

Abstract
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disease characterized by the motor neuron degeneration that eventually leads to complete paralysis and death within 2-5 years after disease onset. One of the major pathological hallmark of ALS is abnormal accumulation of inclusions containing TAR DNA-binding protein-43 (TDP-43). TDP-43 is normally found in the nucleus, but in ALS, it localizes in the cytoplasm as inclusions as well as in the nucleus. Loss of nuclear TDP-43 functions likely contributes to neurodegeneration. TBPH is the Drosophila ortholog of human TDP-43. In the present study, we confirmed that Drosophila models harboring TBPH knockdown develop locomotive deficits and degeneration of motoneurons (MNs) due to loss of its nuclear functions, recapitulating the human ALS phenotypes. We previously suggested that ter94, the Drosophila ortholog of human Valosin-containing protein (VCP), is a modulator of degeneration in MNs induced by knockdown of Caz, the Drosophila ortholog of human FUS. In this study, to determine the effects of VCP on TDP-43-assosiated ALS pathogenic processes, we examined genetic interactions between TBPH and ter94. Overexpression of ter94 suppressed the compound eye degeneration caused by TBPH knockdown and suppressed the morbid phenotypes caused by neuron-specific TBPH knockdown, such as locomotive dysfunction and degeneration of MN terminals. Further immunocytochemical analyses revealed that the suppression is caused by restoring the cytoplasmically mislocalized TBPH back to the nucleus. In consistent with these observations, a loss-of-function mutation of ter94 enhanced the compound eye degeneration caused by TBPH knockdown, and partially enhanced the locomotive dysfunction caused by TBPH knockdown. Our data demonstrated that expression levels of ter94 influenced the phenotypes caused by TBPH knockdown, and indicate that reagents that up-regulate the function of human VCP could modify MN degeneration in ALS caused by TDP-43 mislocalization.
Keywords: Amyotrophic lateral sclerosis, TDP-43, VCP, Drosophila, neuron
Introduction
ALS is a progressive neurodegenerative disease characterized by degeneration of motoneurons (MNs). Clinical phenotypes of this disease are progressive muscle weakness and eventually fatal paralysis typically within 2-5 years after disease onset. The prevalence of ALS is 4-6 per 100,000 in most countries, and more than 15 million people currently succumb to the disease each year [1-3]. No fundamental treatment for ALS has been established. Pathologically, ALS is characterized by cytoplasmic protein aggregates containing transactivating response element DNA binding protein-43 (TDP-43) [4]. Brains from patients with frontotemporal lobar degeneration (FTLD) also show cytoplasmic protein aggregates containing TDP-43. About 20% of people with ALS meet the clinical criteria for a concomitant diagnosis of FTLD [3]. Some patients with ALS and FTLD have overlapping clinical and neuropathological features [4-7].
The DNA/RNA-binding protein TDP-43 (gene TARDBP) has been identified as the major constituent of the ubiquitin-positive neuronal inclusion bodies observed in patients with FTLD and ALS [4,8-12], and an association between mutations in TDP-43 and an inherited form of ALS was subsequently found [13-20]. The discovery of TDP-43 aroused much interest in molecules related to RNA processing in the pathogenesis of ALS [9]. Subsequently, mutations in another gene, fused in sarcoma (protein FUS, gene FUS) was identified as a genetic cause of both familial ALS and FTLD [10,21-24]. TDP-43 and FUS are RNA-binding proteins implicated in multiple aspects of RNA processing including splicing of mRNA precursors and shuttling of mRNAs between the nucleus and the cytoplasm [25,26]. Recent studies have indicated that C9orf72 may be involved in that process [27]. The di-peptide repeats arising from the hexanucleotide expansion mutation interferes with nucleocytoplasmic shuttling [28].
We previously developed a fly model of ALS by introducing knockdown of Cabeza (Caz) which is the Drosophila ortholog of human FUS [44], and demonstrated a genetic link between Caz and ter94, the Drosophila ortholog of human Valosin-containing-protein/p97 (VCP) [29]. Flies with neuron-specific Caz knockdown develop locomotive and anatomical deficits in MNs at neuromuscular junctions (NMJs) [30]. Overexpression of wild-type ter94 on the background of Caz knockdown remarkably suppressed those phenotypes and the nuclear deficiency of Caz [29]. Ter94 is predicted to share ~83% amino acid sequence identity with human VCP. VCP is a member of the family of ATPases associated with a variety of cellular activities. These proteins are implicated in a large variety of biological functions including the regulation of ubiquitin-dependent protein degradation, vesicle-mediated transport, autophagy/mitophagy, DNA repair and nucleocytoplasmic shuttling, and others [31,32]. VCP mutations may account for ~1%-2% of familial ALS [33]. Loss of function of the Drosophila ortholog for human TDP-43, TBPH, induces locomotive deficits, reduces life span, and leads to anatomical defects at NMJs [34,35]. These phenotypes resemble those of our Caz knockdown fly models. Most human patients with ALS have ubiquitinated inclusions that stain for TDP-43 rather than FUS [1]. In the study using Drosophila models, it is reported that FUS and TDP-43 function together in vivo [36]. Therefore, our previous findings regarding Caz and ter94 should be further generalized with TBPH and ter94.
In this study, we therefore examined the genetic interaction between TBPH and ter94. Overexpression of ter94 effectively rescued those phenotypes, whereas mutation in ter94 partially exacerbated locomotive disabilities induced by neuron-specific TBPH knockdown. These results suggest that Up-regulated VCP may suppress the pathogenic processes that lead to the degeneration of MNs in TDP-43-associated ALS/FTLD.
Materials and methods
Fly stocks
Fly stocks were maintained at 25°C on standard food containing 0.65% agar, 10% glucose, 4% dry yeast, 5% cone flour and 3% rice bran. The fly stock ter94K15502 (BL10454) was obtained from the Bloomington Drosophila Stock Center. Establishment of the lines carrying GMR-GAL4 was described previously [37]. RNAi lines used in this study were obtained from the Vienna Drosophila Resource Center (VDRC) and BDSC. The RNAi of the fly lines we obtained was targeted to the region corresponding to residues (aa) 81-aa181 (UAS-TBPH-IR81-181; VDRC number 38377), aa517-aa531 (UAS-TBPH-IR517-531; VDRC number 104401), aa516-aa531 (UAS-TBPH-IR 516-531; BDSC number 29517), and aa564-aa570 (UAS-TBPH-IR564-570; BDSC number 39014) of Drosophila TBPH. TBPH double-stranded RNA (dsRNA; inverted repeats, IRs) targeted to different regions of the TBPH mRNA is expressed in those fly lines.
We crossed transgenic UAS-TBPH-IR flies with Act5C-GAL4, GMR-GAL4 or elav-GAL4 flies to drive expression of TBPH dsRNA throughout the whole body of flies, specifically in eye imaginal discs, or specifically in neuronal tissues, respectively. We generated eye-specific TBPH knockdown flies (GMR-GAL4; UAS-TBPH-IR/+; +) (GMR>UAS-TBPH-IR/+) and neuron-specific TBPH knockdown flies (yw; UAS-TBPH-IR/+; elav-GAL4/+) (elav>UAS-TBPH-IR/+). Each transgenic strain showed a consistent phenotype (Table 1).
Table 1.
Associated phenotypes of fly strains carrying UAS-TBPH-IR crossed with different GAL4 driver strains (at 28°C)
| Chromosome linkage | Act5C-GAL4> | GMR-GAL4> | elav-GAL4 | |
|---|---|---|---|---|
| UAS-TBPH-IR81-181 | II | Lethal | Mild rough eye | LD |
| UAS-TBPH-IR517-531 | II | Lethal | Mild rough eye | LD |
| UAS-TBPH-IR516-531 | III | ND | Mild rough eye | LD |
| UAS-TBPH-IR564-570 | II | ND | Mild rough eye | LD |
We used four independent TBPH-RNAi constructs, UAS-TBPH-IR81-181, UAS-TBPH-IR517-531, UAS-TBPH-IR516-531, and UAS-TBPH-IR564-570 obtained from VDRC and BDSC. To drive expression of TBPH double-stranded RNA in the whole body of the flies, or specifically in the eye imaginal discs or neuronal tissues, we crossed UAS-TBPH-IR flies with Act5C-GAL4, GMR-GAL4, or elav-GAL4 flies, respectively. Phenotypes associated with the resulting genotypes are summarized. Each transgenic strain showed a consistent phenotype. ND, not determined; LD, locomotive dysfunction.
The UAS-ter94 flies were kindly provided by Dr. Akira Kakizuka [38]. The lines generated in this study are as follows: GMR-GAL4; UAS-GFP-IR/+; + (GMR>UAS-GFP-IR/+), GMR-GAL4; UAS-TBPH-IR517-531/+; + (GMR>UAS-TBPH-IR/+), GMR-GAL4; UAS-TBPH-IR517-531/UAS-TBPH-IR517-531; + (GMR>UAS-TBPH-IR/UAS-TBPH-IR), GMR-GAL4; UAS-TBPH-IR517-531/ter94K15502; + (GMR>UAS-TBPH-IR/ter94K15502), GMR-GAL4; UAS-TBPH-IR517-531/ter9403775; + (GMR>UAS-TBPH-IR/ter9403775), GMR-GAL4; UAS-TBPH-IR517-531/UAS-GFP; + (GMR>UAS-TBPH-IR/UAS-GFP), GMR-GAL4; UAS-TBPH-IR517-531/UAS-ter94; + (GMR>UAS-TBPH-IR/UAS-ter94), yw; UAS-GFP-IR/+; elav-GAL4/+ (elav>UAS-GFP-IR/+; a driver control), yw; UAS-TBPH-IR517-531/+; elav-GAL4/+ (elav>UAS-TBPH-IR/+), yw; ter94K15502/+; +, yw; UAS-TBPH-IR517-531/ter94K15502; elav-GAL4/+ (elav>UAS-TBPH-IR/ter94K15502), yw; UAS-TBPH-IR517-531/UAS-GFP; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-GFP), yw; UAS-TBPH-IR517-531/UAS-ter94; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-ter94), yw; UAS-ter94/+; elav-GAL4/+ (elav>UAS-ter94/+).
Production of rabbit anti-TBPH antibodies
The TBPH peptide, N-CRKGPNNPNNNPAANGIKTD-C, corresponding to aa485-aa503 (the underlined N-terminal C residue was an added residue) was conjugated to keyhole limpet hemocyanin and mixed with Freund’s complete adjuvant to provide a suspension, which was injected subcutaneously into rabbits (Female Japanese White) kept under specific pathogen-free conditions. The rabbits were then boosted with inoculations of an immunogen of the same quality once a week for 7 weeks, and a terminal bleed was performed to collect the maximum amount of serum (Sigma-Aldrich). The IgG was purified from the serum using a protein A Mag Sepharose Xtra (GE Healthcare).
