitory effects of arginine deprivation because of the re-expression of ASS1 and reacquired de novo arginine biosynthesis capabilities (Figures 1A and 1B). Although ADI-PEG20-induced arginine deprivation resulted in the inhibition of cellular proliferation in wild-type (WT) cells, proliferation rates of LTAT cell lines differed compared with their parental lines but did not follow a consistent pattern (Figure 1B).
Figure 1. Effects of Acute and Chronic Arginine Deprivation on Cell Growth and Metabolism.

(A) Immunoblot measuring ASS1 and c-Myc expression in untreated WT, short-term ADI-PEG20-treated, and LTAT samples (representative of n = 3).
(B) Viable cell count normalized to plating density after short- and long-term ADI-PEG20 treatment (n = 3). Data are represented as mean ± SD.
(C) Metabolite levels in SKLMS1 WT NT, WT + ADI-PEG20, and LTAT cells (n = 2). Data are represented as mean ± SD.
G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; F1,6P, fructose-1,6,bisphosphate; 3-PG, 3-phosphoglycerate; 2-PG, 2-phosphoglycerate; α-KG, α-ketoglutarate. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figures S1 and S2 and Table S1.
To investigate the metabolic changes resulting from both short- and long-term arginine deprivation, we profiled the metabolome of the SKLMS1 wild-type cell line with and without ADI-PEG20 treatment as well as the SKLMS1 LTAT cell line using CE-MS. Profiling the major metabolites of purine and glutathione metabolism, glycolysis, the TCA and urea cycle, and other pathways (Table S1) identified global metabolomic alterations upon arginine deprivation and confirmed the depletion of intracellular arginine and increase in citrulline expected with ADI-PEG20 treatment (Figure S2A). A heatmap of relative metabolite abundances displayed the three unique metabolic profiles of the untreated WT, WT + ADI-PEG20, and SKLMS1 LTAT cells (Figure S2B). Because of negligible metabolic changes between untreated SKLMS1 LTAT and SKLMS1 LTAT + ADI-PEG20 cells, the untreated SKLMS1 LTAT line was dropped from further studies, and all samples denoted “LTAT” were carried out under continuous ADI-PEG20 treatment (Figure S2B). Principle component analysis (PCA) confirms the three unique metabolite profiles identified in the heatmap (Figure S2C). These results demonstrate that both acute and chronic arginine deprivation significantly alter cellular metabolism of the SKLMS1 cell line.
Treatment of SKLMS1 cells with ADI-PEG20 altered upper and lower glycolysis, glutamine, and serine metabolism (Figure 1C). Additionally, the SKLMS1 LTAT line demonstrated an accumulation of pyruvate and a depletion of glutamine (Figure 1C). We hypothesized that these metabolic alterations are required for adaptation and eventual escape to the arginine starvation-induced growth arrest and, thus, may be clinically actionable targets.
Loss of PKM2 and Changes in Glucose Biology upon Arginine Deprivation
Because PKM2 is an important regulatory enzyme in cancer glycolysis, changes in total PKM2 levels and phosphorylation status were determined in WT untreated, ADI-PEG20-treated, and LTAT cell lines. A substantial decrease in PKM2 levels was observed upon ADI-PEG20 treatment (Figure 2A). Levels of phospho-PKM2 also decreased upon acute ADI-PEG20 treatment, demonstrating that altered PKM2 levels and phospho-driven changes in enzyme kinetics occur upon ADI-PEG20 treatment. The ratio of phospho-PKM2 to PKM2 with acute ADI-PEG20 treatment demonstrates that both protein levels and phosphorylation states are altered simultaneously (Figure 2A). The ratio in the LTAT cell lines follows no discernable pattern (Figure 2A). The level of PKM1 remained constant (Figure 2A). Together, these data suggest that alterations in PKM2 kinetics may play a role in the changes in glycolysis seen upon arginine starvation.
Figure 2. Loss of PKM2 and Alterations in Glycolysis after Arginine Deprivation.

(A) Protein expression in untreated WT, WT + ADI-PEG20, and LTAT cell lines. Densitometry was performed on p-PKM2/PKM2 and PKM2/tubulin, with relative expression normalized to untreated levels shown (representative of n = 3),
(B) Normalized proliferation rates of cells grown in various glucose concentrations after 36 hr (n = 3). Data are represented as mean ± SD.
(C) Changes in glucose uptake upon short- and long-term ADI-PEG20 treatment (n = 6). Data are represented as mean ± SD.
(D) Oligomycin-induced cell death in WT and LTAT cell lines as assayed by propidium iodide (PI) fluorescence-activated cell sorting (FACS) (n = 3). Data are represented as mean ± SD.
*p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
Next, we sought to determine whether the adaptation to arginine starvation altered cellular glucose dependence. Therefore, WT and LTAT cell lines were cultured in low-glucose medium (Figure 2B). When normalized to the proliferation rate of samples grown in normal medium, there was no significant difference in the proliferation rate of WT and LTAT samples in low-glucose concentrations (Figure 2B). To further determine changes in glucose biology upon arginine starvation, extracellular glucose uptake was measured in cell lines treated with ADI-PEG20 as well as LTAT cell lines and demonstrated lower glucose uptake upon short- and long-term arginine deprivation (Figure 2C). Taken together, this suggests that the underlying glucose biology differs between WT and LTAT cell lines. Therefore, we examined the ATP measurements from the CE-MS data and found an acute decrease and long-term increase in ATP levels in short-term ADI-PEG20-treated and LTAT cell lines, respectively (Figure S3A). Thus, we sought to determine whether arginine deprivation altered the cellular dependence on mitochondrial ATP generation via OxPhos.
To test the OxPhos dependence of each cell line, samples were treated with oligomycin, an inhibitor of complex V in the electron transport chain. The LTAT lines were significantly more sensitive to cell death by oligomycin, indicating an increased reliance on mitochondrial OxPhos ATP generation upon long-term ADI-PEG20 treatment (Figure 2D). Although some of the wild-type cell lines experienced minor growth inhibition in the presence of oligomycin (data not shown), LTAT cell lines invariably showed higher levels of cell death. The wild-type cell lines, therefore, appear to more closely follow the phenotype described as the Warburg effect, whereby tumor cells preferentially ferment glucose into lactate regardless of the oxygen abundance available for glucose oxidation. LTAT cell lines did not follow the classical definition of the Warburg effect as closely as their respective WT cell lines, as illustrated by the increased sensitivity to cell death by disruption of mitochondrial OxPhos (Figure 2D). Taken together, the data indicate that the LTAT cell lines adapt to long-term ADI-PEG20 treatment by alterations in glucose metabolism and additionally suggest that the response to arginine starvation in ASS1-deficient cell lines is an increased reliance upon OxPhos over aerobic glycolysis.
U13C Glucose Tracing Shows Upregulated Glucose-Dependent Serine Biosynthesis
Next, we sought to identify the changes in glucose utilization upon short- and long-term ADI-PEG20 treatment by utilizing U13C glucose tracings and subsequent mass spectrometry. The cellular abundance of 54 metabolites as well as all possible 13C isotopomers was determined after short- and long-term ADI-PEG20 treatment. ADI-PEG20-treated cells exhibited a marked decrease in 13C-labeled metabolites in the PPP and downstream ATP/ADP/AMP, lactate, as well as TCA cycle intermediates (6-phosphogluconolactone [6PG] and citrate, respectively, in Figure 3A and ATP/ADP/AMP in Figure S3B). The decrease in 13C-labeled lactate upon ADI-PEG20 treatment indicates a shift away from aerobic glycolysis and is likely due to the concomitant decreases observed in both lactate dehydrogenase A (LDHA) levels and the activating phosphorylation of Tyr10 (Figure 2A; Fan et al., 2011). The decreased carbon labeling of lactate and PPP metabolites upon arginine deprivation is consistent with inhibition of the Warburg effect.
Figure 3. Glucose and Serine Metabolism after Arginine Deprivation.

(A) Average relative and normalized isotopomer abundance from U13C glucose in SKLMS1 WT NT, WT + ADI-PEG20, and SKLMS1 LTAT cell lines. Red arrows indicate increased 13C flux upon short-term treatment (n = 3).
(B) Cell death induction in WT cell lines after treatment with ADI-PEG20 and/or CBR-5884 as measured by PI FACS (n = 3). Data are represented as mean ± SD.
(C and D) PI FACS measurement of cell death in WT and LTAT cell lines after treatment with CBR-5884 (n = 3) (C) or treatment with MTX (n = 3) (D). Data are represented as mean ± SD.
*p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
Immunoblot analysis shows a significant decrease of pyruvate dehydrogenase subunit E1α upon arginine deprivation as well as a simultaneous increase in the inactivating phosphorylation of Ser300 on E1α (Figure 2A; Rardin et al., 2009). These alterations would decrease the contribution of glucose-derived carbons to acetyl-CoA and TCA cycle intermediates and increase the need for TCA cycle anaplerosis via other carbon sources.
Upon ADI-PEG20 treatment, glucose-derived carbons are shunted into the serine biosynthesis pathway, as demonstrated by U13C-glucose tracing (Figure 3A). Immunoblotting for phosphoglycerate dehydrogenase (PHGDH), the first and rate-limiting enzyme in the biosynthesis of serine, showed a significant upregulation upon acute arginine deprivation, with either stable levels or further increases in LTAT cell lines (Figure 2A). Direct inhibition of PHGDH with the small-molecule inhibitor CBR-5884 was found to cause significantly greater levels of cell death when paired with ADI-PEG20 treatment (Figure 3B; Mullarky et al., 2016). Although 13C labeling of serine from glucose in LTAT cell lines returned to levels similar to wild-type cell lines, LTAT lines demonstrated increased sensitivity to PHGHD inhibition with CBR-5884 compared with the WT lines as well as increased 13C labeling in glycine, indicating continued dependence on the serine biosynthesis pathway (Figure 3C).