Immunohistochemistry
For immunohistochemical analysis, CNS tissues were dissected from third instar larvae, fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) for 15 min at 25°C, washed with PBS containing 0.3% Triton X-100, and incubated with Alexa 488-conjugated phalloidin (1 unit/200 µl) in PBS containing 0.3% Triton X-100 for 20 min at 25°C. The samples were then blocked with blocking buffer (PBS containing 0.15% Triton X-100 and 10% normal goat serum) for 30 min at 25°C, and then incubated with rabbit anti-TBPH antibody (1:100) in blocking buffer for 20 h at 4°C. After extensive washing with PBS containing 0.3% Triton X-100, samples were incubated in the dark with secondary antibodies labeled with Alexa 546 (1:400; Invitrogen) diluted in the blocking buffer for 3 h at 25°C. After washing with PBS containing 0.3% Triton X-100 and PBS, the samples were stained with DAPI (0.5 µg/ml)/PBS/0.1% Triton X-100. After extensive washing with PBS containing 0.1% Triton X-100 and PBS, the samples were mounted in Vectashield (Vector Laboratories Inc.) and inspected under a confocal laser scanning microscope (OLYMPUS FLUOVIEW FV10i). TBPH protein signal was excited by 557-572 nm. Images of CNS immunostaining were analyzed with the program Meta Morph Imaging System 7.7 (Molecular Devices Inc.). Intensities of TBPH signals in nuclei were analyzed using this program. Individual nucleus marked with DAPI were used to estimate the TBPH signals located in nucleus. This program allows quantification of the average and standard error of fluorescence emission from nuclei of each fly strain.
For NMJ staining, third instar larvae were dissected in HL3 saline [39], and then fixed in 4% paraformaldehyde/PBS for 30 min. The blocking buffer contained 2% bovine serum albumin and 0.1% Triton X-100 in PBS. Fluorescein isothiocyanate-conjugated goat anti-horseradish peroxidase (HRP) (1:1000, MP Biochemicals) was used as the detection antibody. The samples were mounted and observed under a confocal laser scanning microscope (Carl Zeiss LSM 510, Jena, Germany). MN 4 (Ib) in muscle 4 in abdominal segment 2 was quantified. Images were acquired using a Zeiss LSM 510 confocal laser scanning microscope by merging 1-µm interval z-sections onto a single plane. The Meta Morph imaging system was used to measure nerve terminal branch lengths and Ib bouton sizes.
Immunoblotting analysis
Protein extracts from the adult head of Drosophila carrying elav>UAS-GFP-IR/+, elav>UAS-TBPH-IR81-181/+, and elav>UAS-TBPH-IR517-531/+ flies were prepared. Briefly, the head was excised from adult flies and homogenized in sample buffer containing 1 M Tris-HCl (pH 6.8), 10% sodium dodecyl sulfate, 50% glycerol, 1% bromophenol blue, and 1.2% β-mercaptoethanol using a pestle, sonicated, boiled at 95°C for 3 min, and centrifuged at 10,000×g for 10 min at 4°C. The supernatants (extracts) were run on a 4-12% gradient polyacrylamide gel (Invitrogen, Thermo Fisher Scientific) and then transferred onto an Immuno-Blot PVDF membrane (Novex, Life Technologies). The blotted membranes were blocked with 5% skim milk in PBS containing 0.1% Tween 20 for 30 min at 25°C, followed by incubation with rabbit polyclonal anti-TBPH antibody (1:2000) for 16 h at 4°C. After washing, the membranes were incubated with HRP-conjugated anti-rabbit IgG (Thermo Scientific, IL, USA: 1:10000) for 1 h at 25°C. Antibody binding was detected using ECL Western blotting detection reagents (GE Healthcare, Tokyo, Japan), and images were analyzed using an Image QuantTM LAS 4000 image analyzer (GE Healthcare Bioscience, Tokyo, Japan). Comparison of TBPH protein levels in each 58-kDa TBPH protein band was performed using the Meta Morph Imaging system 7.7 (Molecular Pertis Inc.).
Scanning electron microscopy
For compound eye observation, adult flies were anesthetized with 99% diethyl ether, mounted on stages, and observed under an SEM V-7800 (Keyence Inc.) in the low vacuum mode [40]. In every experiment, at least five adult flies were chosen from each line for scanning electron microscopy to assess the eye phenotype. For each experiment, we found no significant variation in eye phenotype among the five individuals from the same strain.
Climbing assay
Climbing assays were performed as described previously [34,41]. Flies carrying elav>UAS-GFP-IR/+, elav>UAS-TBPH-IR/+, elav>UAS-TBPH-IR/ter94K15502, ter94K15502/+, elav>UAS-TBPH-IR/UAS-GFP, elav>UAS-TBPH-IR/UAS-ter94, and elav>UAS-ter94/+ were placed at 28°C. Newly eclosed adult male flies were separated and placed in vials at a density of 20 flies per vial. Flies were transferred, without anesthesia, to a conical tube. The tubes were tapped to collect the flies at the bottom, and they were allowed to climb the wall for 30 s. After 30 s, the flies were collected at the bottom by tapping of the tube and were again allowed to climb for 30 s. Similar procedures, all of which were videotaped, were repeated five times in total. For each climbing experiment, the height that each fly climbed was determined by a score (height climbed); 0 (less than 2 cm), 1 (between 2 and 3.9 cm), 2 (between 4 and 5.9 cm), 3 (between 6 and 7.9 cm), 4 (between 8 and 9.9 cm) or 5 (greater than 10 cm). The climbing index for each fly strain was calculated as follows: each score was multiplied by the number of flies for which that score was recorded, and the products were summed and divided by five (the total number of flies examined). These climbing assays were carried out every 7 days until the 28th day after eclosion.
Data analysis
For statistical analysis of the climbing assay, SPSS software (IBM) was used. Graph Pad Prism version 6.0 was used to perform each statistical analysis. The Mann-Whiney test was used to assess the statistical significance of comparisons between two groups of data. For other assays, one-way analysis of variance (ANOVA) was used to determine the statistical significance of comparisons between groups of data. When the one-way ANOVA showed significant variation among groups, a subsequent Dunnett’s test was used for pairwise comparisons between groups. All data are shown as the mean ± standard error (SE). For the statistical analysis of the viability assay, Graph Pad Prism version 6.0 software was used.
Results
Phenotypes of RNAi flies for TBPH derived from several different GAL4 driver strains
Drosophila TBPH (CG10327) shows 54.4% identity and 80.6% similarity to TDP-43 (Figure 1A). Both proteins share two RNA recognition motifs (RRMs) and an unstructured Gly-rich C-terminal region where most of the ~50 different ALS mutations reside. FUS also has an RRM and a Gly-rich C-terminal region. From this point of view, TDP-43 resembles FUS [5].
Figure 1.

Comparison of human TDP-43 and Drosophila TBPH. A. Sequence alignment of human TDP-43 (414 amino acids) and Drosophila TBPH (531 amino acids). Identical amino acids are dark-shaded, and similar or related amino acids are light-shaded. The amino acid sequence of TBPH was retrieved from FlyBase and compared with that of human TDP-43 using FASTA and BLAST. B. Schematic drawings of the domain structures of human TDP-43 and Drosophila TBPH proteins. TDP-43 and TBPH comprise two RNA recognition motifs (RRM1 and RRM2) and a C-terminal Gly-rich region. The human TDP-43 domain is highly conserved in Drosophila TBPH. The solid line under the schematic of Drosophila TBPH shows the target genomic sequence of each of the two RNAi transgenes employed in this study, UAS-TBPH-IR81-181 and UAS-TBPH-IR517-531.
In the previous studies, two RNAi fly lines targeted to the region corresponding to amino acid residues (aa) 81-aa181 (UAS-TBPH-IR81-181/+) and aa517-aa531 (UAS-TBPH-IR517-531/+) of TBPH were mainly used [34,35,42-44]. In those studies, the GAL4-UAS targeted expression system was commonly used [45]. Eye imaginal disc-specific knockdown of TBPH by GMR-GAL4 induced the rough-eye phenotype accompanied by fusion of ommatidia and loss of mechanosensory bristles [43]. In the present study, we confirmed the rough-eye phenotype induced by knockdown of TBPH by crossing GMR-GAL4 with RNA lines, not only UAS-TBPH-IR81-181 and UAS-TBPH-IR517-531 (Figure 2B, 2C) but also UAS-TBPH-IR516-531 and UAS-TBPH-IR564-570 (Table 1). Both pan-neuron-specific knockdown of TBPH by elav-GAL4 and pan-glial-specific knockdown of TBPH by repo-GAL4 result in locomotive deficits [34,41,42]. Other studies reported that pan-neuron-specific knockdown of TBPH by Nsyb-GAL4 and C155-GAL4 or CCAP/bursicon-neuron-specific knockdown of TBPH by CCAP-GAL4 result in neuronal loss and wing inflation deficits [35,44]. In addition, a deletion mutant of TBPH results in changes in NMJ morphology and locomotive deficits [46,47]. Here we further confirmed the locomotive deficits induced by knockdown of TBPH by crossing elav-GAL4 with four different RNAi lines: UAS-TBPH-IR81-181, UAS-TBPH-IR517-531 (Figure 3A), UAS-TBPH-IR 516-531, and UAS-TBPH-IR564-570 (Table 1). We also confirmed the reduced life span and anatomical defects at NMJs by crossing elav-GAL4 with UAS-TBPH-IR81-181 or UAS-TBPH-IR517-531 (Figures 3B, 4). In addition, we found that knockdown of TBPH in all cells of the whole body of flies by Act5C-GAL4 resulted in lethality (Table 1). This is consistent with previous observations in which homozygous TBPH mutations result in pupal lethality [35,44,47]. Because all of these phenotypes were observed with RNAi lines targeted to at least two different regions of the TBPH mRNA, the possibility of off-target effects can be excluded. Based on these results, we decided to use the flies carrying UAS-TBPH-IR81-181 and UAS-TBPH-IR517-531 in the subsequent experiments (Figure 1B).
Figure 2.