Serine is converted into glycine by removal of a single carbon in a folate-dependent reaction, where both glycine and the single carbon transferred to the folate cofactor are required for the biosynthesis of purines and pyrimidines (Lane and Fan, 2015; Locasale, 2013). We therefore assayed cellular susceptibility to methotrexate (MTX), an anti-folate analog that inhibits dihydrofolate reductase and stops folate cycle-dependent single-carbon metabolism. Upon methotrexate treatment, LTAT cell lines showed a significantly higher level of cell death than WT cell lines (Figure 3D). The significant increase in cell death with methotrexate and CBR-5884 in LTAT lines is consistent with the upregulation in serine and glycine biosynthesis seen in the 13C tracing data by the increased label seen in glycine, indicating the continued upregulation of glucose flux through the serine biosynthesis pathway upon short- and long-term ADI-PEG20 treatment.
Changes in Glutamine Dependence and Utilization after Arginine Deprivation
To determine cellular reliance on glutamine, WT and LTAT cell lines were cultured in glutamine-free medium. LTAT lines were significantly more susceptible to glutamine withdrawal than their WT counterparts (Figure 4A). From the CE-MS data, cellular abundance of glutamine decreases significantly in a time-dependent fashion upon ADI-PEG20 treatment; longer treatment is associated with more profound reductions in glutamine levels (Figure 4B). This loss reflects increased usage of glutamine as opposed to decreased biosynthesis because the cell lines were cultured in excess glutamine. A significant increase in extracellular uptake of radiolabeled glutamine ([3H]-Gln) after short-term ADI-PEG20 treatment and further increases in uptake upon long-term treatment demonstrate the increased utilization of glutamine upon arginine deprivation (Figure 4C). Glutamine is shunted into the TCA cycle upon arginine deprivation, with each of the cell lines showing either increased glutaminase (GLS) and/or glutamate dehydrogenase (GDH) protein levels upon ADI-PEG20 treatment (Figure 4D). Additionally, short hairpin RNA (shRNA)-mediated GLS knockdown decreases cell viability under conditions of glucose deprivation, demonstrating the dependence on the metabolic shift to glutamine utilization in the absence of glucose (Figure S4). Taken together, these data suggest that arginine deprivation increases the rate of glutamine anaplerosis and oxidation through the TCA cycle.
Figure 4. Glutamine Metabolism after Arginine Deprivation.

(A) Normalized viable cell counts of WT and LTAT cell lines grown in glutamine-free medium after 24, 48, or 72 hr (n = 3). Data are represented as mean ± SD.
(B) Metabolite levels in SKLMS1 WT NT, WT + ADI-PEG20, and LTAT cell lines (n = 2). Data are represented as mean ± SD.
(C) Normalized measurements of [3H]-Gln uptake after short- and long-term ADI-PEG20 treatment (n = 12). Data are represented as mean ± SEM.
(D) Immunoblot analysis of enzymes involved in glutamine metabolism and asparagine biosynthesis upon treatment with ADI-PEG20 (representative of n = 3).
(E) Average relative and normalized isotopomer abundance from stable isotope tracing of U13C glutamine in SKLMS1 WT NT, WT + ADI-PEG20, and SKLMS1 LTAT cells (n = 3).
*p < 0.05, **,p < 0.01, ***p < 0.001. See also Figures S4 and S5.
The decrease in 13C glucose-derived TCA cycle intermediates upon ADI-PEG20 treatment (Figure 3A) suggests an increased flux of other carbon sources into the TCA cycle, with data suggesting an ADI-PEG20-induced upregulation of glutamine anaplerosis. To demonstrate an increased rate of glutamine flux into the TCA cycle and alterations in the fate of glutamine upon short- and long-term ADI-PEG20 treatment, SKLMS1 lines were cultured with U13C glutamine and subjected to metabolite extraction and CE-MS. Tracing the five 13C-labeled carbons from glutamine demonstrates entrance into the TCA cycle, subsequent oxidative decarboxylation, oxidation, and hydration to form malate, with further oxidation to oxaloacetate and cataplerosis for the biosynthesis of aspartate and asparagine (Figure 4E). The increase in the four 13C labels seen in malate upon ADI-PEG20 treatment informs this directionality. Additionally, dramatic upregulation in asparagine synthetase (ASNS) occurred concurrently with the significant increase in asparagine labeling (Figure 4D). The total relative amount of 13C-labeled citrate decreased, with small increases in five 13C labeling in citrate suggesting a slight increase in the rate of reductive decarboxylation. Finally, no significant increase in 13C accumulation in cytidine triphosphate (CTP) or uridine triphosphate (UTP) was seen (Figure S5), and a small component of glutamine-derived 13C was found to be utilized in glutathione biosynthesis (Figure 4E). Taken together, we find that glutamine anaplerosis is upregulated upon ADI-PEG20 treatment, with the major benefactor being amino acids, specifically aspartate and asparagine.