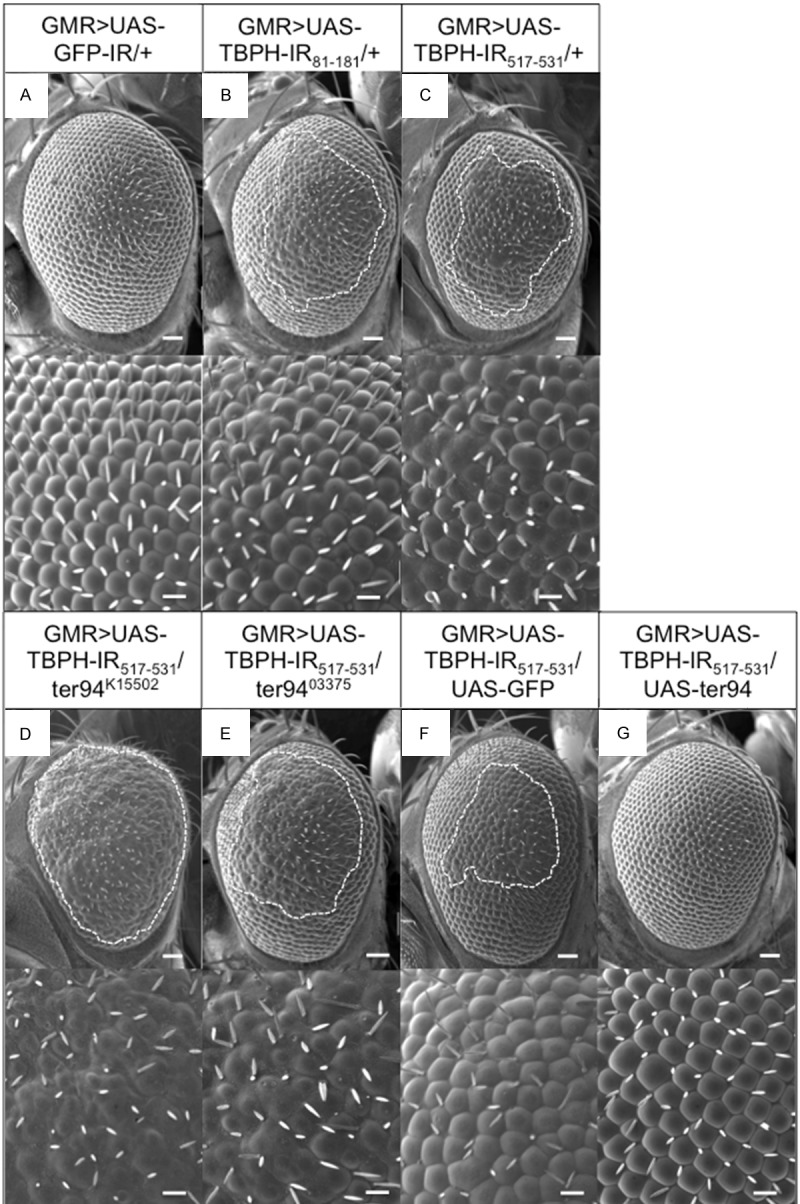
The rough-eye morphology induced by TBPH knockdown is modified by genetic changes in ter94. Each panel shows a scanning electron micrograph of the adult compound eye. Each lower panel shows a higher magnification image of the corresponding upper panel. (A) GMR-GAL4/Y; UAS-GFP-IR/+; + (GMR>UAS-GFP-IR/+). The eyes of control flies exhibited apparently normal eye morphology including an organized ommatidial architecture. (B) GMR-GAL4/Y; UAS-TBPH-IR81-181/+; + (GMR>UAS-TBPH-IR81-181/+); (C) GMR-GAL4/Y; UAS-TBPH-IR517-531/+; + (GMR>UAS-TBPH-IR517-531/+). Adult eyes from two independent fly lines with eye-specific TBPH knockdown. Flies carrying GMR>UAS-TBPH-IR81-181/+ (B) or GMR>UAS-TBPH-IR517-531/+ (C) had essentially the same rough-eye morphology and exhibited ommatidial degeneration. (D) GMR-GAL4/Y; UAS-TBPH-IR517-531/ter94K15502; + (GMR>UAS-TBPH-IR517-531/ter94K15502), (E) GMR-GAL4/Y; UAS-TBPH-IR517-531/ter9403775; + (GMR>UAS-TBPH-IR517-531/ter9403775). Each of the flies carrying GMR>UAS-TBPH-IR517-531/ter94K15502 (D) and GMR>UAS-TBPH-IR517-531/ter9403775 (E) showed rough-eye morphology that was enhanced relative to that observed in flies with GMR>UAS-TBPH-IR517-531/+ alone (C). (F) GMR-GAL4/Y; UAS-TBPH-IR517-531/UAS-GFP; + (GMR>UAS-TBPH-IR517-531/UAS-GFP); (G) GMR-GAL4/Y; UAS-TBPH-IR517-531/UAS-ter94; + (GMR>UAS-TBPH-IR517-531/UAS-ter94). The flies carrying GMR>UAS-TBPH-IR517-531/UAS-ter94 (G) suppressed rough-eye morphology that observed in flies with GMR>UAS-TBPH-IR517-531/UAS-GFP (F). Posterior is to the right, and dorsal is to the top. The flies developed at 28°C. Scale bars in each upper panel indicate 50 µm and in the bottom panel indicate 14.2 µm. The rough areas of the compound eyes are marked with dotted lines.
Figure 3.
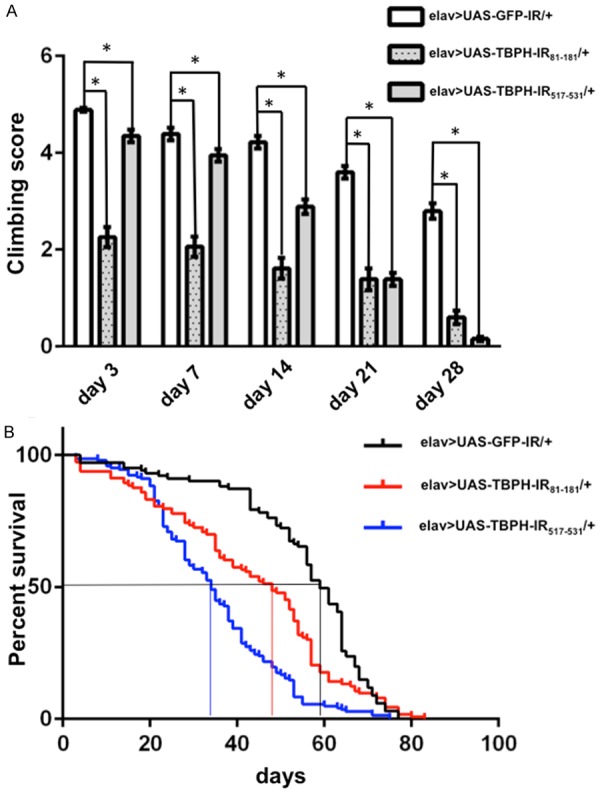
Climbing assays and life span analyses. A. Climbing ability in the neuron-specific TBPH knockdown flies. The locomotive ability of the neuron-specific TBPH knockdown flies carrying yw/Y; UAS-TBPH-IR81-181/+; elav-GAL4/+ (elav>UAS-TBPH-IR81-181/+, n = 100, dotted gray columns) and carrying yw/Y; UAS-TBPH-IR517-531/+; elav-GAL4/+ (elav>UAS-TBPH-IR517-531/+, n = 100, gray columns) was significantly decreased as seen by assessing the climbing ability, compared with flies carrying yw/Y; UAS-GFP-IR/+; elav-GAL4/+ (elav>UAS-GFP-IR/+, n = 100, white columns) for every age. *P < 0.001. B. Life span analysis of neuron-specific TBPH knockdown flies. The life span of flies carrying elav>UAS-TBPH-IR81-181/+ (n = 100) and elav>UAS-TBPH-IR517-531/+ (n = 100) was significantly decreased compared to the control flies carrying elav>UAS-GFP-IR/+ (n = 100) (P < 0.001). The average life span of the control flies was 59.0 days, whereas that of flies carrying elav>UAS-TBPH-IR81-181/+ and elav>UAS-TBPH-IR517-531/+ was 48.0 days and 34.0 days, respectively. We found no significant differences in life span between flies carrying elav>UAS-TBPH-IR81-181/+ and those carrying elav>UAS-TBPH-IR517-531/+.
Figure 4.
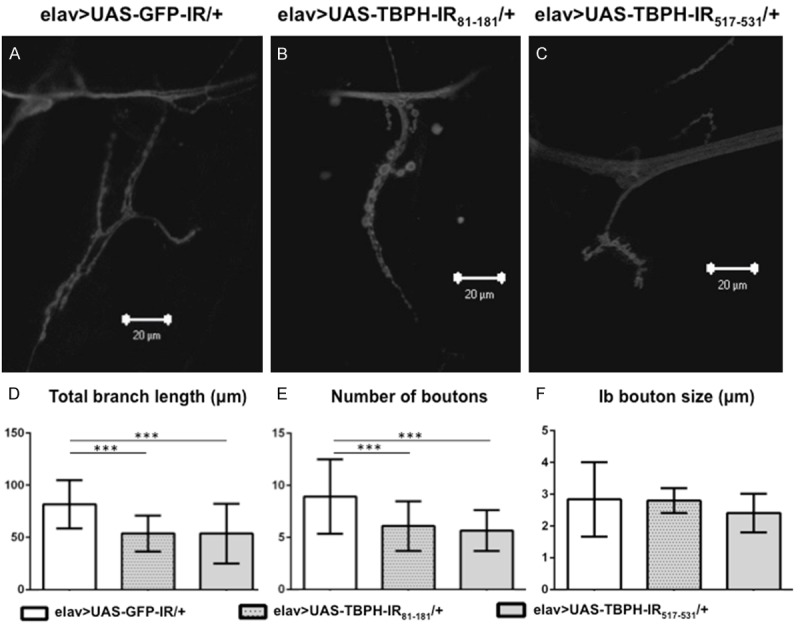
Confocal images of anti-HRP staining of muscle 4 synapses in neuron-specific TBPH knockdown larvae. Images of flies of the following genotypes are shown: (A) yw/Y; UAS-GFP-IR/+; elav-GAL4/+ (elav>UAS-GFP-IR/+, driver control), (B) yw/Y; UAS-TBPH-IR81-181/+; elav-GAL4/+ (elav>UAS-TBPH-IR81-181/+) and (C) yw/Y; UAS-TBPH-IR517-531/+; elav-GAL4/+ (elav>UAS-TBPH-IR517-531/+). (D) Total branch length of the NMJs from muscle 4 for each of the indicated genotypes is shown. Compared to the total length of synaptic branches of MNs in driver control larvae (A), the lengths in the two neuron-specific TBPH knockdown larvae (B and C) were significantly decreased (P < 0.001, n = 10, D). These decreases in branch length of the two neuron-specific TBPH knockdown larvae (B and C) were almost the same (P = 0.9, n = 10, D). (E) The number of synaptic boutons for each of the indicated genotypes is shown. The number of synaptic boutons of MNs in the two neuron-specific TBPH knockdown larvae (B and C) was also significantly decreased compared to driver control larvae (A) (P < 0.001, n = 10, E). These decreases in the number of synaptic boutons in the two neuron-specific TBPH knockdown larvae were almost the same (P = 0.8, n = 10, E). (F) The size of synaptic boutons for each of the indicated genotypes is shown. The size of synaptic Ib boutons was measured (n = 10 for elav>UAS-GFP-IR/+, n = 10 for elav>UAS-TBPH-IR81-181/+ and n = 10 for elav>UAS-TBPH-IR517-531/+). We found no significant differences in the size of synaptic boutons among driver control larvae, larvae with elav>UAS-TBPH-IR81-181/+, and larvae with elav>UAS-TBPH-IR517-531/+ (P = 0.8). ***P < 0.001. The scale bars indicate 20 µm (A to C).
A loss-of-function mutation and overexpression of ter94 conversely modified the compound eye degeneration caused by TBPH knockdown
To identify factors that influence TBPH knockdown pathogenesis, we searched for interacting genes that suppress or enhance the TBPH knockdown phenotype in the compound eye. Modifier screening of the rough-eye phenotype has been commonly used to identify the interacting genes [29,30]. First, we searched for mutations in several genes related to ALS and identified ter94 as a genetic interactant with TBPH. Heterozygous loss-of-function mutation of ter94 remarkably enhanced the rough-eye phenotype caused by eye-specific TBPH knockdown (GMR>UAS-TBPH-IR517-531/ter94K15502, and GMR>UAS-TBPH-IR517-531/ter9403375) (Figure 2D, 2E). These results demonstrate that the loss-of-function mutation of ter94 acts as a dominant enhancer of the TBPH knockdown. Conversely, the overexpression of wild-type ter94 (GMR>UAS-TBPH-IR517-531/UAS-ter94) (Figure 2G) effectively suppressed the rough-eye phenotype induced by eye-specific TBPH knockdown compared to the responder control of UAS-ter94 flies (GMR>UAS-TBPH-IR517-531/UAS-GFP) (Figure 2F). In addition, we found no apparent effect on the compound eye morphology in ter94K15502/+ flies (data not shown).
Evaluation of the efficiency of neuron-specific TBPH knockdown
We next examined the genetic interaction between TBPH and ter94 in neurons. First, we clarified the efficiency of neuron-specific TBPH knockdown. We raised a polyclonal antibody against a peptide corresponding to aa485-aa503 of Drosophila TBPH and used it for immunoblotting analyses of extracts prepared from adult heads carrying elav>UAS-GFP-IR/+ (yw/Y; UAS-GFP-IR/+; elav-GAL4/+), elav>UAS-TBPH-IR81-181/+ (yw/Y; UAS-TBPH-IR81-181/+; elav-GAL4/+), and elav>UAS-TBPH-IR517-531/+ (yw/Y; UAS-TBPH-IR517-531/+; elav-GAL4/+).
A major band with an apparent molecular weight of 58 kDa was detected on immunoblots of all the flies using the anti-TBPH antibody (Figure 5A). The intensity of this band was reduced to 83.8% in flies carrying elav>UAS-TBPH-IR81-181/+ and to 83.9% in flies carrying elav>UAS-TBPH-IR517-531/+ compared with its intensity in control flies carrying elav>UAS-GFP-IR/+ (Figure 5B). The intensity of a minor band corresponding to 57.7 kDa was also reduced in flies carrying elav>UAS-TBPH-IR81-181/+ and elav>UAS-TBPH-IR517-531/+, suggesting that this band is very likely a splicing variant of TBPH as described (FlyBase, http://flybase.org). These data confirmed that TBPH is modestly knocked down in flies carrying both elav>UAS-TBPH-IR81-181/+ and elav>UAS-TBPH-IR517-531/+. Although the knockdown efficiency is apparently low, the adult head contains not only neuronal cells but also non-neuronal cells in which TBPH is not knocked down by elav-GAL4. We used these modestly knocked-down fly models in the following studies. These fly models may be suitable for evaluation of genetic interactions with TBPH, because TBPH is so essential for Drosophila development that a null allele or a severe hypomorph of TBPH induces lethality during pupal development [35,44,47].
Figure 5.
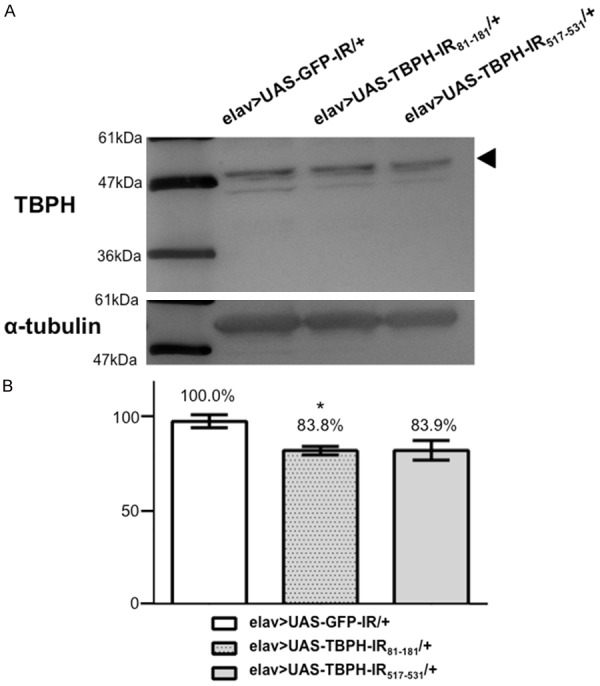
Western immunoblotting analyses. (A) Proteins were extracted from 1-day-old adult heads carrying yw/Y; UAS-GFP-IR/+; elav-GAL4/+ (elav>UAS-GFP-IR/+), yw/Y; UAS-TBPH-IR81-181-IR/+; elav-GAL4/+ (elav>UAS-TBPH-IR81-181/+), and yw/Y; UAS-TBPH-IR517-531-IR/+; elav-GAL4/+ (elav>UAS-TBPH-IR517-531/+). These blots were probed with the polyclonal anti-TBPH antibody. α-tubulin was used as a loading control. A 58-kDa band corresponding to the TBPH protein was detected. Densitometric quantification of the 58-kDa bands was performed with triplicate immunoblot analyses of each fly strain in (A). (B) The intensity of the 58-kDa bands, which indicate the expression level of TBPH protein, was reduced in flies carrying elav>UAS-TBPH-IR81-181/+ (P < 0.05) and elav>UAS-TBPH-IR517-531/+ (P = 0.06) compared to driver control flies. The columns and horizontal bars indicate the mean values and the standard errors, respectively. *P < 0.05.
The effects of overexpression or loss-of-function mutation of ter94 on the mobility defects caused by neuron-specific TBPH knockdown
Based on the results with the compound eye system, we explored the effects of overexpression or loss-of-function of ter94 on locomotive deficits induced by neuron-specific TBPH knockdown by performing the well-established climbing assay [32]. An age-dependent decline in the climbing ability was observed in all examined flies (Figure 6). Neuron-specific TBPH knockdown flies carrying elav>UAS-TBPH-IR/+ exhibited a significantly decreased climbing ability compared to control flies carrying elav>UAS-GFP-IR/+ at the following days of age: day 7; -9.5%, P < 0.001, day 14; -22%, P < 0.01, day 21; -56.7%, P < 0.001, and day 28; -80.1%, P < 0.001 (Figure 6B, gray columns). Flies overexpressing wild-type ter94 on the background of neuron-specific TBPH knockdown (elav>UAS-TBPH-IR/UAS-ter94, Figure 6A, black columns) showed significantly better climbing ability than did flies with neuron-specific TBPH knockdown (elav>UAS-TBPH-IR/UAS-GFP, Figure 6A, white columns) on day 3 and day 28 (day 3; +13.4%, P < 0.01 and day 28; +70.2%, P < 0.01).
Figure 6.
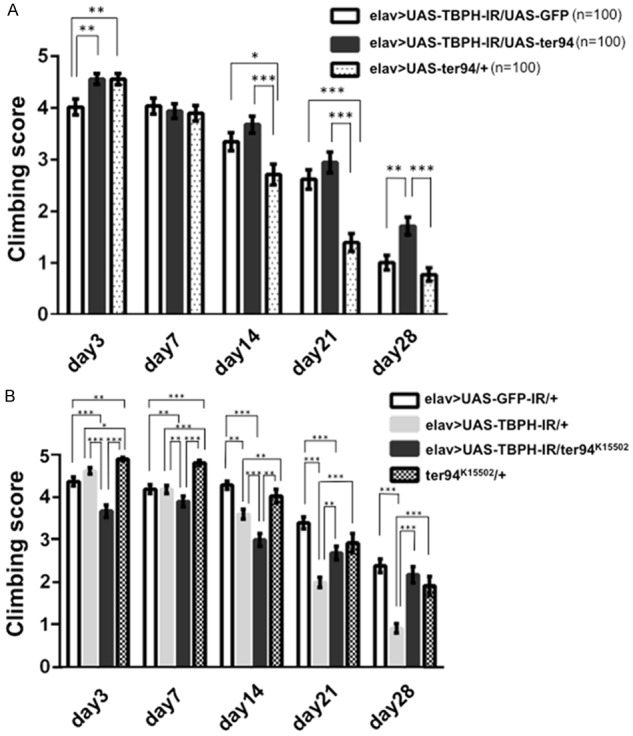
Neuron-specific TBPH knockdown flies show the locomotive defect. Overexpression of wild-type ter94 partially rescued those defects. Conversely, a loss-of-function ter94 mutation partially exacerbated the climbing defect in TBPH knockdown flies. A. The climbing ability of flies carrying yw/Y; UAS-TBPH-IR517-531/UAS-GFP; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-GFP; a responder control) (n = 100, white columns) was decreased. Adult flies carrying yw/Y; UAS-TBPH-IR517-531/UAS-ter94; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-ter94) (n = 100, black columns) showed significantly better climbing ability than those carrying elav>UAS-TBPH-IR/UAS-GFP on days 3 and 28. On days 14 and 21, climbing scores of flies carrying elav>UAS-TBPH-IR/UAS-ter94 were slightly better than those carrying elav>UAS-TBPH-IR/UAS-GFP. Flies carrying elav>UAS-TBPH-IR/UAS-ter94 showed also significantly better climbing ability than those carrying yw/Y; UAS-ter94/+; elav-GAL-4/+ (elav>UAS-ter94/+) (n = 100, hatched columns). B. The climbing ability of flies carrying yw/Y; UAS-TBPH-IR517-531/+; elav-GAL4/+ (elav>UAS-TBPH-IR/+) (n = 210, gray columns) was significantly decreased compared to that of flies carrying yw/Y; UAS-GFP-IR/+; elav-GAL4/+ (elav>UAS-GFP-IR/+; a driver control) (n = 185, white columns) and yw/Y; ter94K15502/+; + (ter94K15502/+) (n = 95, hatched columns) for every age examined except day 3. On each day after eclosion until day 21, flies carrying yw/Y; UAS-TBPH-IR517-531/ter94K15502+; elav-GAL4/+ (elav>UAS-TBPH-IR/ter94K15502) (n = 100, black columns) exhibited a significantly decreased climbing ability compared to neuron-specific TBPH knockdown flies carrying elav>UAS-TBPH-IR/+. Columns and horizontal bars show the mean and SE of the measurements, respectively. ***P < 0.001, **P < 0.01, and *P < 0.05.