Inhibition of the Warburg Effect after Arginine Deprivation
Because arginine deprivation caused an increase in glutamine utilization (Figure 4), an increase in susceptibility to cell death by oligomycin treatment (Figure 2D), and a decrease in glucose fermentation into lactic acid (Figure 3A), we hypothesized that the changes in the metabolic network upon ADI-PEG20 treatment resulted in an inhibition of the Warburg effect. To test this hypothesis, we used the Seahorse XF96 to profile the metabolic phenotypes of cells under normal conditions and in the context of arginine starvation. When measuring the extracellular acidification rate (ECAR), an indicator of lactic acid fermentation, we observed a significant increase in ECAR of untreated cells upon addition of glucose to the medium. However, when cells were pretreated with ADI-PEG20, the increase in extracellular acidification upon addition of glucose was significantly blunted (Figure 5A). This suggests that untreated cells preferentially ferment glucose into lactic acid at a higher rate than cells treated with ADI-PEG20, which have a decreased rate of glucose fermentation.
Figure 5. Inhibition of the Warburg Effect after Arginine Deprivation.

(A and B) Normalized measurements of ECAR (A) and OCR (B) upon ADI-PEG20 treatment (WT, n = 11; ADI, n = 12).
(C) Percent of ATP generated from glycolysis in WT and LTAT cell lines (WT, n = 11; LTAT, n = 12).
(D) Plot of ECAR versus OCR measurements upon short- and long-term ADI-PEG20 treatment (WT, n = 11; ADI, n = 12; LTAT, n = 12).
All data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***,p < 0.001. See also Figure S6.
Mitochondrial respiration was profiled by measurements of the oxygen consumption rate (OCR) to determine changes in the level of OxPhos-dependent ATP generation upon arginine deprivation. The spare respiratory capacity, or the ability of the mitochondria to generate additional ATP via the electron transport chain when under conditions of energy stress, was significantly decreased in cells treated with ADI-PEG20 (Figure 5B). This indicates that the mitochondria in these cells are less capable of upregulating OxPhos ATP generation upon increased energy demand than the mitochondria of untreated cells. The increased sensitivity of LTAT cell lines to cell death by oligomycin further suggests an increased reliance on OxPhos under conditions of arginine deprivation (Figure 2D). Additionally, the percentage of total cellular ATP generated from glycolysis is significantly decreased in LTAT cell lines compared with WT cell lines (Figure 5C). The decrease in lactic acid fermentation and increased rate of OxPhos indicate that arginine deprivation can inhibit the Warburg effect. As shown by the energetic profiling (Figure 5D), glycolytic WT cell lines decrease the ECAR and increase the OCR upon short- and long-term ADI-PEG20 treatment as they downregulate the Warburg effect and upregulate OxPhos in response to arginine starvation.
Previous studies in breast cancer have shown a decrease in mitochondrial OxPhos upon treatment with ADI-PEG20 because of decreased Cox4 expression, in contrast to the cellular response to arginine deprivation we identified (Qiu et al., 2014). When our samples were examined for Cox4 expression using the same antibody, no consistent pattern emerged, with SKLMS1 demonstrating increased Cox4 expression (Figure S6). This may reflect a difference in the biology of breast cancer compared with melanoma and sarcoma or a difference in the breast cancer cell line response to ADI-PEG20 treatment.
Targeting Upregulation of Glutamine Metabolism Causes Synthetic Lethality
The increased reliance on glutamine anaplerosis and OxPhos upon arginine deprivation identifies a potential synergy in arginine deprivation and inhibition of glutamine metabolism. To test the potential for a synthetic lethal interaction between ADI-PEG20 treatment and inhibition of glutamine metabolism, cell lines were treated with either ADI-PEG20, a GLS inhibitor (bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide [BPTES]), or the two drugs in combination. The combination of ADI-PEG20 and BPTES resulted in a significantly greater reduction in viable cell number than either drug individually (Figure 6A) and a greater increase in cell death upon simultaneous treatment than either drug individually (Figure 6B). To ensure that these results were not a consequence of an off-target effect, the synthetic lethality was tested in cell lines after GLS shRNA knockdown (Figure 6C). In the GLS knockdown cells, treatment with ADI-PEG20 reduced the viable cell number to the same degree as treatment with ADI-PEG20 and BPTES in combination, as opposed to the control cell lines, where only the combination of treatments demonstrated a synergetic interaction, indicating minimal off-target effects (Figure 6C). We then tested a broad range of ASS1-deficient cell lines identified from the NCI-60 cell line (including breast, colon, lung, glioblastoma, and head and neck cancer cell lines), a patient-derived xenograft breast cancer cell line, and an osteosarcoma cell line with ADI-PEG20 and BPTES and found the combination to similarly result in a greater reduction in viable cell count than either drug individually (Figure 6D).