Flies carrying the loss-of-function allele of ter94 and neuron-specific TBPH knockdown (elav>UAS-TBPH-IR/ter94K15502) had significantly worse locomotive ability than flies with neuron-specific TBPH knockdown alone (elav>UAS-TBPH-IR/+) until day 14 (day 3; -20.5%, P < 0.001, day 7; -6.7%, P < 0.01, and day 14; -16.8%, P < 0.001, Figure 6B, black columns). However, after day 21, flies carrying the loss-of-function allele of ter94 and neuron-specific TBPH knockdown (elav>UAS-TBPH-IR/ter94K15502) showed better locomotive ability than flies with neuron-specific TBPH knockdown alone (elav>UAS-TBPH-IR/+) (day 21; +34.5%, P < 0.001 and day 28; +137.5%, P < 0.001, Figure 6B, black columns).
These results demonstrate that overexpression of wild-type ter94 could partially rescue the locomotive defect induced by TBPH knockdown, whereas the loss-of-function ter94 mutation partially exacerbated those phenotypes at young ages. Unaccountably, overexpression of wild-type ter94 and the loss-of-function allele of ter94 could partially rescue the locomotive defect induced by TBPH knockdown at older ages (Figure 6A, black columns and Figure 6B, black columns).
Effects of overexpression or loss-of-function mutation of ter94 on the morphology of MN presynaptic terminals in the NMJs of neuron-specific TBPH knockdown flies
Locomotive deficits are sometimes accompanied by abnormal NMJ morphology as reported with other neurodegenerative disease model flies including ALS [29,30,48-52]. We therefore next examined the effects of ter94 on the morphology of NMJs. The Drosophila NMJ is a valuable model for investigating fundamental questions about synapses. Development of synapses in both Drosophila and vertebrates is similar at the cellular and molecular levels [51]. We examined the NMJ structure including the total length of synaptic branches, the number of synaptic boutons, and the size of synaptic boutons in larvae from TBPH knockdown strains.
First, we examined the effects of overexpression of ter94 on the morphology of NMJs in TBPH knockdown flies. The total branch length in larvae carrying elav>UAS-TBPH-IR/UAS-GFP was significantly decreased compared with those carrying elav>UAS-GFP-IR/+ (100.8 ± 8.52 µm versus 42.9 ± 6.5 µm, P < 0.001, Figure 7D). Meanwhile, the total branch length was significantly longer in the larvae with ter94 overexpression on the background of neuron-specific TBPH knockdown (elav>UAS-TBPH-IR/UAS-ter94, Figure 7C) than in responder control larvae (elav>UAS-TBPH-IR/UAS-GFP, Figure 7B) (77.8 ± 11.2 µm versus 42.9 ± 6.5 µm, P < 0.05, Figure 7D). The number of MN synaptic boutons was significantly decreased in larvae carrying elav>UAS-TBPH-IR/UAS-GFP compared to driver control larvae carrying elav>UAS-GFP-IR/+ (6 ± 0.8 versus 9.1 ± 1.2, P < 0.05, Figure 7E). Meanwhile, the number of MN synaptic boutons was significantly increased in larvae carrying elav>UAS-TBPH-IR/UAS-ter94 (10.8 ± 1.9, Figure 7C) compared to those carrying elav>UAS-TBPH-IR/UAS-GFP (6 ± 0.8, Figure 7B) (P < 0.05, Figure 7E). The number of MN synaptic boutons may be increased in larvae carrying elav>UAS-TBPH-IR/UAS-ter94 due to the growth of synaptic terminals. The size of boutons in larvae carrying elav>UAS-TBPH-IR/UAS-ter94 and in larvae carrying elav>UAS-TBPH-IR/UAS-GFP was smaller than that in those carrying elav>UAS-GFP-IR/+ (2.8 ± 0.1 µm, 2.1 ± 0.3 µm, and 4.4 ± 0.4 µm, respectively, P < 0.05, Figure 7F). However, we found no significant difference in the size of synaptic boutons between larvae carrying elav>UAS-TBPH-IR/UAS-GFP and elav>UAS-TBPH-IR/UAS-ter94 (Figure 7F).
Figure 7.
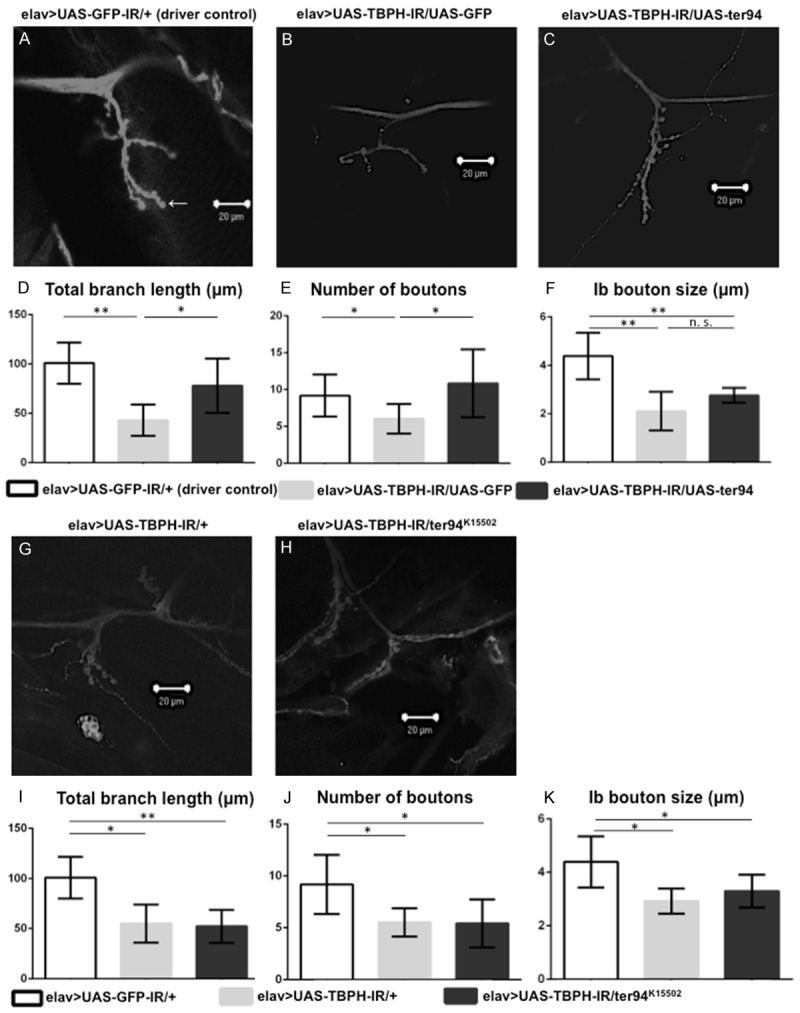
Overexpression of wild-type ter94 improves the morphology of NMJs in TBPH knockdown larvae. A loss-of-function ter94 mutation did not change the morphology of the MN presynaptic terminals at NMJs in neuron-specific TBPH knockdown larvae. A representative image of anti-HRP staining of muscle 4 synapses in third instar larvae carrying yw/Y; UAS-GFP-IR/+; elav-GAL4/+ (elav>UAS-GFP-IR/+, A; a driver control), yw/Y; UAS-TBPH-IR517-531/UAS-GFP; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-GFP, B; a responder control), yw/Y; UAS-TBPH-IR517-531/UAS-ter94; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-ter94, C), yw/Y; UAS-TBPH-IR517-531/+; elav-GAL4/+ (elav>UAS-TBPH-IR/+, G), and yw/Y; UAS-TBPH-IR517-531/ter94K15502+; elav-GAL4/+ (elav>UAS-TBPH-IR/ter94K15502, H). (D and I) Total branch length of the NMJ from muscle 4 for each of the indicated genotypes is shown. Compared to the total length of synaptic branches of MNs in driver control larvae (A), the length in larvae carrying elav>UAS-TBPH-IR/UAS-GFP (B) was significantly decreased (P < 0.001, n = 6, D). Conversely, the total branch length of MNs in larvae with ter94 overexpression on the background of neuron-specific TBPH knockdown (C) was significantly longer than that in larvae carrying elav>UAS-TBPH-IR/UAS-GFP (B) (P < 0.05, n = 6, D). The total branch length in neuron-specific TBPH knockdown larvae (G) was significantly decreased (P < 0.001, n = 6, I). This decrease in branch length observed in the neuron-specific TBPH knockdown larvae (G) was almost the same as in larvae carrying the neuron-specific TBPH knockdown crossed with the loss-of-function allele of ter94 (H) (P = 0.7, n = 7, I). (E and J) The number of synaptic boutons for each of the indicated genotypes is shown. The number of synaptic boutons in larvae carrying wild-type ter94 overexpression on the background of neuron-specific TBPH knockdown (C) was significantly increased compared to larvae carrying elav>UAS-TBPH-IR/UAS-GFP (B) (P < 0.05, n = 6, E). Compared to larvae carrying elav>UAS-GFP-IR/+, the number of synaptic boutons is significantly decreased in larvae carrying elav>UAS-TBPH-IR/UAS-GFP (P < 0.001, n = 6, E). The number of synaptic boutons in larvae carrying elav>UAS-TBPH-IR/UAS-ter94 is almost the same as that in driver control larvae. The number of synaptic boutons of MNs in neuron-specific TBPH knockdown larvae (G) was also significantly decreased compared to driver control larvae (A) (P < 0.05, n = 6, J). This decrease in the number of synaptic boutons in neuron-specific TBPH knockdown larvae was almost the same in larvae carrying the neuron-specific TBPH knockdown crossed with the loss-of-function allele of ter94 (H) (P = 0.9, n = 7, J). (F and K) The size of synaptic boutons for each of the indicated genotypes is shown. The size of synaptic Ib boutons (indicated with an arrow in A; the Ib boutons measured in this paper are the largest boutons in the NMJs) was measured (n = 6 for elav>UAS-GFP-IR, n = 6 for elav>UAS-TBPH-IR/UAS-GFP, n = 6 for elav>UAS-TBPH-IR/UAS-ter94, n = 6 for elav>UAS-TBPH-IR, and n = 7 for elav>UAS-TBPH-IR/ter94K15502). We found no significant differences in the size of synaptic boutons between larvae with elav>UAS-TBPH-IR/UAS-GFP (B) and elav>UAS-TBPH-IR/UAS-ter94 (C), or between those with elav>UAS-TBPH-IR (G) and elav>UAS-TBPH-IR/ter94K15502 (H). The size was significantly smaller in neuron-specific TBPH knockdown larvae (2.9 ± 0.2 µm, G) than in control larvae (4.3 ± 0.4 µm, A) (P < 0.01, K). Columns and horizontal bars show the mean and SE of the measurements, respectively. **P < 0.01, and *P < 0.05. The scale bars indicate 20 µm (A, B, C, G, and H).
Next, we explored the effects of loss-of-function mutation of ter94 on the morphology of NMJs induced by TBPH knockdown. Compared to the total length of synaptic branches of MNs in driver control larvae carrying elav>UAS-GFP-IR/+ (100.8 ± 8.52 µm, Figure 7A), the length was significantly decreased in neuron-specific TBPH knockdown larvae carrying elav>UAS-TBPH-IR/+ (54.89 ± 7.7 µm, Figure 7G) (P < 0.01, Figure 7I). This decreased branch length was not enhanced by crossing with the loss-of-function allele of ter94 (elav>UAS-TBPH-IR/ter94K15502, 52.1 ± 3.9 µm, Figure 7H) (P = 0.74, Figure 7I). The average number of synaptic boutons per MN was also significantly smaller in neuron-specific TBPH knockdown larvae (5.5 ± 0.5, Figure 7G) than in control larvae (9.2 ± 1.2, Figure 7A) (P < 0.01, Figure 7J). This decrease in the number of synaptic boutons did not change following crossing with the loss-of-function allele of ter94 (5.3 ± 0.7, Figure 7H) (P = 0.82, Figure 7J). Similarly, we found no significant difference in the size of synaptic boutons between neuron-specific TBPH knockdown larvae and those with neuron-specific TBPH knockdown combined with the loss-of-function allele of ter94 (P = 0.25, Figure 7K).
These results indicated that wild-type ter94 overexpression improved synaptic terminal growth and the number of synaptic boutons, which were impaired by TBPH knockdown. Although the loss-of-function mutation of ter94 did not change the decrease in branch length or the number of synaptic boutons caused by TBPH knockdown, we suspect that the decrease in branch length and the number of synaptic boutons caused by TBPH knockdown are too severe to be further affected by an additional ter94 loss-of-function.
Effect of overexpression or loss-of-function mutation of ter94 on the nuclear TBPH levels in the larval CNS
To monitor TBPH expression and localization, we immunostained brain-ventral ganglia complexes (BVGCs) of third instar larvae with the anti-TBPH antibody. In driver control larvae, anti-TBPH immunoreactivity was evident in the nucleus of the neuronal cells. Anti-TBPH immunoreactivity did not colocalize with actin filaments stained with phalloidin (Figure 8B1, 8B2, 8B1+B2), but colocalized with nuclei stained with diamino-2-phenylidole dehydrochloride (DAPI; Figure 8B1, 8B3, 8B1+B3). These results confirmed that TBPH is localized in the nucleus as reported previously [6,18].
Figure 8.
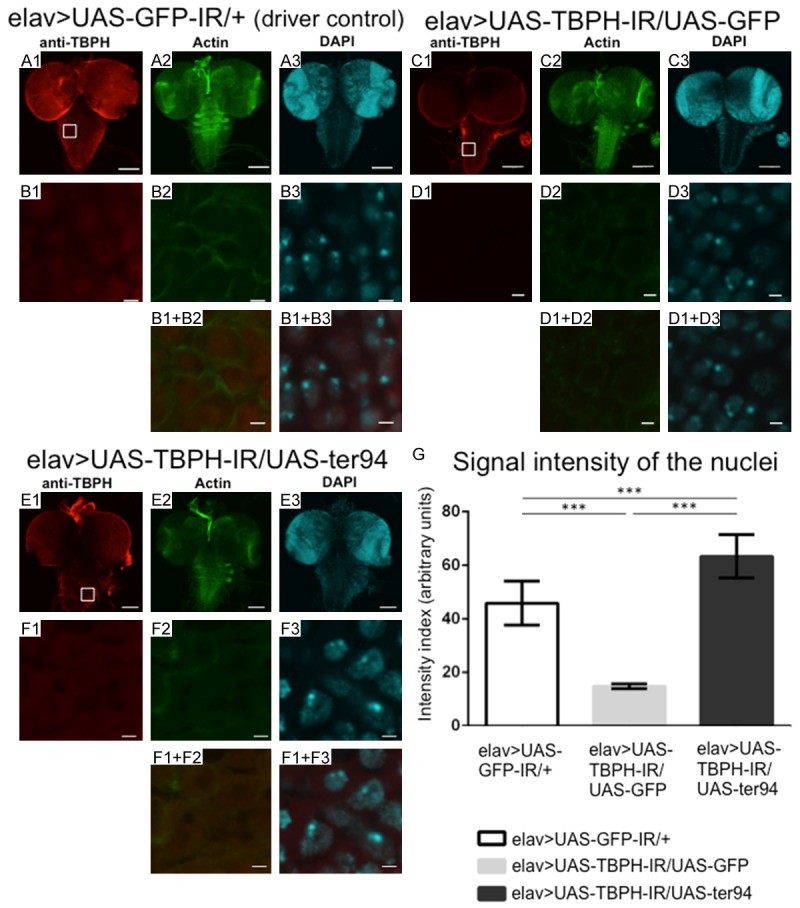
Overexpression of wild-type ter94 restored nuclear TBPH signal intensities in the larval CNS with neuron-specific TBPH knockdown. (A-F) Are representative images of corresponding genotypes. (A1-A3) Are immunofluorescent images of the larval CNS taken from driver control larvae carrying yw/Y; UAS-GFP-IR/+; elav-GAL4/+ (elav>UAS-GFP-IR/+). The larval CNS comprises the brain-ventral ganglia complex (BVGC). The BVGC of driver control larvae carrying elav>UAS-GFP-IR/+ showed nuclear signals from endogenous TBPH (A1). Anti-TBPH antibody immunoreactivity was evident in the nucleus of neuronal cells (B1). The TBPH signal did not colocalize with phalloidin-stained actin filaments (B2, B1+B2). (C1-C3) Are the BVGCs of TBPH knockdown larvae carrying yw/Y; UAS-TBPH-IR517-531/UAS-GFP; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-GFP). (E1-E3) Are the BVGCs of larvae that overexpress wild-type ter94 on the background of TBPH knockdown carrying yw/Y; UAS-TBPH-IR517-531/UAS-ter94; elav-GAL4/+ (elav>UAS-TBPH-IR/UAS-ter94). Panels (B1) to (B3), (D1) to (D3), and (F1) to (F3) are higher magnification images of the boxed areas in (A1), (C1), and (E1), respectively. (B1+B2, B1+B3, D1+D2, D1+D3, F1+F2, and F1+F3) Are merged images. The indirect immunofluorescence in (A1, B1, C1, D1, E1, and F1) is the signal from the polyclonal anti-TBPH antibody (based on Alexa Fulor 546 channel). The fluorescence in (A2, B2, C2, D2, E2, and F2) is from phalloidin (based on Alexa Fulor 488 channel), which labels actin. The fluorescence in (A3, B3, C3, D3, E3, and F3) is from DAPI (based on DAPI channel). (G) This graph plots the mean (± SE) of the intensity of the nuclear TBPH signal in BVGCs from third instar larvae as fluorescence emission in arbitrary units for each genotype. Columns and horizontal bars show the mean and SE of 15 nuclei, respectively. ***P < 0.001. Compared to the signal intensity of nuclear TBPH in the BVGCs of the control larvae (A1), the TBPH signal intensity in the BVGCs of larvae carrying elav>UAS-TBPH-IR/UAS-GFP (C1) was decreased (P < 0.001, G). The signal intensity of nuclear TBPH was significantly higher in larvae carrying elav>UAS-TBPH-IR/UAS-ter94 (F1) than in larvae carrying elav>UAS-TBPH-IR/UAS-GFP (D1) (P < 0.001, G). Neuron-specific TBPH knockdown reduces nuclear TBPH signal intensities in the larval CNS. And overexpression of wild-type ter94 restored nuclear TBPH signal intensities in the larval CNS with neuron-specific TBPH knockdown. The scale bars indicate 100 µm (A1 to A3, C1 to C3, and E1 to E3) and 2 µm (B1 to B3, C1 to C3, and F1 to F3).
First, we examined TBPH expression and localization in the CNS of larvae carrying overexpression of ter94 and neuron-specific TBPH knockdown. The intensity of nuclear TBPH signals was reduced to 37.4% in the BVGCs of neuron-specific TBPH knockdown larvae carrying elav>UAS-TBPH-IR/+ [intensity units = 28.68±0.60 (arbitrary units), measured in Figure 9B1] compared with that of driver control larvae (intensity units = 45.85±2.74, measured in Figure 8B1; P < 0.001, Figure 9E). These results indicate that neuron-specific TBPH knockdown effectively reduced nuclear TBPH level. In comparison with the TBPH signal intensities in BVGCs of responder control larvae carrying yw/Y; UAS-TBPH-IR/UAS-GFP; elav-GAL4/+ (Figure 8, elav>UAS-TBPH-IR/UAS-GFP, C1), the TBPH signals in BVCGs of larvae carrying yw/Y; UAS-TBPH-IR/UAS-ter94; elav-GAL4/+ (Figure 8, elav>UAS-TBPH-IR/UAS-ter94, E1) were remarkably stronger. Quantification analysis of the TBPH signal showed that the intensity of the nuclear TBPH signal in elav>UAS-TBPH-IR/UAS-ter94 larvae (intensity units = 63.32±2.54, measured in Figure 8F1) was 4.3-fold higher than that in elav>UAS-TBPH-IR/UAS-GFP larvae (intensity units = 14.71±0.29, measured in Figure 8B1; P < 0.001, Figure 8G). These results indicate that overexpression of wild-type ter94 restored the reduced TBPH signal in the nucleus induced by neuron-specific TBPH knockdown. Taken together, our results suggest that overexpression of ter94 can rescue the TBPH knockdown phenotype.
Figure 9.