Figure 6. ADI-PEG20 Treatment and Glutamine Metabolism Inhibition Induce Synthetic Lethality.

(A and B) Normalized viable cell counts (n = 3) (A) and PI FACS measurement of cell death (B) after treatment with ADI-PEG20 and/or BPTES (n = 3). Data are represented as mean ± SD.
(C) Normalized viable cell counts after ADI-PEG20 and/or BPTES treatment in shGFP and shGLS cell lines (n = 3). Data are represented as mean ± SD.
(D) Normalized viable cell count after treatment with ADI-PEG20 and/or BPTES in a variety of cancer types. Statistics represent significance between samples treated with ADI-PEG20 and BPTES and samples from other treatment conditions (n = 3). Data are represented as mean ± SD.
(E) Tumor volume of SKMEL2 WT shGFP and shGLS xenografts left untreated and treated with ADI-PEG20. Data are represented as mean ± SEM. Statistics represent significance between shGFP and shGLS tumors treated with ADI-PEG20 (n = 5).
(F) Immunoblot analysis of SKMEL2 WT shGFP and shGLS tumors harvested following treatment with ADI-PEG20 for 30 days.
*p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S7.
Unfortunately, in vivo experiments using BPTES in mice were prevented by crystallization of the compound upon injection. Therefore, as an alternative, shRNA targeting the coding sequence for glutaminase (shGLS) and shRNA targeting the coding sequence for green fluorescent protein (shGFP) SKMEL2 xenografts were utilized to recapitulate the synthetic lethality in vivo. Xenografts with shGLS constructs demonstrated approximately a 12-day delay in growth to 100 mm3 compared with the shGFP control tumors; when established, all tumors grew at approximately the same rate (Figure 6E). When experimental tumors reached approximately 100 mm3, ADI-PEG20 was injected intramuscularly once every 3 days. Although ADI-PEG20 treatment caused the shGFP control tumors to remain static for a short period of time before gaining resistance to the drug and resuming growth, the shGLS tumors shrank significantly upon treatment with ADI-PEG20 (Figure 6E). These results indicate that treatment with ADI-PEG20 and simultaneous GLS inhibition decreases the tumor burden in vivo. Analysis of xenografts on day 30 showed ASS1 re-expression or selection for a subpopulation of cells with less effective GLS knockdown, likely mechanisms of acquired resistance to ADI-PEG20 growth inhibition (Figure 6F), further supporting in vivo synthetic lethality. Finally, LTAT shGFP and shGLS tumors treated with ADI-PEG20 grew at the same rate as the untreated WT shGFP tumors; selection for LTAT shGLS cells likely induced further reprogramming of cellular metabolism to compensate for the increased reliance on glutamine seen in the LTAT lines (Figure S7).
DISCUSSION
We demonstrate that the removal of arginine from ASS1-deficient, arginine auxotrophic cancers rapidly alters metabolism, downregulates the Warburg effect, and upregulates glutamine anaplerosis (Figure 7). Interestingly, targeting a defect in the urea cycle can lead to the upregulation of OxPhos, glutamine metabolism, and serine biosynthesis. This shift renders cells vulnerable to synthetic lethal metabolic strategies in which ADI-PEG20 induces arginine starvation, whereas glutaminase inhibitors suppress the compensatory reliance on glutamine and PHGDH inhibitors prohibit the upregulation of serine biosynthesis. Although neither GLS inhibition nor arginine deprivation alone is sufficient to induce high levels of cell death because of the rapid metabolic reprogramming that occurs upon single-agent exposure, together they are therapeutically effective at killing tumor cells and suppressing tumor growth. ADI-PEG20 also diverts glucose into serine biosynthesis and subsequent downstream single-carbon folate metabolism, sensitizing cells to anti-folate metabolites such as methotrexate.
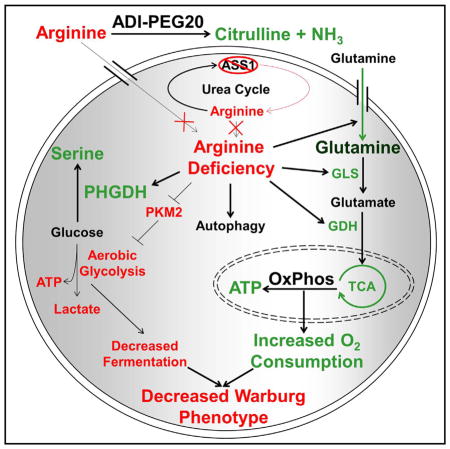
Figure 7. Model of Arginine Deprivation-Induced Metabolic Changes in ASS1-Deficient Cancers.

Model of metabolic changes upon ADI-PEG20-induced arginine deprivation causing the shift of glucose from aerobic glycolysis to glucose-dependent serine biosynthesis and the simultaneous upregulation of glutamine anaplerosis and aspartate and asparagine biosynthesis. THF, tetrahydrofolate; meTHF, 5,10-methylenetetrahydrofolate.
Many defects in metabolism have been identified in cancer, including ASS1 deficiency, isocitrate dehydrogenase (IDH) mutations, overexpression of PKM2, succinate dehydrogenase and fumarate hydratase deficiencies, and mutations in various mitochondrial complex proteins (Rohle et al., 2013). With the exception of IDH mutations in leukemia being sensitive to AG-221 (an inhibitor of IDH) monotherapy, single-agent metabolic targeting usually leads to rapid tumor metabolic re-networking and therapeutic resistance (Allen et al., 2014; Gaude and Frezza, 2014; Sasaki et al., 2012). Here we demonstrate that, by measuring the global metabolic changes in response to arginine deprivation, we can not only identify adaptive pathways but also develop therapies to target them. Importantly, this is the first reported dual metabolic synthetic lethal therapeutic strategy for cancer that relies solely on targeting metabolic pathways as opposed to layering a metabolic strategy on top of the current standard of care, such as chemotherapy, in an attempt to improve efficiency.
Another key observation is the realization that the Warburg effect can be exploited for therapeutic benefit. ADI-PEG20 treatment of glycolytic, ASS1-deficient cell lines resulted in inhibition of the Warburg effect. These changes were accompanied by a simultaneous redirection of glucose through the serine biosynthetic pathway and compensatory engagement of a survival program utilizing glutamine anaplerosis to harness the TCA cycle for energy and biomass generation. By altering how cancer cells utilize glucose, we can disrupt the ability of tumor cells to build the biomass necessary to sustain rapid cellular division.
Finally, using a global metabolomic approach to study the acute and long-term effects of arginine starvation in ASS1-deficient tumors, we develop a paradigm in which changes in metabolic pathways can be used to develop multidrug, biomarker-driven cancer therapies that exploit the unique metabolism of cancers. The ability to acutely alter the metabolome and thereby induce shifts in nucleotide synthesis, amino acid composition, and lipid metabolism has the potential to uncover other metabolic vulnerabilities specific to cancer cells. It is our hope that we can utilize this type of biomarker-directed approach to selectively starve and then kill cancer cells with minimal effect on normal tissues.
EXPERIMENTAL PROCEDURES
Metabolomic Analysis
SKLMS1 WT and LTAT were plated in 10-cm dishes and either left untreated or treated with 1 μg/mL ADI-PEG20 for 72 hr. Methanol metabolite extraction was performed according to the human metabolome technologies (HMT) metabolite extraction method for adherent cells. Metabolite concentrations were normalized to viable cell counts.
For U13C tracings, SKLMS1 lines were plated with or without 1 μg/mL ADI-PEG20 for 48 hr, and then U13C-labeled medium was added. U13C glucose and U13C glutamine (Cambridge Isotope Laboratories) medium contained glucose at 4.5 g/L and glutamine at 2 mM and was incubated with or without 1 μg/mL ADI-PEG20 for 48 hr. After an additional 24-hr incubation period, methanol metabolite extraction was performed according to the HMT metabolite extraction for adherent cells protocol. Data were corrected for the natural abundance of the stable isotope.
Extracellular Flux Analysis
Samples were plated in a 10-cm dish with or without ADI-PEG20. After 48 hr, cells were collected, and 20,000 cells were plated per well in a 96-well Seahorse plate with normal or ADI-PEG20 pretreated medium. Assays were performed 18–24 hr after plating with a XF96 analyzer. Oligomycin, rotenone, and antimycin A were used at 10 μM, and carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) was used at 5 μM in Mito tests. Glucose was used at 10 mM, oligomycin at 5 μM, and 2-Deoxy-D-glucose (2-DG) at 100 mM for glycolysis tests. OCR measurements from mitochondrial stress tests are normalized to basal respiration rate, and ECAR measurements from glycolysis stress tests are normalized non-glycolytic ECARs. Percent glycolytic ATP was calculated with values of glycolytic ATP obtained from the glucose-induced proton production rate and OxPhos ATP obtained from oligomycin decrease in OCR. All materials were obtained from Agilent Technologies.
Statistics
Comparisons were performed with a Student’s t test with p values denoted as *p < 0.05, **p < 0.01, and ***p < 0.001 (NS, not significant).