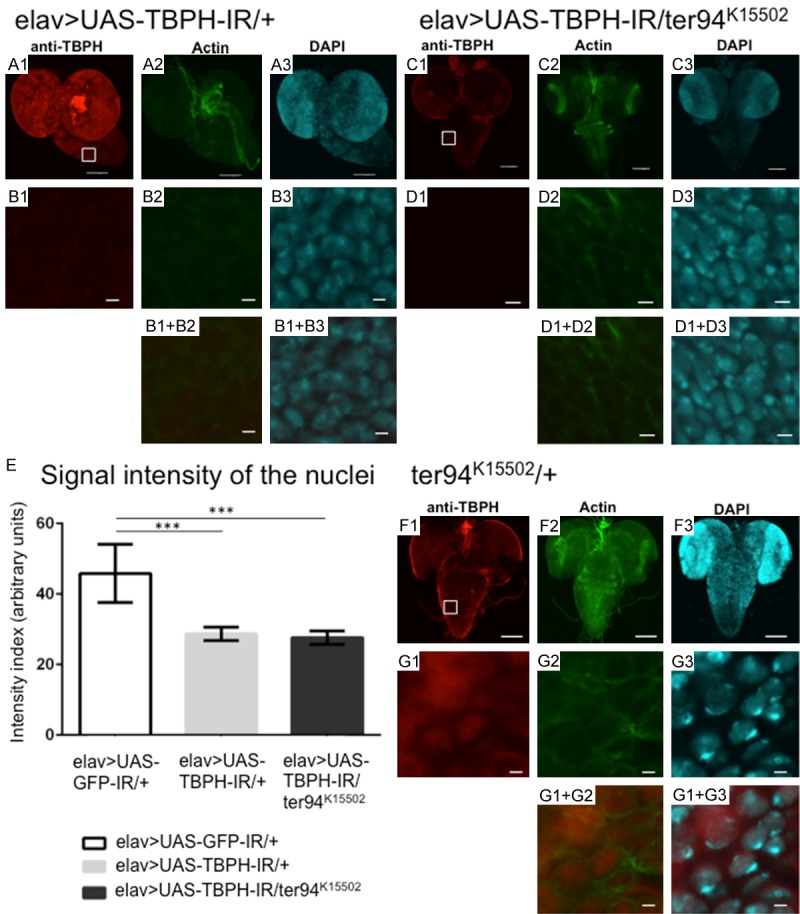
A loss-of-function ter94 mutation does not change the decreased TBPH signal intensities in nuclei. (A-G) Are representative images of corresponding genotypes. (A1) to (A3) are the BVGCs of TBPH knockdown larvae carrying yw/Y; UAS-TBPH-IR517-531/+; elav-GAL4/+ (elav>UAS-TBPH-IR/+). (C1-C3) Are the BVGCs of larvae that co-express ter94K15502 on the background of TBPH knockdown carrying yw/Y; UAS-TBPH-IR517-531/ter94K15502; elav-GAL4/+ (elav>UAS-TBPH-IR/ter94K15502). (F1-F3) Are the BVGCs of larvae carrying yw/Y; ter94K15502/+; + (ter94K15502/+) in the heterozygous state. Panels (B1) to (B3), (D1) to (D3), (G1) to (G3) are higher magnification images of the boxed area in (A1) and (C1), respectively. (B1+B2, B1+B3, D1+D2, D1+D3, G1+G2 and G1+G3) Are merged images. The indirect immunofluorescence in (A1, B1, C1, D1, and F1) is signal from the polyclonal anti-TBPH antibody. The fluorescence in (A2, B2, C2, D2 and F2) is from phalloidin, which labels actin. The fluorescence in (A3, B3, C3, D3 and F3) is from diamino-2-phenylidole dihydrochloride (DAPI), which labels DNA. The signal intensity from TBPH in the BVGC was remarkably reduced in larvae carrying elav>UAS-TBPH-IR/+ (A1). The intensity of the nuclear TBPH signal was significantly reduced in the BVGCs of larvae carrying elav>UAS-TBPH-IR/+ (B1) compared to that of driver control larvae (Figure 8B1) (P < 0.001, E). (E) This graph plots the mean (± SE) of the intensity of the nuclear TBPH signal in BVGCs from third instar larvae as fluorescence emission in arbitrary units for each genotype. Larvae carrying the loss-of-function allele of ter94 and neuron-specific TBPH knockdown (elav>UAS-TBPH-IR/ter94K15502) (C1) also showed remarkably reduced TBPH signal intensities in the BVGCs. The intensity of the nuclear TBPH signal did not change in larvae with the loss-of-function allele of ter94 and TBPH knockdown (D1), even compared to that of the TBPH knockdown larvae (B1) (P = 0.2, E). TBPH signal intensities in the nuclei of the CNS of larvae carrying a loss-of-function ter94 mutation (G1) were almost the same as TBPH signal intensities in the nuclei of the CNS of larvae carrying elav>UAS-GFP-IR/+ (Figure 8B1). The scale bars indicate 100 μm (A1 to A3, C1 to C3, and F1 to F3) and 5 μm (B1 to B3, D1 to D3, and G1 to G3). Columns and horizontal bars show the mean and SE of 15 nuclei, respectively. ***P < 0.001.
Next, we analyzed TBPH expression and localization in the CNS of larvae carrying a heterozygous loss-of-function ter94 mutation and neuron-specific TBPH knockdown. Larvae carrying the loss-of-function allele of ter94 and neuron-specific TBPH knockdown, yw; UAS-TBPH-IR/ter94K15502; elav-GAL4/+ (Figure 9, elav>UAS-TBPH-IR/ter94K15502, Figure 9C1) (intensity units = 27.63±0.61, measured in Figure 9D1) exhibited almost the same TBPH signal intensities in the BVGCs as larvae carrying elav>UAS-TBPH-IR larvae (Figure 9B1) (P = 0.23, Figure 9E). Larvae carrying the loss-of-function allele of ter94, yw; ter94K15502/+; + (Figure 9, ter94K15502/+, Figure 9G1) without TBPH knockdown exhibited almost the same TBPH signal intensities in the BVGCs as driver control larvae (Figure 8B1). Thus, the loss-of-function allele of ter94 did not affect the protein levels of TBPH in the nucleus of TBPH knockdown flies, presumably because TBPH knockdown alone caused a sufficiently severe decrease in expression of TBPH protein to cause neuronal dysfunction in those flies.
Discussion
In the present study, we demonstrated for the first time overexpression of ter94 improved the phenotypes of fly lines with neuron-specific TBPH knockdown. Overexpression of wild-type ter94 significantly suppressed the morbid phenotypes induced by TBPH knockdown such as rough-eye phenotypes, locomotive disabilities, and degeneration of MN synaptic terminals. On the other hand, the loss-of-function allele of ter94 (ter94K15502) enhanced the rough-eye phenotypes and locomotive dysfunction induced by TBPH knockdown in the early stage of adult flies. Considering the molecular basis of the interaction between TBPH and ter94, we demonstrated that overexpression of wild-type ter94 restored the reduction of intranuclear TBPH caused by TBPH knockdown. These observations are similar to the effects of overexpression and loss-of-function of ter94 on phenotypes due to Caz knockdown as we previously reported [29].
TDP-43 (TBPH) and FUS (Caz) share many structural similarities. Both proteins are involved in various aspects of mRNA metabolism including splicing, nucleocytoplasmic shuttling, transcription, mRNA stability, and stress granule dynamics [25]. TDP-43 is normally present in the cytoplasm at low levels, where it is found in a large heterogeneous nuclear ribonucleoprotein (hnRNP) complex as a nucleocytoplasmic shuttling molecule [53-55]. On the other hand, VCP (ter94) is a hexamer ATPase that is required for protein degradation during endoplasmic reticulum stress. VCP regulates diverse cellular functions including autophagy, DNA repair, membrane fusion, ubiquitin-mediated protein degradation, nucleocytoplasmic shuttling, and others [31-33,56]. Recently, the GGGGCC (G4C2) repeat expansion in a noncoding region of C9orf72 has been demonstrated as the most common cause of familial forms of ALS. Substantial evidence shows that a primary consequence of G4C2 repeat expansion is compromised nucleocytoplasmic shuttling through the nuclear pore, revealing a novel mechanism of neurodegeneration [55,57]. Recent studies also provided a list of C9orf72 modifiers, some of which are related to nucleocytoplasmic shuttling [53]. In recent studies, restoration of nucleocytoplasmic shuttling rescues neurodegeneration in yeast, fly, and mouse models of ALS/FTLD [53,54,58]. Thus, nucleocytoplasmic shuttling may be a novel and promising therapeutic target for treating ALS/FTLD [59].
Our results demonstrate a genetic link between TBPH and ter94, the Drosophila orthologs of TDP-43 and VCP, respectively. Genetic interaction between human TDP-43 and ter94 was previously reported. The combination of ter94 with TDP-43 resulted in mild enhancement of rough eye phenotype induced by expression of TDP-43. In immunofluorescence analysis in larval salivary glands, Drosophila carrying fkh-GAL4>UAS-TDP-43 with ter94 mutation decreased the protein level of TDP-43 in nucleus [43]. However, these studies with artificially expressed human TDP-43 provided only a limited information in a limited tissue. We therefore examined not only the compound eye phenotypes but also neuron-specific phenotypes. Furthermore, we examined not only loss-of-function mutation of ter94 but also overexpression of wild-type ter94. Our data thus provide further information on the genetic interaction between TDP-43 and VCP in neurons. We propose a working hypothesis for the effect of VCP on the cellular localization of TDP-43 as summarized in the schematic in Figure 10. VCP may directly participate in importing TDP-43 into the nucleus (Figure 10A) and extracting TDP-43 from RNP complexes to permit recycling (Figure 10B) [43]. Our results demonstrate that overexpression of ter94 restores the reduction in TBPH in the nucleus. Wild-type ter94 overexpression may translocate TBPH from the cytoplasm to the nucleus when the amount of TBPH in the nucleus is decreased.
Figure 10.
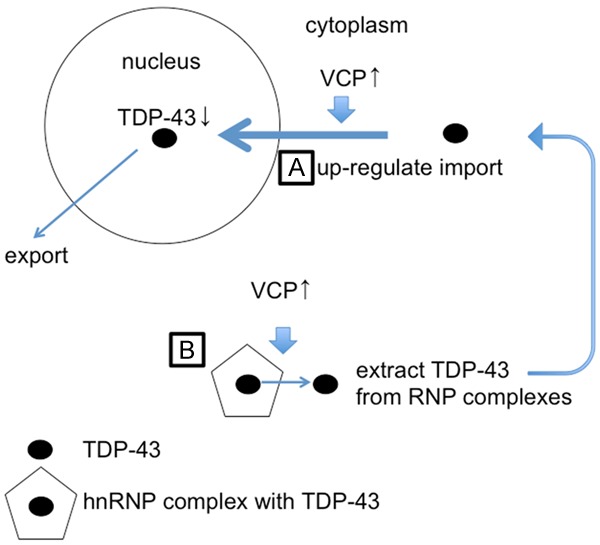
A schematic of the MN cell body showing modified nucleocytoplasmic shuttling. We propose two hypotheses: VCP may directly participate in importing TDP-43 into the nucleus (A), and in extracting TDP-43 from RNP complexes to permit recycling (B).