Supplementary Material
Highlights.
ADI-PEG20 upregulates glutamine anaplerosis and oxidative phosphorylation
Targeting glutamine metabolism with ADI-PEG20 induces synthetic lethality
Arginine deprivation causes ASS1 deficient cells to upregulate serine biosynthesis
Acquired resistance to ADI-PEG20 sensitizes to inhibition of serine metabolism
Acknowledgments
The authors would like to thank Laura Luecking for assistance with the Seahorse experiments. Funding was received from CJ’s Journey, The Sarcoma Foundation of America, a Sarcoma Alliance for Research and Collaboration (SARC) Career Development Award (to B.A.V.T.), and Polaris Pharmecueticals (to B.A.V.T.). J.S.B. is employed at Polaris with stock ownership.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.12.077.
AUTHOR CONTRIBUTIONS
J.C.K., B.C.P., C.B.B., S.E.S.L., M.B.S., N.R., A.L., S.T., M.D.R., Z.M., J.M., S.C.T., and B.A.V.T. assisted with the experiments. J.C.K., K.K., L.S., J.S.B., J.M., T.T., and B.A.V.T. assisted with data analysis. T.T. assisted with chemical synthesis of BPTES. J.C.K., G.R.B., and B.A.V.T. assisted with hypothesis design. J.C.K., B.C.P., J.M., J.M.H., L.S.M., and B.A.V.T. designed the experiments and prepared the manuscript with help from all co-authors.
References
- Allen MD, Luong P, Hudson C, Leyton J, Delage B, Ghazaly E, Cutts R, Yuan M, Syed N, Lo Nigro C, et al. Prognostic and therapeutic impact of argininosuccinate synthetase 1 control in bladder cancer as monitored longitudinally by PET imaging. Cancer Res. 2014;74:896–907. doi: 10.1158/0008-5472.CAN-13-1702. [DOI] [PubMed] [Google Scholar]
- Bean GR, Kremer JC, Prudner BC, Schenone AD, Yao JC, Schultze MB, Chen DY, Tanas MR, Adkins DR, Bomalaski J, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death Dis. 2016;7:e2406. doi: 10.1038/cddis.2016.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci. 2012;37:309–316. doi: 10.1016/j.tibs.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458–462. doi: 10.1038/nature11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changou CA, Chen YR, Xing L, Yen Y, Chuang FYS, Cheng RH, Bold RJ, Ann DK, Kung HJ. Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proc Natl Acad Sci USA. 2014;111:14147–14152. doi: 10.1073/pnas.1404171111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zhang J, Manley JL. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010;70:8977–8980. doi: 10.1158/0008-5472.CAN-10-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30:671–678. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, Szlosarek PW. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 2010;126:2762–2772. doi: 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- Delage B, Luong P, Maharaj L, O’Riain C, Syed N, Crook T, Hatzimichael E, Papoudou-Bai A, Mitchell TJ, Whittaker SJ, et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis. 2012;3:e342. doi: 10.1038/cddis.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer. 2004;100:826–833. doi: 10.1002/cncr.20057. [DOI] [PubMed] [Google Scholar]
- Fan J, Hitosugi T, Chung TW, Xie J, Ge Q, Gu TL, Polakiewicz RD, Chen GZ, Boggon TJ, Lonial S, et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol Cell Biol. 2011;31:4938–4950. doi: 10.1128/MCB.06120-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feun LG, Marini A, Walker G, Elgart G, Moffat F, Rodgers SE, Wu CJ, You M, Wangpaichitr M, Kuo MT, et al. Negative argininosuccinate synthetase expression in melanoma tumours may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase. Br J Cancer. 2012;106:1481–1485. doi: 10.1038/bjc.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014;2:10. doi: 10.1186/2049-3002-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüning NM, Rinnerthaler M, Bluemlein K, Mülleder M, Wamelink MM, Lehrach H, Jakobs C, Breitenbach M, Ralser M. Pyruvate kinase triggers a metabolic feedback loop that controls redox metabolism in respiring cells. Cell Metab. 2011;14:415–427. doi: 10.1016/j.cmet.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumińska M, Ignacak J, Kedryna T, Stachurska MB. Tumor-specific pyruvate kinase isoenzyme M2 involved in biochemical strategy of energy generation in neoplastic cells. Acta Biochim Pol. 1997;44:711–724. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Icard P, Lincet H. A global view of the biochemical pathways involved in the regulation of the metabolism of cancer cells. Biochim Biophys Acta. 2012;1826:423–433. doi: 10.1016/j.bbcan.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V, Cremona F, Ensor CM, Holtsberg FW, Bomalaski JS, et al. Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol. 2004;22:1815–1822. doi: 10.1200/JCO.2004.11.120. [DOI] [PubMed] [Google Scholar]
- Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, Gandour-Edwards R, Chuang FY, Bold RJ, Kung HJ. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009;69:700–708. doi: 10.1158/0008-5472.CAN-08-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi E, Masuda M, Nakayama R, Ichikawa H, Satow R, Shitashige M, Honda K, Yamaguchi U, Shoji A, Tochigi N, et al. Reduced argininosuccinate synthetase is a predictive biomarker for the development of pulmonary metastasis in patients with osteosarcoma. Mol Cancer Ther. 2010;9:535–544. doi: 10.1158/1535-7163.MCT-09-0774. [DOI] [PubMed] [Google Scholar]
- Kung C, Hixon J, Choe S, Marks K, Gross S, Murphy E, DeLaBarre B, Cianchetta G, Sethumadhavan S, Wang X, et al. Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem Biol. 2012;19:1187–1198. doi: 10.1016/j.chembiol.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–2485. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Tsai WB, Wangpaichitr M, Tsukamoto T, Savaraj N, Feun LG, Kuo MT. Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Mol Cancer Ther. 2013;12:2581–2590. doi: 10.1158/1535-7163.MCT-13-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Rathmell JC. PKM2 and the tricky balance of growth and energy in cancer. Mol Cell. 2011;42:713–714. doi: 10.1016/j.molcel.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. 2009;9:1084–1101. doi: 10.2174/138955709788922610. [DOI] [PubMed] [Google Scholar]
- Mattaini KR, Brignole EJ, Kini M, Davidson SM, Fiske BP, Drennan CL, Vander Heiden MG. An epitope tag alters phosphoglycerate dehydrogenase structure and impairs ability to support cell proliferation. Cancer Metab. 2015;3:5. doi: 10.1186/s40170-015-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraki-Moud F, Ghazaly E, Ariza-McNaughton L, Hodby KA, Clear A, Anjos-Afonso F, Liapis K, Grantham M, Sohrabi F, Cavenagh J, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood. 2015;125:4060–4068. doi: 10.1182/blood-2014-10-608133. [DOI] [PubMed] [Google Scholar]
- Mok SC, Elias KM, Wong KK, Ho K, Bonome T, Birrer MJ. Biomarker discovery in epithelial ovarian cancer by genomic approaches. Adv Cancer Res. 2007;96:1–22. doi: 10.1016/S0065-230X(06)96001-1. [DOI] [PubMed] [Google Scholar]
- Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, Wong JC, Christen S, Takahashi H, Singh PK, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci USA. 2016;113:1778–1783. doi: 10.1073/pnas.1521548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson LJ, Smith PR, Hiller L, Szlosarek PW, Kimberley C, Sehouli J, Koensgen D, Mustea A, Schmid P, Crook T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int J Cancer. 2009;125:1454–1463. doi: 10.1002/ijc.24546. [DOI] [PubMed] [Google Scholar]
- Ott PA, Carvajal RD, Pandit-Taskar N, Jungbluth AA, Hoffman EW, Wu BW, Bomalaski JS, Venhaus R, Pan L, Old LJ, et al. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Invest New Drugs. 2013;31:425–434. doi: 10.1007/s10637-012-9862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu F, Chen YR, Liu X, Chu CY, Shen LJ, Xu J, Gaur S, Forman HJ, Zhang H, Zheng S, et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci Signal. 2014;7:ra31. doi: 10.1126/scisignal.2004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature. 2015;527:379–383. doi: 10.1038/nature15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem. 2009;389:157–164. doi: 10.1016/j.ab.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen LJ, Lin WC, Beloussow K, Shen WC. Resistance to the anti-proliferative activity of recombinant arginine deiminase in cell culture correlates with the endogenous enzyme, argininosuccinate synthetase. Cancer Lett. 2003;191:165–170. doi: 10.1016/s030-43835(02)00693-6. [DOI] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162:552–563. doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed N, Langer J, Janczar K, Singh P, Lo Nigro C, Lattanzio L, Coley HM, Hatzimichael E, Bomalaski J, Szlosarek P, et al. Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death Dis. 2013;4:e458. doi: 10.1038/cddis.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szlosarek PW, Klabatsa A, Pallaska A, Sheaff M, Smith P, Crook T, Grimshaw MJ, Steele JP, Rudd RM, Balkwill FR, Fennell DA. In vivo loss of expression of argininosuccinate synthetase in malignant pleural mesothelioma is a biomarker for susceptibility to arginine depletion. Clin Cancer Res. 2006;12:7126–7131. doi: 10.1158/1078-0432.CCR-06-1101. [DOI] [PubMed] [Google Scholar]
- Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, Kuo MT. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012;72:2622–2633. doi: 10.1158/0008-5472.CAN-11-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, Thompson CB. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci USA. 2012;109:6904–6909. doi: 10.1073/pnas.1204176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.