In summary, we have provided the first evidence that the overexpression of ter94 improves the neurodegeneration induced by TBPH knockdown in vivo. Our data suggest that up-regulation of VCP induces translocation of TDP-43 from the cytoplasm to the nucleus. Up-regulated VCP may suppress the pathogenic processes that lead to the degeneration of MNs in TDP-43-associated ALS/FTLD. This study may provide a basis to support the future development of novel disease-modifying therapies for human ALS.
Acknowledgements
We acknowledge our lab members for their technical assistance and discussions. We would like to thank Dr. Akira Kakizuka for generously providing us the UAS-ter94 fly strains used in these experiments. We acknowledge all members of the Chromosome engineering laboratory for helpful discussion, technical support and suggestions. We thank the Bloomington Drosophila stock center, the Vienna Drosophila RNAi center and Kyoto Drosophila Genetic Resource Center for giving us fly lines. This study was supported by Japan Agency for Medical Research and Development (AMED) (17dk0207030h0002 and 17ek0109222h0001; to T.T.) and supported by JSPS KAKENHI Grant Numbers JP26893227, and JP16K19519; by a Grant for Joint Research Project of The Center for Advanced Insect Research Promotion, Kyoto Institute of Technology; and by a grant-in-aid of The Nakabayashi Trust For ALS Research, Tokyo, Japan (Y.A.). Moreover it was supported by JSPS KAKENHI Grant Numbers JP24659438; by the Strategic Research Program for Brain Sciences and the Practical Research Projects for Rare/Intractable Diseases (Y.N.) from Japan Agency for Medical Research and Development (AMED); a Health Labour Sciences Research Grant for Research on Development of New Drugs to Y.N from the Ministry of Health, Labour and Welfare; Intramural Research Grants for Neurological and Psychiatric Disorders (27-7) from the National Center of Neurology and Psychiatry; an IBC Grant from the Japan Amyotrophic Lateral Sclerosis Association.
Disclosure of conflict of interest
None.
References
- 1.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 2.Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 3.Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:530–534. doi: 10.1001/archneur.64.4.530. [DOI] [PubMed] [Google Scholar]
- 5.Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol. 2014;10:337–348. doi: 10.1038/nrneurol.2014.78. [DOI] [PubMed] [Google Scholar]
- 6.Lemmens R, Moore MJ, Al-Chalabi A, Brown RH Jr, Robberecht W. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 2010;33:249–258. doi: 10.1016/j.tins.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 8.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Bioph Res Co. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 9.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackenzie IR, Feldman HH. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. Neuropathol Exp Neurol. 2005;64:730–739. doi: 10.1097/01.jnen.0000174335.27708.0a. [DOI] [PubMed] [Google Scholar]
- 11.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 12.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 13.Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–474. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- 14.Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 16.Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ, Budka H, Ghetti B, Spina S. TARDBP variation associated with frontotemporal dementia, supranuclear gazepalsy, and chorea. Mov Disord. 2009;24:1843–1847. doi: 10.1002/mds.22697. [DOI] [PubMed] [Google Scholar]
- 17.Pesiridis GS, Lee VM, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:R156–R162. doi: 10.1093/hmg/ddp303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, Lee VM, Schellenberg GD, Yu CE. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- 21.Hewitt C, Kirby J, Highley JR, Hartley JA, Hibberd R, Hollinger HC, Williams TL, Ince PG, McDermott CJ, Shaw PJ. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2010;67:455–461. doi: 10.1001/archneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- 22.Kwiatkowski TJ Jr, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vande MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 23.Rademakers R, Stewart H, DeJusus-Hernandez M, Krieger C, Graff-Radford N, Fabros M, Briemberg H, Cashman N, Eisen A, Mackenzie IR. FUS gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve. 2010;42:170–176. doi: 10.1002/mus.21665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110:1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- 27.Prpar MS, Darovic S, Kovanda A, Bajc CA, Zupunski V, Rogelj B. Nuclear trafficking in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain. 2016;140:13–26. doi: 10.1093/brain/aww197. [DOI] [PubMed] [Google Scholar]
- 28.Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, Guharoy M, De Decker M, Jaspers T, Ryan VH, Janke AM, Baatsen P, Vercruysse T, Kolaitis RM, Daelemans D, Taylor JP, Kedersha N, Anderson P, Impens F, Sobott F, Schymkowitz J, Rousseau F, Fawzi NL, Robberecht W, Van Damme P, Tompa P, VanDen Bosch L. Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell. 2017;65:1044–1055. doi: 10.1016/j.molcel.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azuma Y, Tokuda T, Shimamura M, Kyotani A, Sasayama H, Yoshida T, Mizuta I, Mizuno T, Nakagawa M, Fujikake N, Ueyama M, Nagai Y, Yamaguchi M. Identification of ter94, Drosophila VCP, as a strong modulator of motor neuron degeneration induced by knockdown of Caz, Drosophila FUS . Hum Mol Genet. 2014;23:3467–3480. doi: 10.1093/hmg/ddu055. [DOI] [PubMed] [Google Scholar]
- 30.Sasayama H, Shimamura M, Tokuda T, Azuma Y, Yoshida T, Mizuno T, Nakagawa M, Fujikake N, Nagai Y, Yamaguchi M. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS One. 2012;7:e39483. doi: 10.1371/journal.pone.0039483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braun RJ, Zischka H. Mechanisms of Cdc48/VCP-mediated cell death: from yeast apoptosis to human disease. Biochim Biophys Acta. 2008;1783:1418–1435. doi: 10.1016/j.bbamcr.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 33.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurro MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G The ITALSGEN Consortium. Galassi G, Scholz SW, Taylor JP, Rescagno G, Chio A, Traynor BJ. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583:1586–1592. doi: 10.1016/j.febslet.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 35.Vanden Broeck L, Naval-Sanchez M, Adachi Y, Diaper D, Dourlen P, Chapuis J, Kleinberger G, Gistelinck M, Van Broeckhoven C, Lambert JC, Hirth F, Aerts S, Callaers P, Dermaut B. TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila . Cell Rep. 2013;3:160–172. doi: 10.1016/j.celrep.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 36.Wang JW, Brent JR, Tomlinson A, Shneider NA, Mc Cabe BD. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Invest. 2011;121:4118–4126. doi: 10.1172/JCI57883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi Y, Hirose F, Matsukage A, Yamaguchi M. Identification of three conserved regions in the DREF transcription factors from Drosophila melanogaster and Drosophila virilis . Nucleic Acids Res. 1999;27:510–516. doi: 10.1093/nar/27.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Higashiyama H, Hirose F, Yamaguchi M, Inoue YH, Fujikake N, Matsukage A, Kakizuka A. Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ. 2002;9:264–273. doi: 10.1038/sj.cdd.4400955. [DOI] [PubMed] [Google Scholar]
- 39.Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 40.Ly LL, Suyari O, Yoshioka Y, Tue NT, Yoshida H, Yamaguchi M. dNF-YB plays dual roles in cell death and cell differentiation during Drosophila eye development. Gene. 2013;520:106–118. doi: 10.1016/j.gene.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 41.Romano G, Appocher C, Scorzeto M, Klima R, Baralle FE, Megighian A, Feiguin F. Glial TDP-43 regulates axon wrapping, GluRIIA clustering and fly motility by autonomous and non-autonomous mechanisms. Hum Mol Genet. 2015;24:6134–6145. doi: 10.1093/hmg/ddv330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin MJ, Cheng CW, Shen CK. Neuronal function and dysfunction of Drosophila dTDP. PLoS One. 2011;6:e20371. doi: 10.1371/journal.pone.0020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, Tang W, Winton MJ, Neumann M, Trojanowski JQ, Lee VM, Forman MS, Taylor JP. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010;30:7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanden Broeck L, Kleinberger G, Chapuis J, Gistelinck M, Amouyel P, Broeckhoven C, Lambert JC, Callaerts P, Dermaut B. Functional complementation in Drosophila to predict the pathogenicity of TARDBP variants: evidence for a loss-of-function mechanism. Neurobiol Aging. 2015;36:1121–1129. doi: 10.1016/j.neurobiolaging.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 46.Godena VK, Romano G, Romano M, Appocher C, Klima R, Buratti E, Baralle FE, Feiguin F. TDP43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PLoS One. 2011;6:e17808. doi: 10.1371/journal.pone.0017808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hazelett DJ, Chang JC, Lakeland DL, Morton DB. Comparison of parallel high-throughput RNA sequencing between knockout of TDP-43 and its overexpression reveals primarily nonreciprocal and nonoverlapping gene expression changes in the central nervous system of Drosophila . Genes Genomes Genetics. 2012;2:789–802. doi: 10.1534/g3.112.002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang HC, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, Artavanis-Tsakonas S. Modeling spinal muscular atrophy in Drosophila. PLoS One. 2008;3:e3209. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chee F, Mudher A, Newman TA, Cuttle M, Lovestone S, Shepherd D. Overexpression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Biochem Soc Trans. 2006;34:88–90. doi: 10.1042/BST0340088. [DOI] [PubMed] [Google Scholar]
- 50.Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila . Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 51.Lloyd TE, Taylor JP. Flightless flies: Drosophila models of neuromuscular disease. Ann N Y Acad Sci. 2010;1184:E1–E20. doi: 10.1111/j.1749-6632.2010.05432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shcherbata HR, Yatsenko AS, Patterson L, Sood VD, Nudel U, Yaffe D, Baker D, Ruohola-Baker H. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. EMBO J. 2007;26:481–493. doi: 10.1038/sj.emboj.7601503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ward ME, Taubes A, Chen R, Miller BL, Sephton CF, Gelfand JM, Minami S, Boscardin J, Martens LH, Seeley WW, Yu G, Herz J, Filiano AJ, Arrant AE, Robertson ED, Kraft TW, Farese RV Jr, Green A, Gan L. Early retinal neurodegeneration and impaired Ran-mediated nuclear import of TDP-43 in progranulin-deficient FTLD. J Exp Med. 2014;211:1937–1945. doi: 10.1084/jem.20140214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu S, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang T, Xu W, Qin M, Yang Y, Bao P, Shen F, Zhang Z, Xu J. Pathogenic mutations in the valosin-containing protein/p97 (VCP) N-domain inhibit the SUMOylation of VCP and lead to impaired stress response. J Biol Chem. 2016;291:14373–14384. doi: 10.1074/jbc.M116.729343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016;132:159–173. doi: 10.1007/s00401-016-1586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: Implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1249:202–211. doi: 10.1016/j.brainres.2008.10.021. [DOI] [PubMed] [Google Scholar]